

Abstract
A new strategy to prepare antimicrobial surfaces by a simple dip-coating procedure is reported. Amphiphilic polycations with different mole ratios of monomers containing dodecyl quaternary ammonium, methoxyethyl, and catechol groups were synthesized by free-radical polymerization. The polymer coatings were prepared by immersing glass slides into a polymer solution and subsequent drying and heating. The quaternary ammonium side chains endow the coatings with potent antibacterial activity, while the methoxyetyhyl side chains enable tuning the hydrophobic/hydrophilic balance and the catachol groups promote immobilization of the polymers into films. The polymer coated surfaces displayed bactericidal activity against Escherichia coli and Staphylococcus aureus in a dynamic contact assay and prevented accumulation of viable E. coli, S. aureus, and Acinetobacter baumannii for up to 96 hours. Atomic force microscopy (AFM) images of coating surfaces indicated that the surfaces exhibit virtually the same smoothness for all polymers except the most hydrophobic. The hydrophobic polymer without methoxyethyl side chains showed clear structuring into polymer domains, causing high surface roughness. Sum-frequency generation (SFG) vibrational spectroscopy characterization of the surface structures demonstrated that the dodecyl chains are predominantly localized at the surface-air interface of the coatings. SFG also showed that the phenyl groups of the catechols are oriented on the substrate surface. These results support our hypothesis that the adhesive or cross-linking functionality of catechol groups discourages leaching of polymers, allowing tuning of the amphiphilic balance by incorporating hydrophilic components into the polymer chains to gain potent biocidal activity.
Keywords: polycation, antimicrobial, catechol, coating, amphiphilic
Introduction
The development of new materials that can effectively inhibit the growth of microbes and prevent bacterial accumulation on surfaces has received increasing attention because 60-70% of nosocominal microbial infections are associated with medical devices and implants.1 A common strategy to prepare such surfaces is to incorporate biocidal agents such as antibiotics,2 triclosan,3 surfactants,4 and silver nanoparticles5 in coating matrices. These active compounds are then released from the surface to kill bacteria in solution or a proxy of the surface.6 Alternatively, certain biocides such as quaternary ammonium salts can be permanently immobilized to create surfaces, which kill bacteria on contact.7-12 Synthetic polymers containing alkyl quaternary ammonium groups in their side chains have been widely applied as on-contact killing surfaces on substrates including glass,13 plastics,14 and cellulosic filter papers.15
Covalent attachment of these polycations to the surface can be achieved by grafting polymer chains from initiators fixed on surfaces (surface-initiated polymerization)7, 8, 16 or grafting polymer chains to functional groups on the surfaces by coupling reactions7. Using these strategies with defined covalent bonding, the effect of polymer compositions and conformations on the activity have been systematically examined.7 Although these methods have been useful for the application of existing polymers,8 laborious and costly surface modification is required for polymer attachment. On the other hand, a one-step paint-like coating procedure was reported by Klibanov and co-workers using highly hydrophobic polycations.17 These water-insoluble N-alkyl-poly(ethyleneimine)s (PEIs) were obtained by the modification of PEIs with long alkyl chains, which were coated onto glass or polyethylene slides simply by dipping the substrates into the organic solutions of the polymers and drying.17,18 The strong hydrophobicity of polymers prevented leaching of polymers in aqueous solutions, and the polycations on the surface killed bacteria on contact. Although this approach provides a facile and inexpensive coating procedure, the method is strictly limited to highly hydrophobic polymers. This limits their utility because fine-tuning the balance between hydrophilic and hydrophobic properties is essential to optimize the activity of antimicrobial polymers on surfaces and in solutions.19 Hence, it would be highly desirable to extend the one-step coating procedure to include hydrophilic components.
To that end, we investigated amphiphilic polycations containing a catechol derivative, which is known to adhere to substrates readily, for the facile preparation of permanent biocidal coatings. The catechol derivative is a synthetic mimic of the natural amino acid 3,4-dihydroxyphenylalanine (DOPA) found in the mussel adhesive protein.20 It is generally accepted that the catechol group can form covalent bonds, hydrogen bonds, or strong physical interactions with surfaces, as well as can cross-link polymer chains, affording robust coatings.20-27 Synthetic polymers containing DOPA and DOPA analogues have shown strong interfacial adhesion strength for a variety of material surfaces and conditions including wet surfaces.21-23,28 Jiang and coworkers reported non-fouling polyampholytes from initiators with DOPA, which form polymer films and prevent adsorption of proteins.29 Based on these previous reports, we hypothesized that the catechol groups would promote immobilization of amphiphilic polycation chains onto surfaces without requiring any prior surface modifications. We further hypothesized that this approach would enable incorporation of hydrophilic segments into the polymers, which might otherwise leach from the surface. This would allow optimizing the amphiphilic balance of polycations for maximizing the antimicrobial activity without compromising the adhesion properties of polymers or stability of coatings. To test these hypotheses, we have synthesized random terpolymers composed of monomers with alkyl quaternary ammonium salts (QA) as the biocidal component, methoxyethyl side chains as a hydrophilic group, and catechols as the anchoring moieties. In this study, we tested coatings on glass surfaces as an initial assessment of feasibility of this polymer design. Their wettability, surface morphology, polymer orientation, and antibacterial activity of the polymer coatings on glass slides establish a proof of principle for the facile preparation of biocidal coatings with optimal antimicrobial effectiveness.
Experimental Section
Materials
Dopamine-HCl, 1-bromododecane, and methoxyethyl acrylate (MEA) were purchased from Acros. 2, 2′-Azobisisobutyronitrile (AIBN) and 2-(dimethylamino)ethyl methacrylate (DMAEMA) were purchased from Sigma-Aldrich (St. Louis, MO). Methacrylate anhydride was purchased from Polysciences (Warrington, PA). Reagent grade solvents were purchased from Fisher (Hanover Park, IL). These chemical agents and solvents were used without further purification. Pre-cleaned glass slides (cat. 12-552-3) were purchased from Fisher. A live/dead Baclight bacterial viability kit was purchased from Invitrogen (L-7007, Carlsbad, CA). Mueller-Hinton (MH) broth and agar were obtained from Difco Laboratories (Franklin Lakes, NJ). The monomer dopamine methacrylamide (DMA) was prepared by the procedure described by Lee et al. 21 (Supporting Information). The dodecyl QA monomer (DMAEMAC12) was prepared by the procedure described by Ravikumar et al. 30 (Supporting Information). Gel permeation chromatography (GPC) was carried out using a Waters 440 GPC, a Wyatt Optilab Refractive Index detector, and Waters’ Millennium software for GPC data acquisition and processing with an HT-4, HT-3, and HT-2 3 columns placed in series. The molecular weights were determined relative to narrow molecular weight polystyrene standard.
Synthesis of copolymers Poly(DMA-MEA-DMAEMAC12)
A series of random copolymers were prepared by altering the mole ratio of DMA, MEA and DMAEMAC12 (Scheme 1). The total amount of monomers was kept constant at 2 mmol. The mol % of DMA was fixed at 15% in the feed monomer compositions. All monomers (typically, 665 mg for DMA, 0-1882 mg for MEA, and 1221-6910 mg for DMAEMAC12) were dissolved in DMF (10 mL) and then 2, 2′-azobisisobutyronitrile (AIBN) (33 mg, 0.2 mmol) was added. The solution was bubbled with N2 for 5 minutes and heated at 60 °C overnight. The resultant polymers were precipitated in diethyl ether (400 mL). The polymer was dissolved in a small amount of methanol and precipitated again in diethyl ether. This procedure was repeated three times. The molecular weight of polymers was determined by GPC after hydrolysis of side chains in the presence of acid or base (Supporting Information).
Scheme 1.

Polymer Synthesis
Preparation of polymer coatings
Glass slides were cut into small pieces (1.0 cm × 2.5 cm) and rinsed with deionized water and isopropanol. These glass slides were immersed into a polymer solution (50 mg/mL, ethanol) for 1 min and then dried by air. In this procedure, the both sides of slides were coated, giving a coated area of total 5.0 cm2. The coated glass slides were heated at 70 °C overnight.
Static contact angle measurement
Static contact angle on the coated or non-coated glass surfaces was measured by a Cam-100 Optical Contact Angle Goniometer (KSV Instruments Ltd., Monroe, CT) at room temperature using deionized water (2.0 μL). For each polymer coating sample, five to ten measurements from different spots in the same surface were taken, and three to five different samples were tested. The average of these values was reported as a contact angle.
Atomic force microscopy
Topographic images were obtained using a PicoSPM atomic force microscope (Molecular Imaging, Inc). Measurements were taken in tapping mode using silicon cantilevers (K-Tek International, Inc). A typical force constant was 0.12N•m−1 and a scanning rate of 2.0 lines per second was used. Images were obtained under ambient conditions at room temperature and were processed by the Scanning Probe Imaging Processor software (Image Metrology).
SFG vibrational spectroscopy
The details of experimental setup and procedures for SFG measurements have been described in the previous reports.31-35 Thin polymer films were prepared by spin-coating a 2 wt% solution of P1, P2, P3 or P4 dissolved in ethanol onto fused silica substrates using a spin-coater from Specialty Coating Systems (Indianapolis, IN). The samples were subsequently placed in a 110 °C oven for at least 1 hr prior to analysis to ensure removal of any residual solvent. The visible and infrared (IR) input beams travel through the fused silica substrate to overlap spatially and temporally on the polymer-air interface or the polymer-water interface at input angles of 60° and 54°, respectively. The pulse energies were approximately 200 and 100 μJ respectively, with beam diameters of 500 μm. SFG spectra were collected using the ssp (s-polarized sum frequency output, s-polarized visible input and p-polarized IR input) polarization. Signals were normalized by the intensities of the input visible and IR beams.
Dynamic contact antimicrobial assay
Antimicrobial activity of polymer coatings was determined by a modified standard protocol: ASTM E2149-01 Standard Test Method for Determining the Antimicrobial Activity of Immobilized Agents Under Dynamic Contact Conditions 9,36. The Escherichia coli ATCC 25922 or Staphylococcus aureus ATCC 25923 was grown in Mueller-Hinton (MH) broth (5 mL, pH 7.4) at 37°C overnight. The cell culture was diluted with MH broth to give OD600 = 0.1, which was incubated at 37°C with orbital rotation at 200 rpm for 90 minutes for E. coli or 140 minutes for S. aureus. The bacterial culture in the mid-logarithmic phase (OD600 = 0.5-0.6) was diluted to OD600 = 0.1 by MH broth, and further diluted by Phosphate Buffer Saline (PBS, pH 7.4, without calcium and magnesium) serially (OD600 = ~0.001, corresponding to ~2×105 CFU/mL). The polymer-coated glass slides were completely immersed into this bacterial suspension (5mL) in a sterile 15 mL conical tube. The conical tubes were incubated with gentle shaking (200rpm) at 37 °C for 1 hr. A non-coated glass slide was used as a control. An aliquot of the bacterial sample solution (100μL) was diluted serially, plated on the agar plates, and incubated at 37 °C overnight to count the number of bacterial colonies. Three samples for the polymer coating or control glass slide were tested, each in triplicate, and three independent experiments were performed. The average numbers of viable cells (colony-forming unit, cfu) in solution and standard deviations were calculated from the results of all experiments.
Live/Dead BacLight viability stain
A LIVE/DEAD BacLight Bacterial Viability kit (L-7007, Invitrogen, Carlsbad, CA) was used to determine bacterial cell viability. Polymer-coated glass slides were prepared by the same procedure as described above, except glass slides with an original size (7.5 cm × 2.5 cm) were used. A solution of the mixed SYTO 9 and PI dyes was prepared according to the manufacturer’s instruction. The bacterial suspension (1 mL, ~108 cfu/mL) was mixed with the fluorescent dyes (10 μL) for 15 min. This bacterial suspension with the dyes (10 μL) was dropped onto the polymer-coated glass slide or un-treated glass slide (control), and a glass coverslip (2.5 cm × 2.5 cm) was placed on the droplet. This slide sample was incubated with shaking at 200 rpm for 1 hour at 37°C. The slide was also place in dark to avoid bleaching of fluorescent dyes by ambient light. The slide was examined using a fluorescence microscope (Olympus 1X71, Center Valley, PA) equipped with Fluorescence Illumination System (X-Cite 120, EXFO) and appropriate filter sets. Images were obtained using an oil immersion 60 × objective len.
Biofilm development on coatings
A polymer (50 mg/mL) solution in ethanol (50μL) was dropped into glass bottom wells of 96-well microtiter plate (MatTek Corp. Ashland, MA) and dried by air to remove the solvent. Overnight bacterial culture was diluted 100 times with fresh 10% Luria Bernati (LB) medium, and the diluted bacterial culture (100μL) was transferred to the polymer-coated wells of the microtiter plate. The microtiter plate was incubated at 30 °C. At the incubation time points of 12, 24, 48, and 96 hours, the bacterial culture in the well was removed by a pipette, and the well surface was washed gently 3 times with PBS buffer (150 μL) to remove unattached bacterial cells. The remaining attached biofilm cells were scraped from the well and resuspended in 100 μL of PBS buffer. A serial of 10-fold dilutions of this bacterial suspension were plated onto a LB agar plates and incubated at 37 °C overnight for colony counting.
3D imaging of biofilm development on coatings
For visualizing bacteria attached to surfaces or in biofilms, the same bacterial culture was incubated in polymer-coated wells of microtiter plate as described above. The supernatant was removed, and the well surface was rinsed briefly with PBS buffer three times. PBS buffer (100μL) containing 1 μM of SYTO 9 and 10 μM of propidium iodide (PI) was added into the well. The plate was incubated for 15 min in dark. Fluorescent images were acquired with an Olympus Fluoview™ FV1000 confocal microscope (Olympus, Markham, Ontario) with Melles Griot Laser supply and detectors and filter sets for monitoring SYTO 9 and PI fluorescence. Images were obtained using an oil immersion 60 × objective len. Three dimensional images were reconstructed using the Amira software package (Amira, San Diego, CA) from a stack of 33 (control) or 8 (P2) sectional images of biofilm samples.
Results and discussion
Polymer design and synthesis
We have synthesized amphiphilic, cationic copolymers (P0-P4) with different mole ratios of monomers containing alkyl quaternary ammonium (DMAEMAC12), methoxyethyl (MEA), and catechol (DMA) groups by free-radical polymerization. (Scheme 1, Table 1). A series of random copolymers without DMA (P5-P8) were also prepared for comparison, to elucidate the role of the catechol groups (Table 1). The mole percentage of DMA groups in the polymers range from 11 to 13 %, which are comparable to the adhesive random copolymers previously reported by Lee et. al (11 mol %).21 For antimicrobial activity, we functionalized these copolymers with dodecyl quaternary ammonium (QA) groups, which have been previously used for antibacterial9,37,38 and antifungal30 coatings. The hydrophilic MEA monomer has also been used for preparation of anti-fouling and hemocompatible polymer coatings because of its hydrophilic and neutral properties, which are expected to reduce non-specific adhesion of proteins and accumulation of bacterial cells on the surfaces.39, 40 The amphiphilic balance of copolymers can be controlled by altering the ratio of the DMAEMAC12 and MEA monomers, enabling examination of their effect on the activity of the polymers. Similar to the polymer structures reported here, polyurethane random copolymers with dodecyl QA and methoxyethyl groups in side chains have been prepared as a polymer-surface modifier; these soft block polyurethanes are concentrated at the surfaces of bulk polyurethanes. The coatings of these polymers also displayed the ability to kill bacteria on contact.37, 38
Table 1.
Polymer characterization
| Polymer | Monomer compositiona (mol. %) | ||||
|---|---|---|---|---|---|
| DMA | MEA | DMAEMAC12 | Yield (%) |
Mnb (×103) |
|
| w/ catechol | |||||
| P0 | 11 | 89 | 0 | 69 | n.d. |
| P1 | 12 | 72 | 16 | 70 | 39 c |
| P2 | 12 | 45 | 43 | 75 | 30c |
| P3 | 13 | 31 | 56 | 72 | 28c |
| P4 | 12 | 0 | 88 | 80 | 30c |
| w/o catechol | |||||
| P5 | 0 | 73 | 27 | 79 | 29d |
| P6 | 0 | 44 | 56 | 70 | 20d |
| P7 | 0 | 26 | 74 | 77 | 28d |
| P8 | 0 | 0 | 100 | 80 | 33d |
The monomer compositions were determined by 1H NMR analysis.
The molecular weight of polymers hydrolyzed by acid or base. Mn was determined by GPC relative to polystyrene standards.
The polymers were hydrolyzed by acid.
The polymers were hydrolyzed by base.
The molecular weights of the polymers could not be determined by gel permeation chromatography due to the low solubility in GPC solvents (DMF and THF). Therefore, the side chains of polymers were hydrolyzed in the presence of acid or base, and then the molecular weights of resultant poly(acid)s were determined by GPC (Table 1). All hydrolyzed polymers have the similar molecular weights in the range of 20-40 kDa (Table S1 in Supporting Information), indicating that the molecular sizes of polymer chain backbone are comparable for all polymers studied here. For preparation of coatings, glass slides were immersed into a polymer solution in ethanol, dried by air, and then heated at 70 °C overnight.
Water static contact angle
In order to quantify the effect of comonomer composition on the hydrophobicity of the obtained surfaces, we measured their static water contact angles. The contact angles of coating surfaces exceeded that of the control (unmodified glass, contact angle = 42.3±0.1°) by more than two-fold for all polymers (Fig. 2). The static contact angles of the polymers lacking catechol groups of DMA (P5-8) increased with increasing DMAEMAC12 content. The P8 coating, which has no hydrophilic MEA groups, displayed the largest contact angle. The polymers with catechol groups showed only slight differences as the DMAEMAC12 content was adjusted between 64 and 0 mol% (Fig. 1). This result indicates that the wettability of the coatings containing DMA is not strongly dependent on the comonomer compositions in this range. The P4 coating, which lacks MEA, however, displayed a significantly higher contact angle than the others, indicating some amount of hydrophilic units are necessary to tune the surface wettability. It is interesting that, above 50% DMAEMAC12, the polymers without DMA displayed larger contact angles than the polymers containing DMA, indicating the coatings of polymers without the catechol groups have more hydrophobic surfaces.
Figure 2.

AFM images of polymer P1-P4 coatings and surface roughness. Polymers were spin-coated onto a fused silica window substrate and cured for 1 hr at 120 °C. Images are 5 μm by 5 μm.
Figure 1.
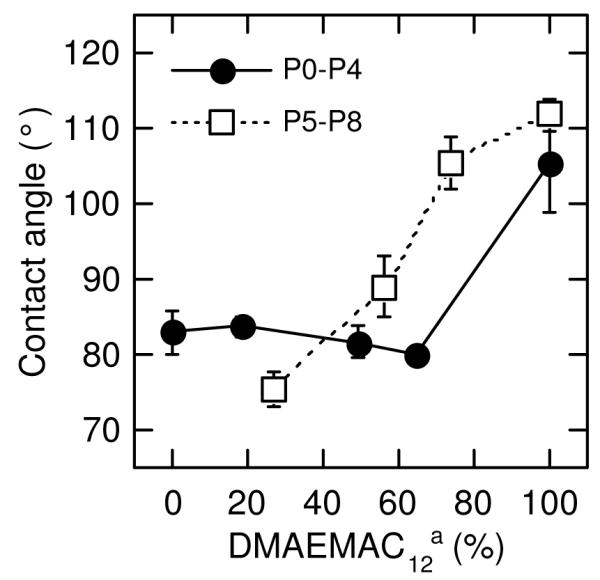
Water static contact angle of polymer coatings. The static contact angle of control surface (uncoated glass slides) was 42.3±0.1°. a DMAEMAC12 percentages were calculated based on the mol.% of DMAEMAC12 and MEA using the equation of 100 × DMAEMAC12 (%) /( DMAEMAC12 (%)+ MEA (%))
The dependence of surface wettability of the polymers containing DMA (P0-P4) on monomer compositions might be due to the catechol functionality. The catechol side chains could anchor the polymers onto glass surfaces and cross-link them, and allowing swelling of the polymer network at the polymer-water interface. The methoxyethylene groups may be exposed to water at the polymer-water interface and the dodecyl chains of QA groups may be aggregated and embedded in the coatings. Therefore, the MEA could be responsible for hydrophilicity of the coatings P0-P3, which is not dependent on the amount of dodecyl QA that are removed from the interface.7 On the other hand, the polymer chains lacking the catechol groups (P5-P8) could adapt their confirmations without restriction of cross-linking by the catechol groups and also leach into solution, which is proven later, although the polymer samples appeared to be insoluble to water. Therefore, the wettability of these polymers lacking the catechol groups is closely related to the ratio of DMAEMAC12 to MEA, that is, the amphiphilicity or solubility of the polymers. At high DMAEMAC12 ratios, the dodecyl QA groups are likely to dominate the wettability, giving hydrophobic surfaces.
Surface morphology of polymer coatings
To investigate the surface morphology of polymer coatings, AFM images were obtained for each of the polymers spin-coated onto fused silica window substrates (Fig. 2). The surfaces of P1, P2 and P3 are similarly smooth, with no structures of significant note. In contrast, P4, which contains only DMA and DMAEMAC12, showed clear structuring into polymer domains. The analysis of the images indicate that the mean root square roughness of the coating surface is 1-2 nm for P1-3 and ~4 nm for P4. The higher roughness of P4 coating is likely due to the close packing of dodecyl chains of QA groups, forming the segregated domains. It has been demonstrated that smooth and hydrophilic surfaces prevent bacterial adhesion.41 Accordingly, incorporation of hydrophilic groups into the polymer chains would be advantageous for discouraging strong bacterial adhesion to the coating surfaces. Accumulation of bacteria on the surface would be expected to diminish the antimicrobial effectiveness after prolonged exposure. The antimicrobial activity of surfaces discussed later appears to confirm that the smooth, hydrophilic surfaces (P2, P3) enable prolonged antimicrobial activity.
Surface structures and organization of polymer chains
SFG vibrational spectroscopy was used to characterize the surface structures and organization of polymer chains on the surface (Fig. 3).42, 43 The peak at ~2880 cm−1 is attributed to the symmetric stretch of the terminal CH3 group of the long alkyl chain. The peak at ~2945 cm−1 is attributed to the CH3 Fermi resonance. The third peak, located at ~2850 cm−1 in each spectrum, is attributed to the symmetric stretch of a single CH2 in the dodecyl chain of the QA groups. Because of the symmetry requirements of SFG, only one CH2 group of dodecyl chains of QA groups can generate observable SFG signal. The presence of these clear peaks indicates that the dodecyl chains are predominantly localized at the surface-air interface. The relative intensities of the methyl and methylene peaks increased as the content of dodecyl QA groups in DMAEMAC12 was increased from 16 to 88% (P1 to P4). This seems to suggest that the orientation of dodecyl chains was enhanced by increasing the dodecyl QA content due to closer packing of the dodecyl chains. This result was also supported by the AFM images, which indicated segregation for polymers with high dodecyl QA content (P4) into domains. On the other hand, when the coating is submerged in water, the SFG signals at the water-coating interface disappeared, indicating the randomization of polymer chains on the surface, which is likely due to the swelling of amphiphilic polymer coatings in contact with water (Fig. 4). This process is reversible upon drying of the surfaces, recovering the SFG signals. This reversibility may be facilitated by the catechol groups which anchor and cross-link the polymer chains. As such, these connections may enable the films to recover to their initial state even after swelling in contact with water. The result also suggests that the polymers are not leached or removed from the coatings by wetting.
Figure 3.
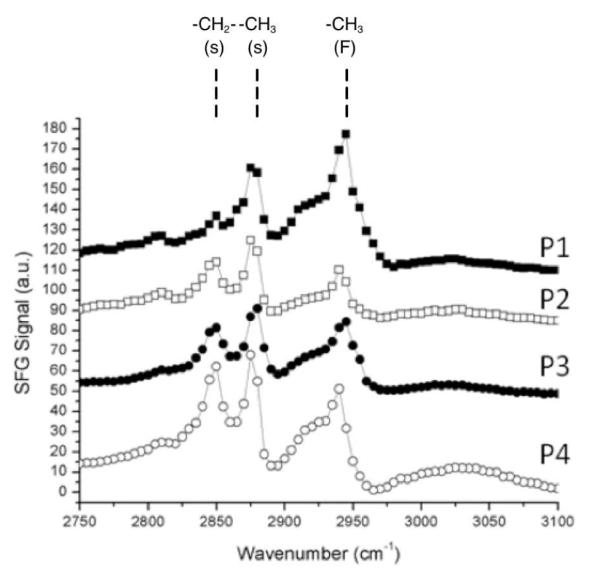
SFG spectra of the polymer/air interface in ssp polarization. Spectra are offset for comparison. s: symmetric stretch; F: Fermi resonance.
Figure 4.

SFG spectra of P2 coating surface. a) in air; b) in contact with water; and c) in air, after removal from water contact.
Another small peak at ~3040 cm−1 is both weak and quite broad, and can be attributed to the catechol group in DMA.44 Although phenyl rings such as these are inherently low-SFG signal-bearing moieties, a clear signal relative to the other peaks indicates that the phenyl rings could be orienting in a predictable manner on the substrate (fused silica). This could be due to anchoring of the phenyl OH groups to the fused silica substrate, causing the phenyl rings to orient roughly parallel to the surface normal. The details of adhesive mechanism of the catechol groups in the polymers studied here are, however, unclear at this point.
Antimicrobial activity of polymer coatings
The antimicrobial activity of the polymer coatings was examined in a dynamic solution condition, in which the coated glass-slides were submerged in bacterial dispersions and incubated with gentle shaking (Table 2). In this assay, the bacterial dispersion was prepared in PBS buffer, which is a bacterial non-growth medium. The P0 coating, which contains no DMAEMAC12 did not cause any reduction of E. coli and S. aureus populations as compared to the negative control (uncoated glass slides). On the other hand, the coatings which contain the biocidal DMAEMAC12 groups killed both strains of bacteria in the assay, indicating that the dodecyl QA groups are essential for the antimicrobial activity of the coatings. The activity against E. coli depends on the ratio of DMAEMAC12 and MEA. No detectable E. coli cells remained in suspension for the P2 and P3 coatings, which implies complete killing. The P1 and P4 coatings induced only a fraction of reduction in the number of viable E. coli cells. Hence, it appears that an optimal ratio of cationic, amphiphilic units (DMAEMAC12) to hydrophilic units (MEA) is necessary to achieve potent antimicrobial activity. P2 and P3 appear to give the optimal balance of hydrophilic/hydrophobic nature for excellent biocidal activity. The polymers also showed the same trend in their activity against S. aureus although they appear to be somewhat less effective as compared to E. coli. The coatings of the polymers containing no DMA (P5-8) displayed no viable E. coli or S. aureus cells detected in the solutions, suggesting complete killing.
Table 2.
Dynamic contact antimicrobial activity of polymer coatings
| Polymer coatings |
Viable cells (105 cfu/mL) |
|
|---|---|---|
| E. colic | S. aureusc | |
| Control a | 3.6±0.4 | 3.8±0.6 |
| w/ catechol | ||
| P0 | 3.8±3.0 | 3.9±0.7 |
| P1 | 1.7±0.4 | 3.5±0.4 |
| P2 | 0 b | 1.2±0.4 |
| P3 | 0 b | 0.3±0.2 |
| P4 | 2.3±0.5 | 1.1±0.5 |
| w/o catechol | ||
| P5 | 0 b | 0 b |
| P6 | 0 b | 0 b |
| P7 | 0 b | 0 b |
| P8 | 0 b | 0 b |
Uncoated glass slide.
A portion of assay solution was plated without dilution, and no colony formation was observed.
The initial concentration of bacteria was ~2 × 105 cfu/mL.
To determine whether any antimicrobials leached from the coatings to kill bacteria in solution, or the coatings kill bacteria on contact, the coated glass slides were soaked in PBS buffer without bacteria, and this solution was added into an E. coli suspension. If the polymers were leached from the coatings into solution, the biocidal activity of buffer containing the polymers could be detected. The PBS buffer solution incubated with the coatings of polymers P1-4 showed no activity, although the same assay showed 100% killing of E. coil for P5-8, which lack the catechol groups (Table S3 in Supporting information). This suggests that the biocidal activity of P5-P8 coatings proceeds by leaching and killing in solution. In contrast, it would seem plausible that the P1-P4 coatings kill bacteria on contact. In addition, we examined the 1H NMR spectra of deuterated water in which the surfaces were submerged, to identify any leached polymers. The results indicate that the polymers P5-P8, which lack catechol groups, are released from the surfaces. In contrast, the NMR spectra indicated that little or no polymer was released from the coatings P1-P4 (Figures S1-S3 in Supplemental Information). These results support the notion that the polymers with the catechol groups form antimicrobial coatings which kill bacteria on contact, whereas the polymer lacking catechol groups leach from the surfaces.
Zones of inhibition were also measured to determine the activity of any leached polymers which diffuse on agar. No zone of inhibition in agar plates was detected for all polymers including the polymers lacking catechol groups although they leached when they are submerged in buffer solution (data not shown. See Supporting Information for the experimental procedure). This indicates that the constant exposure of coatings to aqueous solution causes the release of polymers or the leached polymers may not be able to diffuse though the agar matrix due to their low solubility into water or aggregation.
To determine the maximum antimicrobial capacity of the P2 and P3 coatings in terms of the number of bacterial cells, we further tested the activity of the surfaces against increasing numbers of E. coli cell challenges. The P2 and P3 coatings showed complete killing of E. coli cells up to 1.2×108 and 1.2×107 cfu/mL, respectively (Table S4 in Supporting information). Above these bacterial concentrations, the coatings were able to kill only a fraction of cells. This might be due to saturation of the surfaces by a large number of E. coli cells, above these bacterial concentrations, effectively shielding the antimicrobial activity of polymer chains.
In corroboration with the SFG measurement, the antimicrobial activity of the polymer coatings and NMR measurement of leached polymers support the notion that the catechol functionality discourages or minimizes the release of polymers from their coatings. Since no surface modifications were employed, the polymers seem to leach into solution when they lack the catechol groups. Furthermore, the catechol groups allow introducing hydrophilic components into the polymers without compromising the stability of polymer coatings, to tune the amphiphilic balance of polymers for maximizing their surface activity.
Killing of bacteria on coated surfaces
The viability of bacterial cells on the surface was further examined by staining using the Bacterial Live/Dead fluorescence dyes. For the non-coated glass surface (control), most bacterial cells fluoresced green, indicating these cells were viable (Fig. 5). On the other hand, almost all of the bacterial cells on the polymer coatings fluoresced red, indicating these bacteria cells were membrane damaged or killed. Most of the bacterial cells on the control and polymer-coated surfaces could be removed by rising with buffer solution (Fig. 5). This shows that the bacteria were not able to strongly adhere onto these surfaces, which is advantageous for the surface to retain antimicrobial activity. However, at high bacterial concentrations, the adhesion of a large number of E. coli cells reduces the potency of coatings (Table S4 in Supporting Information).
Figure 5.

Fluorescence micrograph of E. coli (A) and S. aureus (B) on an uncoated (control) and polymer coated slides. The suspensions of bacterial cultures mixed with Bacterial LIVE/DEAD dyes were dropped on slides and examined after 1-hour incubation in dark.
Antimicrobial action of polymer coatings
The antimicrobial mechanism of long-alkyl chain modified poly(vinyl pyridines)s (PVPs) and PEIs immobilized onto surfaces has been postulated. Putatively, the cationic side chains of polymers interact with negatively charged bacterial cell surfaces, and the long-alkyl chains with ammonium salt groups insert to the cell membranes, disrupting the membrane integrity and causing cell death.12, 45 Another mechanism in the literature involves replacement of divalent cations, which stabilize the outer membrane of Gram-negative bacteria, by the cations of the polymer, resulting in membrane disruption. 7,9,13 Although the mechanism of on-contact killing exerted by the polymers in this study remains unexplored, it is likely that membrane disruption is an essential feature of their action.
P4 has the largest percentage of biocidal alkyl ammonium groups of the polymers in this study, however the coating did not display potent antimicrobial activity. Based on the results of water contact angle and SFG measurements, this is likely due to the hydrophobic aggregation of polymer chains at the interface with the aqueous environment, preventing polymer chains from interacting strongly with the bacterial cells. The other copolymer coatings, which contain hydrophilic MEA units, might be sufficiently hydrophilic to extend the polymer chains into the water-coating interface and hence to interact with cell membranes efficiently.46 A similar mechanism was speculated by Tiller et al for poly(vinyl pyridine)s modified with long alkyl chains (C12-C16).12 In addition, the rough surface of P4 coating in the AFM image (Fig. 2) also may enhance the attachment of bacteria onto the surface,41 reducing the antimicrobial efficacy of the polymer while hydrophilic surfaces of other polymers might be unfavorable for the adhesion of bacteria. These results demonstrate that the balance of hydrophilic and hydrophobic moieties is necessary to optimize the activity of the polymers. The inclusion of adhesive catechol groups enables such optimization because polymers with well-balanced amphiphilicity can be coated onto surfaces without surface modifications.
Anti-biofilm development on coatings
We also investigated bacterial biofilm development on the coatings. The polymer coatings were exposed to bacterial solutions of E. coli, S. aureus, or Acinetobacter baumannii for an extended period of time (6-96 hours). It should be noted that the bacterial dispersions in this assay were prepared in a bacterial growth medium (LB broth) in contrast to the non-growth medium (PBS buffer) used for the short-term dynamic contact assay (Table 2). We determined the number of viable cells on the surfaces of polymer coatings. The densities of viable bacteria on the polymer coatings (<106 cfu/cm2) were significantly lower for all polymers up to 96 hours as compared to the control (107-109 cfu/cm2) (Fig. 6). The P2 and P3 coatings are the most potent; few or no viable bacterial cells were detected on the surfaces for up to 96 hours. This corroborates our findings from the dynamic contact killing assay, which indicate that fine tuning the amphiphilic balance of the coatings enables optimization of the antimicrobial potency. Although the dynamic solution testing indicated that the activity of coatings to kill S. aureus on contact is lower than E. coli, these results show that the surfaces more effectively resist the accumulation of viable S. aureus. Together with the results from the dynamic contact assay, the inhibition of bacterial accumulation seems to result from the rapid biocidal activity of the polymer coatings. The rapid bactericidal action (within 1 hour) reduces the number of viable cells available for microcolony formation. However, the surfaces might be eventually saturated by cells, debris, and components of LB broth for 42-96 hours, which deactivate the polymer chains, allowing bacterial growth and accumulation on the coatings.
Figure 6.

Number of viable E. coli (A), S. aureus (B), and A. baumannii (C) cells on an uncoated (control) and polymer-coated glass surfaces after incubation for 6, 12, 24, 48 and 96 hours. * No colony formation was detected.
The bacterial cells on the coating were stained with the Bacterial LIVE/DEAD dyes (Fig. 7). E. coli developed a thick layer of viable cells (green fluorescence) on the control glass surface after 96-hr incubation. In contrast, on the P2-coating surface, only dead cells (red fluorescence) were observed, and the number of cells on the surfaces was significantly lower (p < 0.01). The density and thickness of accumulated bacterial layer on the P2 coating are evidentially much lower than that of the control. This indicates that the coating surface inhibited the accumulation of viable cells efficiently up to 96 hours.
Figure 7.

Three dimensional images of E. coli biofilm on the uncoated (control) and P2-coated glass surface after incubation for 96 hours. The Images were reconstructed from a stack of 33 (control) or 8 (P2) confocal microscopic sectional images using the Amira software package
Conclusions
In this study, amphiphilic polycations containing catechol adhesive groups were synthesized and used to prepare biocidal coatings by a simple one-step coating procedure. The catechol groups promoted the immobilization of the hydrophilic polymers onto glass surfaces. The incorporation of hydrophilic comonomers enhanced the antimicrobial activity of the coatings, possibly due to tuning the amphiphilic balance to facilitate the interactions of polymer chains with bacterial cells, leading to efficient bacteria killing. The coatings prevented biofilm development of E. coli, S. aureus, and A. baumannii for up to 96 hours. These results support our hypotheses that the catechol adhesive groups prevent leaching of hydrophilic polymers from the surface coatings, allowing tuning amphiphilic balance of polycations for potent biocidal activity of the coatings. In this study, only glass slides were tested as a coating substrate. It would be of great interest to investigate the properties and antimicrobial activity of polymer coatings on different substrates including plastics and titanium oxide. As future perspectives, this new approach for preparing polycations with the catechol adhesive groups may be useful for permanent antimicrobial coatings on many materials. The one-step coating method without any surface modification, as demonstrated in this study, will facilitate applications of antimicrobial polymers for coatings on existing biomedical devices.
Supplementary Material
Acknowledgments
This work was supported by the Departments of Biologic and Materials Sciences, School of Dentistry, and Department of Environmental Health Sciences, School of Public Health at the University of Michigan, Michigan Universities Commercialization Initiative (MUCI) Grant “TruEnamel - Synthetic Dental Enamel”, and the NIH (1R21DE020908-01 to KK and CX). ZC acknowledges the support from the Office of Naval Research (N00014-08-1-1211). M.M. was supported by the JSPS Institutional Program for Young Researcher Overseas Visits and the Global COE Program “Elucidation of Materials and Molecular Functions” at Nagoya University in Japan. We thank Edmund Palermo at the University of Michigan for help and discussion.
Footnotes
Supporting Information Available: Synthesis of monomers, molecular weight and 1HNMR characterization of polymers, SFG spectra, antimicrobial assay protocols, determination of leached polymers, killing ability of coatings, and assay protocol for zone of inhibition. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.James DB. Biotechnol. Bioeng. 2008;100(1):1–18. doi: 10.1002/bit.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruggeri V, Francolini I, Donelli G, Piozzi A. J. Biomed. Mater. Res. A. 2007;81A(2):287–298. doi: 10.1002/jbm.a.30984. [DOI] [PubMed] [Google Scholar]
- 3.Zurita R, Puiggali J, Rodriguez-Galan A. Macromol. Biosci. 2006;6(1):58–69. doi: 10.1002/mabi.200500147. [DOI] [PubMed] [Google Scholar]
- 4.Tiller JC, Sprich C, Hartmann L. J. Controlled Release. 2005;103(2):355–367. doi: 10.1016/j.jconrel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Ho CH, Tobis J, Sprich C, Thomann R, Tiller JC. Adv. Mater. 2004;16(12):957–961. [Google Scholar]
- 6.Hetrick EM, Schoenfisch MH. Chem. Soc. Rev. 2006;35(9):780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 7.Huang JY, Koepsel RR, Murata H, Wu W, Lee SB, Kowalewski T, Russell AJ, Matyjaszewski K. Langmuir. 2008;24(13):6785–6795. doi: 10.1021/la8003933. [DOI] [PubMed] [Google Scholar]
- 8.Lee SB, Koepsel RR, Morley SW, Matyjaszewski K, Sun YJ, Russell AJ. Biomacromolecules. 2004;5(3):877–882. doi: 10.1021/bm034352k. [DOI] [PubMed] [Google Scholar]
- 9.Murata H, Koepsel RR, Matyjaszewski K, Russell AJ. Biomaterials. 2007;28(32):4870–4879. doi: 10.1016/j.biomaterials.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Chen YJ, Worley SD, Kim J, Wei CI, Chen TY, Santiago JI, Williams JF, Sun G. Ind. Eng. Chem. Res. 2003;42(2):280–284. [Google Scholar]
- 11.Lin J, Qiu SY, Lewis K, Klibanov AM. Biotechnol. Prog. 2002;18(5):1082–1086. doi: 10.1021/bp025597w. [DOI] [PubMed] [Google Scholar]
- 12.Tiller JC, Liao CJ, Lewis K, Klibanov AM. Proc. Natl. Acad. Sci. U. S. A. 2001;98(11):5981–5985. doi: 10.1073/pnas.111143098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kügler R, Bouloussa O, Rondelez F. Microbiology-Sgm. 2005;151:1341–1348. doi: 10.1099/mic.0.27526-0. [DOI] [PubMed] [Google Scholar]
- 14.Huang JY, Murata H, Koepsel RR, Russell AJ, Matyjaszewski K. Biomacromolecules. 2007;8(5):1396–1399. doi: 10.1021/bm061236j. [DOI] [PubMed] [Google Scholar]
- 15.Roy D, Knapp JS, Guthrie JT, Perrier S. Biomacromolecules. 2008;9(1):91–99. doi: 10.1021/bm700849j. [DOI] [PubMed] [Google Scholar]
- 16.Madkour AE, Dabkowski JA, Nusslein K, Tew GN. Langmuir. 2009;25(2):1060–1067. doi: 10.1021/la802953v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daewon P, Jun W, Alexander MK. Biotechnol. Prog. 2006;22(2):584–589. [Google Scholar]
- 18.Haldar J, An DQ, de Cienfuegos LA, Chen JZ, Klibanov AM. Proc. Natl. Acad. Sci. U. S. A. 2006;103(47):17667–17671. doi: 10.1073/pnas.0608803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palermo E, Kuroda K. Appl. Microbiol. Biotechnol. 2010;87(5):1605–1615. doi: 10.1007/s00253-010-2687-z. [DOI] [PubMed] [Google Scholar]
- 20.Lee H, Dellatore SM, Miller WM, Messersmith PB. Science. 2007;318(5849):426–430. doi: 10.1126/science.1147241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee H, Lee BP, Messersmith PB. Nature. 2007;448(7151):338. doi: 10.1038/nature05968. (4) [DOI] [PubMed] [Google Scholar]
- 22.Westwood G, Horton TN, Wilker JJ. Macromolecules. 2007;40(11):3960–3964. [Google Scholar]
- 23.Burzio LA, Waite JH. Biochemistry (Mosc) 2000;39(36):11147–11153. doi: 10.1021/bi0002434. [DOI] [PubMed] [Google Scholar]
- 24.Sever MJ, Weisser JT, Monahan J, Srinivasan S, Wilker JJ. Angew. Chem. Int. Ed. 2004;43(4):448–450. doi: 10.1002/anie.200352759. [DOI] [PubMed] [Google Scholar]
- 25.Xu CJ, Xu KM, Gu HW, Zheng RK, Liu H, Zhang XX, Guo ZH, Xu B. J. Am. Chem. Soc. 2004;126(32):9938–9939. doi: 10.1021/ja0464802. [DOI] [PubMed] [Google Scholar]
- 26.Zurcher S, Wackerlin D, Bethuel Y, Malisova B, Textor M, Tosatti S, Gademann K. J. Am. Chem. Soc. 2006;128(4):1064–1065. doi: 10.1021/ja056256s. [DOI] [PubMed] [Google Scholar]
- 27.Lee H, Scherer NF, Messersmith PB. Proc. Natl. Acad. Sci. U. S. A. 2006;103(35):12999–13003. doi: 10.1073/pnas.0605552103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J, Tahar N, Kappl M, Tremel W, Metz N, Barz M, Theato P, Butt H. Adv Mater. 2008;20(20):3872–3876. [Google Scholar]
- 29.Li GZ, Xue H, Gao CL, Zhang FB, Jiang SY. Macromolecules. 2010;43(1):14–16. doi: 10.1021/ma902029s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravikumar T, Murata H, Koepsel RR, Russell AJ. Biomacromolecules. 2006;7(10):2762–2769. doi: 10.1021/bm060476w. [DOI] [PubMed] [Google Scholar]
- 31.Chen CY, Wang J, Loch CL, Ahn D, Chen Z. J. Am. Chem. Soc. 2004;126(4):1174–1179. doi: 10.1021/ja0390911. [DOI] [PubMed] [Google Scholar]
- 32.Chen XY, Chen Z. Biochim. Biophy. Acta-Biomembranes. 2006;1758(9):1257–1273. doi: 10.1016/j.bbamem.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 33.Chen Z, Shen YR, Somorjai GA. Annu. Rev. Phys. Chem. 2002;53:437–465. doi: 10.1146/annurev.physchem.53.091801.115126. [DOI] [PubMed] [Google Scholar]
- 34.Loch CL, Ahn D, Chen Z. J. Phys. Chem. B. 2006;110(2):914–918. doi: 10.1021/jp055377b. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Woodcock SE, Buck SM, Chen CY, Chen Z. J. Am. Chem. Soc. 2001;123(38):9470–9471. doi: 10.1021/ja0164071. [DOI] [PubMed] [Google Scholar]
- 36.Annual Book of ASTM Standard 2002. International, A. Ed. West Conshohocken; PA: p. 1597. E2149-2101. [Google Scholar]
- 37.Kurt P, Gamble LJ, Wynne KJ. Langmuir. 2008;24(11):5816–5824. doi: 10.1021/la800203y. [DOI] [PubMed] [Google Scholar]
- 38.Kurt P, Wood L, Ohman DE, Wynne KJ. Langmuir. 2007;23(9):4719–4723. doi: 10.1021/la063718m. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka M, Mochizuki A, Ishii N, Motomura T, Hatakeyama T. Biomacromolecules. 2002;3(1):36–41. doi: 10.1021/bm010072y. [DOI] [PubMed] [Google Scholar]
- 40.Statz AR, Meagher RJ, Barron AE, Messersmith PB. J. Am. Chem. Soc. 2005;127(22):7972–7973. doi: 10.1021/ja0522534. [DOI] [PubMed] [Google Scholar]
- 41.An YH, Friedman RJ. J. Biomed. Mater. Res. 1998;43(3):338–348. doi: 10.1002/(sici)1097-4636(199823)43:3<338::aid-jbm16>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Paszti Z, Even MA, Chen Z. J. Am. Chem. Soc. 2002;124:7016–7023. doi: 10.1021/ja012387r. [DOI] [PubMed] [Google Scholar]
- 43.Snyder RG, Strauss HL, Elliger CA. J. Phys. Chem. B. 1982;86:5145–5150. [Google Scholar]
- 44.Greaves SJ, Griffith WP. Spectrochimica Acta. 1991;47A(1):133–140. [Google Scholar]
- 45.Lin J, Qiu SY, Lewis K, Klibanov AM. Biotechnol. Bioeng. 2003;83(2):168–172. doi: 10.1002/bit.10651. [DOI] [PubMed] [Google Scholar]
- 46.Klibanov AM. J. Mater. Chem. 2007;17(24):2479–2482. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.