

Abstract
The protective impact of exercise on neurodegenerative processes has not been confirmed, and the mechanisms underlying the benefit of exercise have not been determined in human Parkinson’s disease or in chronic animal disease models. This research examined the long-term neurological, behavioral, and mechanistic consequences of endurance exercise in experimental chronic parkinsonism. We used a chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease with moderate neurodegeneration and examined the effects of treadmill exercise on movement and balance coordination, changes in dopamine neuron biomarkers, mitochondrial functions, and neurotrophic factor activities in the nigrostriatal system. The exercise results were compared with that of the control and sedentary chronic parkinsonian animals. After 18 weeks of exercise training in the chronic parkinsonian mice, we observed a significant deterrence in the loss of neuronal dopamine-producing cells and other functional indicators. The impaired movement and balance incoordination in the chronic parkinsonian mice were also markedly reduced following exercise. Mechanistic investigations revealed that the neuronal and behavioral recovery produced by exercise in the chronic parkinsonian mice was associated with an improved mitochondrial function and an increase in the brain region-specific levels of brain-derived and glial cell line-derived neurotrophic factors. Our findings indicate that exercise not only produces neuronal and mitochondrial protection, it also boosts nigrostriatal neurotrophic factor levels in the chronic parkinsonian mice with moderate neurodegeneration. Therefore, modifying lifestyle with increased exercise activity would be a non-pharmacological neuroprotective approach for averting neurodegenerative processes, as demonstrated in experimental chronic parkinsonism.
Keywords: MPTP parkinsonism, endurance exercise, neuroprotection, mitochondrial dysfunction, neurotrophic factor
Introduction
The majority of sporadic Parkinson’s disease (PD) has an onset at mid to late 50 years of age, primarily involving a progressive degeneration of nigrostriatal neurons and loss of dopaminergic transmission. Current pharmacological therapies are used mainly for symptomatic controls and offer only short-term benefits before disease symptoms and drug adverse reactions worsen. As such, it is considered highly relevant to investigate new treatment strategies aimed at neuroprotection and to understand the neuroprotective mechanisms.
There is growing evidence implicating that physical activity and exercise can generally slow down aging, prevent chronic diseases, and promote health (Mattson, 2000; Haskell et al., 2007). While presently there is a lack of effective neuroprotective drugs targeted at interrupting the neurodegenerative processes, numerous reports suggest that exercise and routine physical activities could potentially reduce the risk of further neurological impairment due to stroke, PD and other degenerative diseases (Eldar & Marincek, 2000; Cotman & Berchtold, 2002; Smith & Zigmond, 2003). In several controlled clinical studies, implementing continuous exercise programs to individuals at early stages of PD has resulted in improved daily activity, motor performance, ambulation and overall functional independence (Comella et al., 1994; Reuter et al., 1999; Baatile et al., 2000; Miyai et al., 2000). Aerobic exercise is also shown to trigger the release of dopamine (DA) in human brains (Wang et al., 2000). Although the clinical effect of exercise on physical rehabilitation and improving the quality-of-life performance in PD is quite evident, whether exercise prevents neuronal loss is not confirmed, and the cellular mechanisms underlying the benefit of exercise have not been determined in human PD or in chronic animal disease models.
The neuroprotective impact of exercise and its mechanisms may be better investigated by conducting laboratory experiments with animal models. In the rat model of parkinsonism induced by 6-hydroxydopamine (6-OHDA), both voluntary running and treadmill-paced exercise attenuated DA loss in the striatum with or without significantly demonstrating recovery of behavioral deficits (Tillerson et al., 2003; Mabandla et al., 2004; Poulton & Muir, 2005). In the acute mouse model of parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), while treadmill exercise ameliorates behavioral deficits, striatal dopaminergic loss is reversed in one study (Tillerson et al., 2003) but is not detected in another (Fisher et al., 2004). By reviewing the above published studies (Table S1), it is apparent that differences in the exercise results obtained from animal models of parkinsonism could be due to experimental variabilities, such as the age and species of the animal model, the method, duration and severity of the induced nigrostriatal lesion, and the type and intensity of the applied exercise regimen.
We have previously examined the effects of long-term treadmill exercise in a chronic mouse model of PD (MPD) with severe neurodegeneration that is characteristically comparable to PD at an advanced stage. In chronic MPD exercising at a level in which cardiovascular, respiratory and metabolic outcomes were comparable to a regularly exercising healthy human individual (Al-Jarrah et al., 2007), we demonstrated a significant reduction of movement deficit without showing considerable signs of neurological recovery after 18 weeks (1 week pre-, 5 weeks during and 12 weeks post-MPD induction) of exercise (Pothakos et al., 2009). Based on these observations, it is reasonable not to expect that exercise or other pharmacotherapeutic approaches would effectively protect against or reverse neuronal deficits in the severe chronic MPD or advanced PD, in which the majority of neurons and neurotransmitters are already irreparably lost. We therefore modified our experimental design and elucidated whether long-term exercise would be beneficial in chronic MPD with moderate neurodegeneration that is more comparable to an early stage of PD. We further investigated neuronal mechanisms that may play a role in exercise-induced neuroprotection in the chronic MPD.
Materials and Methods
Chronic Mouse Model of Parkinson’s Disease (MPD)
Six to ten-month old, male, C57BL/6 mice (Harlan Sprague Dawley, Inc., Indianapolis, IN, USA) were housed in single cages with food pellets and water available ad libitum. The room was maintained at a constant temperature and humidity on a 12-h/12-h light/dark cycle. All animal treatments were carried out strictly in accordance to the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23, revised 1996) and were approved by the Institutional Animal Care and Use Committees from the University of Houston. Our experimental procedures did not cause any significant animal suffering. A total of 135 mice were used in the present study.
To prepare the chronic MPD with moderate neurodegeneration, mice were injected with a total of 10 doses of MPTP hydrochloride (15 mg/kg/injection in saline, s.c.) in combination with an adjuvant drug, probenecid (250 mg/kg/injection dissolved in dimethyl sulfoxide, i.p.) that was originally established in our laboratory (Lau et al., 1990). The 10-dose regimen was administered on a five-week schedule with an interval of 3.5 days between injections (Fig. 1). MPTP hydrochloride and probenecid were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Safety precautions for the use of MPTP during chemical preparation and animal injections were taken according to the procedures as previously published (Lau et al., 2005). Control mice were treated with probenecid only. Probenecid was used to inhibit the rapid clearance and excretion of MPTP and its metabolites from the brain and kidney, and it alone did not produce any significant neurotoxic effect; but in combination it potentiated the neurotoxicity of MPTP (Lau, 2005; Lau et al., 2005; Barber-Singh et al., 2009).
Figure 1. Timetable and protocol for animal treatments.
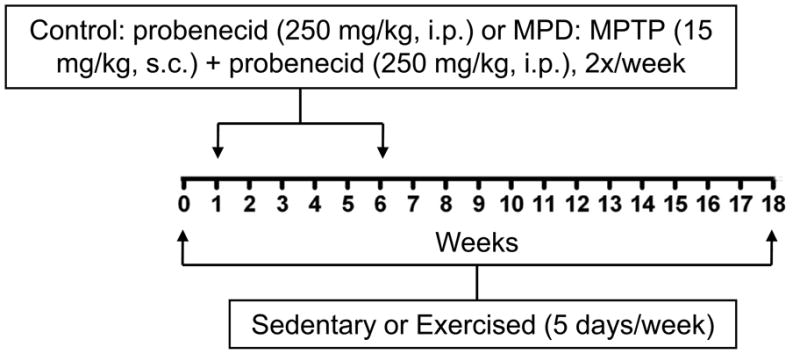
Three groups of animals were treated. Chronic Control group: 18 weeks of sedentation, 5 weeks of 2x probenecid/week; Chronic Sedentary MPD group: 18 weeks of sedentation, 5 weeks of 2x MPTP + probenecid/week; Chronic Exercised MPD group: 18 weeks of exercise, 5 weeks of 2x MPTP + probenecid/week.
The chronic MPD used in this study has been well characterized. In contrast to the most commonly used acute and subacute MPTP mouse models of PD, in which neurological and behavioral deficits are short-lived and spontaneously reversed soon after treatment, the chronic MPD has long-term neurological deficits showing many features resembling that of PD lasting for at least 6 months, long after MPTP has completely dissipated systemically from the animal (Lau, 2005). The observed phenotypic features in the chronic MPD include marked depletion of DA content and terminal DA uptake in association with significant behavioral deficits and loss of DA cells in the substantia nigra pars compacta (SNpc) (Lau et al., 1990; Petroske et al., 2001; Pothakos et al., 2009). Early neuronal apoptosis and delayed appearance of α-synuclein-positive inclusion bodies along with ultrastructural neuronal damage in the SNpc have also been demonstrated (Meredith et al., 2002; Novikova et al., 2006).
Treadmill Exercise in Mice
A six-lane motorized rodent treadmill (Columbus Instruments, Columbus, OH, USA) was utilized for exercise training. For 1 week before, 5 weeks during, and 12 weeks after the completion of chronic MPTP/probenecid treatment (a total of 18 weeks), the exercised group of animals was trained on the treadmill running at 5 days/week, 40 min/day with a speed up to 15 m/min (5 min at 6 m/min, 5 min at 9 m/min, 20 min at 12 m/min, 5 min at 15 m/min, and 5 min at 12 m/min) with 0o of inclination (Fig. 1). Using this treadmill exercise protocol established in our laboratory, the chronic MPD mice were able to complete 450 m per running day with minimal requirement for external stimuli or manual prodding, yet the animals developed physical endurance after one month showing cardiorespiratory and metabolic adaptations comparable to those seen in human subjects undergoing continuous exercise training (Al-Jarrah et al., 2007). Sedentary mice did not exercise; however, they were transported daily to the training room, so that they were exposed to the same environment as the exercised group of animals. All neuronal, biochemical, and behavioral measurements were performed at least 48 hours after the last session of exercise training to avoid any data misinterpretation due to acute exercise residual effects.
Tyrosine Hydroxylase (TH) Immunohistochemistry and Unbiased Stereological Estimates of Cells
Mice were anesthetized with pentobarbital (130 mg/kg; Sigma), transcardially perfused with 40–60 ml of 4% sucrose in 0.1M phosphate-buffered saline (PBS), followed by 70 ml of 4% paraformaldehyde and 4% sucrose in PBS. The brains were removed, placed in the perfusion fixative for 4 h at room temperature and then transferred to PBS and kept at 4°C. The brain samples were sectioned coronally and immunostained with TH antibody. Every fourth section of the SN containing the pars compacta was collected and 6–8 sections per animal were analyzed unilaterally. The modern unbiased, design-based stereology was used to estimate the TH-positive cell number as previously described (Petroske et al., 2001; Scott et al., 2007). Only those cells demonstrating clear visualization of the nucleolus and precise definition of the cell membrane were counted.
Assay for Striatal Dopamine (DA) Content
The striatal DA content was determined as previously described (Lau et al., 1990; Petroske et al., 2001; Lau et al., 2005). Briefly, striata from each animal were isolated, weighed and suspended in 0.5 ml of 0.2N perchloric acid. Each sample was sonicated and centrifuged at 11,000 xg for 15 min at 4°C. The supernatant was filtered through a 4-mm nylon syringe filter with a pore size of 0.45 μm (National Scientific, Rockwood, TN, USA). An aliquot of the filtrate was injected into a high performance liquid chromatography (HPLC, Model 1525, Waters Corporation, Milford, MA, USA) equipped with a C18 reverse phase, 3 μ LUNA column (100 mm × 2.0 mm,Phenomenex, Torrance, CA, USA). The sample was eluted by a mobile phase made of NaH2PO4 (25 mM), Na-citrate (50 mM), EDTA (0.03 mM), diethylamine HCl (10 mM), and sodium octyl sulfate (2.2 mM), at pH of 3.2, plus methanol (30 ml/l) and dimethylacetamide (22 ml/l) at a flow rate of 0.4 ml/min. The DA peak was determined by the Coulometric electrochemical detector (Model Coulochem III, ESA, Inc., Chelmsford, MA, USA) and was calculated by extrapolating the peak area from a standard curve (ranging 0.05-1 ng of each chemical standard) constructed under the same conditions during each run.
Animal Balance and Motor Coordination Performance Test
The ability of the chronic MPD in maintaining balance and motor coordination on a challenging beam was carried out 48 hours after the completion of 18 weeks of endurance exercise training according to the procedures as described by Drucker-Colin & Garcia-Hernandez (1991) and modified by Schallert et al. (2002). The challenging beam was a 1 m long wooden beam suspended 23 cm above a bench top, which was covered with soft pads to protect the mouse in case of a fall. The beam was divided in four gradually narrowing sections (25 cm/section) leading to the mouse's home cage. The beam widths of the four sections were 3.5, 2.5, 1.5, and 0.5 cm in decreasing order. The beam was covered with surgical tape that provided sufficient surface traction for the animals to walk on. There were 1 cm wide ledges hanging 1 cm below each side of the beam to encourage the mice to use their normal gait strategies even when their limbs slipped. All mice were pre-trained for two consecutive days (5 trials/day with inter-trial interval ITI = 10–12 sec) on traversing the beam. On the third day, each mouse was given 5 trials (ITI = 10–12 sec) and the average number of videotaped limb slips per trial and the time latency for returning to the home cage were recorded for statistical analysis. Slips were counted only while the mouse was in forward motion.
Mitochondrial Respiration Assay
Due to tissue limitation, a crude striatal preparation was used for the mitochondrial respiration assay in this study as previously described (Patki et al., 2009). Striata from each animal were isolated and homogenized with a dounce homogenizer in 1 ml of an ice-cold isolation buffer containing mannitol (215 mM), sucrose (75 mM), bovine serum albumin (BSA, 0.1%), EGTA (1 mM), and HEPES (20 mM) at pH of 7.2. All subsequent procedures were carried out at 4°C. The homogenate was centrifuged at 1,300 rpm for 3 min. The supernatant was transferred to a new tube and the pellet was suspended in 0.5 ml of the isolation buffer and centrifuged again at 1,300 rpm for 3 min. The supernatants from both spins were combined and centrifuged at 13,000 rpm for 10 min. This latter supernatant was discarded and the pellet containing the mitochondrial fraction was suspended in 0.1 ml of a respiration buffer containing mannitol (215 mM), sucrose (75 mM), BSA (0.1%), HEPES (20 mM), MgCl2 (2 mM), and KH2PO4 (2.5 mM) at pH of 7.2. The mitochondrial protein concentration was determined with the Pierce micro BCA protein assay kit (Thermo Scientific, Rockford, IL, USA) measured at an absorbance of 595 nm with a Beckman DU 640 spectrophotometer (Fullerton, CA, USA).
The respiratory activity of striatal mitochondria was measured polarographically with a Clark-type oxygen electrode in a sealed, thermo-controlled, and continuously stirred chamber (Oxytherm System, Hansatech Instruments, Norfolk, England) as previously described (Patki et al., 2009). The mitochondrial fraction at a protein concentration of 0.5 mg/ml of respiration buffer (see above) was initially equilibrated in the electrode chamber at 30°C and then reacted with the addition of NADH-linked substrates, pyruvate (5 mM) and malate (2.5 mM). The state 3 respiration was initiated by adding adenosine 5’-diphosphate (ADP, 150 μM) and the state 4 respiration was measured by adding oligomycin (1 μM) after ADP-dependent state 3 respiration had reached completion. The rate of oxygen consumption (nmol/min/mg mitochondrial protein) was calculated based on the slope of polarographic tracings obtained during state 3 and state 4 respiration. In this study, all mitochondrial preparations had demonstrated an average respiratory control ratio (RCR: state 3/state 4 respiration) of at least 4.5.
Measurement of Mitochondrial ATP
Striatal mitochondrial samples were prepared under conditions identical to those of the respiration study. After the completion of respiration experiment, the mitochondrial suspension from the chamber was recovered and mixed with an equal volume of lysis buffer provided in the ATP bioluminescence assay kit (PerkinElmer, Waltham, MA, USA). The mitochondrial ATP content was measured according to the manufacturer’s instruction. Light emitted from luciferase-mediated reaction was captured in a luminometer (Wallac Victor II, PerkinElmer) and the sample ATP content was extrapolated from a standard curve constructed with a series of known ATP concentrations (Patki et al., 2009).
Analysis of Neuronal and Mitochondrial Protein Expression
The expression of mitochondrial antioxidant enzymes, Mn superoxide dismutase (SOD) and Cu-Zn SOD, the striatal carbonylated proteins, DA synthesizing enzyme TH, and DA uptake transporter (DAT) were determined in the present study as before (Patki et al. , 2009). For Western blot analysis of antioxidant protein expression, a crude mitochondrial fraction from the striatum was prepared according to the procedure as described above, except that mitochondria were finally suspended in a Tris–HCl buffer (50 mM, pH 7.4) containing a protease inhibitor cocktail purchased from Sigma Chemical Co. The mitochondrial tissue was suspended in a protein solubilization solution containing SDS (2%), Tris–HCl (62.5 mM, pH 6.8), glycerol (10%), 2-mercaptoethanol (5%), and bromophenol blue (0.001%). The sample was boiled for 5 min and 25 μg of each sample was applied to an SDS polyacrylamide (12%) gel for electrophoresis. The resolved proteins were transferred to a PVDF membrane, which was probed with a polyclonal rabbit anti-Mn-SOD (1:1000, Upstate, Lake Placid, NY, USA) and a polyclonal anti-Cu/Zn-SOD (1:1000, Stressgen Bioreagents, Victoria, BC, Canada).
For evaluating the amount of carbonylated proteins, all reagents were supplied and procedures were followed according to the commercially available OxyBlot kit (Millipore, Billerica, MA, USA). The striatal tissue homogenate in presence of protease inhibitors containing 20 μg of protein was reacted with 10 μl of 2,4-dinitrophenylhydrazine (DNPH) reagent for 15 min at room temperature. The reaction was terminated by injecting 7.5 μl of a neutralization solution. Carbonylated proteins that reacted with DNPH to form DNP-hydrazone were separated on an SDS polyacrylamide (8–16%) gradient gel (Pierce, Rockford, IL, USA). The resolved carbonylated proteins within 43–94 Kd were detected by immunoblotting according to the OxyBlot protocol.
The nigral and striatal TH level was probed with a mouse anti-TH (1:1000, Millipore, Temecula, CA, USA) and the striatal DAT expression was detected with a rat anti-DAT (1:1000, Chemicon) following tissue solubilization, SDS polyacrylamide (12%) gel electrophoresis and Western blot transfer. Immunoreactivity was visualized by a horseradish peroxidase-conjugated goat anti-rabbit (1:2000, Chemicon) or goat anti-rat (1:2000, Chemicon) or goat anti-mouse IgG (1:1500, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) with an enhanced NuGlo chemiluminescent substrate (Alpha Diagnostic International Inc., San Antonio, TX, USA). The exposure time of the blot on film was confined to the linear scale of detection without exceeding the saturation limit. The intensity of the protein bands was measured by densitometry (Fluorochem 8800, Alpha Innotech Corporation, San Leandro, CA, USA) and normalized as a ratio to the expression of glyceraldehyde 3 phosphate dehydrogenase GAPDH with a monoclonal mouse anti-GAPDH (1:3000, Chemicon), or β-tubulin with a monoclonal mouse anti-β-tubulin (1:2000, Upstate, Lake Placid, NY, USA), or β-actin with a monoclonal mouse anti-β-actin (1:2000, Millipore) to ensure that changes of protein level are not simply due to differences in the amount of sample loading.
Measurement of Nigrostriatal Brain-derived Neurotrophic Factor (BDNF) and Glial Cell Line-derived Neurotrophic Factor (GDNF) Levels
The endogenous BDNF and GDNF levels in the SN and striatum were measured with the reagents provided in the BDNF or GDNF Emax ImmunoAssay System and according to the manufacturer’s recommended protocols (Promega Corp., Madison, WI, USA). The nigral or striatal tissue was initially suspended and sonicated in 200 μl of the provided lysis buffer. The homogenate was centrifuged at 14,000 xg for 30 min at 4°C. The supernatant was diluted with Dulbecco’s PBS (1:5), acidified with 1N HCl to an approximate pH of 2.6, and then neutralized with 1N NaOH to an approximate pH of 7.6 before assaying. The BDNF or GDNF content was expressed per mg of protein.
Statistical Analysis
Statistical comparisons of values between two animal groups were carried out by unpaired Student’s t-test and multiple group comparisons were conducted by one-way analysis of variance (ANOVA) with Tukey’s post-hoc test with the Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Data are represented as mean ± S.E.M. In all cases, a P value of <0.05 was considered to be significantly different.
Results
Impact of 18-Week Exercise on SNpc TH-positive Cells in Chronic MPD
Following a total of 18 weeks of treadmill exercise training, we first examined and compared the trait of dopaminergic cells in the SNpc between control, sedentary chronic MPD, and exercised chronic MPD. The sedentary chronic MPD exhibited a marked loss of TH-positive cells in the SNpc (Fig. 2B) when compared with the same area in the control mice (Fig. 2A). Under high power microscopy, in contrast to the control animals (Fig. 2E), the TH-stained neurons in the sedentary chronic MPD appeared to lose their content and integrity; the axons and dendrites clearly showed disarray (Fig. 2F). Similar results were obtained by measuring the TH protein content in SN with Western blot analysis (Fig. 2D) and by counting the TH-positive cell number in the SNpc with random and unbiased stereological estimate (Fig. 2H), which revealed a moderate loss of TH-positive protein expression and cells in the SN of the sedentary chronic MPD [P=0.006 and P=0.002, respectively]. Interestingly, after the chronic MPD had completed 18 weeks of exercise, the immunohistochemical depiction of the SNpc under low power microscopy displayed increased TH-positivity (Fig. 2C) and showed clustering of cells and extensive dendritic arborization under high power microscopy (Fig. 2G). Both TH protein content (Fig. 2D) and total TH-positive cell number in the SNpc (Fig. 2H) were significantly recovered by long-term exercise training when compared with the sedentary chronic MPD or when all groups were analyzed with one-way ANOVA [F (2, 12) = 8.986, P = 0.004 for TH; F (2, 12) = 11.31, P = 0.002 for cell number].
Figure 2. Neuroprotective effect of 18 weeks of exercise in the SN of the chronic MPD.

Representative light microscope photomicrographs illustrating TH immunoreactivity in the SNpc of the control (A), sedentary chronic MPD (B), and 18-week exercised chronic MPD (C). The TH-immunoreactive cells and dendrites in the SNpc of the sedentary chronic MPD were markedly lower than that of the control and the exercise-trained chronic MPD. Similar results were obtained from the Western blot TH-protein analysis as shown in (D), where TH level was expressed as an OD ratio over that of β-tubulin. The TH level in the SN of the sedentary chronic MPD was significantly lower than that in the controls (*P=0.006) or the exercised chronic MPD(*P=0.004); whereas TH levels in the control group and exercised chronic MPD group were not statistically different (**P=0.220). The morphological features of TH-positive neurons in the SNpc were further examined under high power microscopy. The TH-positive neurons in both the control (E) and 18-week exercised chronic MPD mice (G) displayed clustering of cells and extensive dendritic arborization; whereas the cells in the sedentary chronic MPD (F) appeared to lose their integrity and content; furthermore, the axons and dendrites showed morphological disarray. The TH-positive neurons in the SNpc of three groups of animals were counted with a random and unbiased stereological approach (H). The sedentary chronic MPD lost about 55% of the SNpc TH-positive cells when compared with the control or 18-week exercise-trained chronic MPD (*P=0.002). The number of TH-positive cells in the SNpc of control and 18-week exercised chronic MPD were not significantly different (**P=0.487). Scale bar = 250 μm in AC; 10 μm in E-G. N=5 per each group. Statistical comparisons were performed with unpaired Student’s t-test.
Impact of 18-Week Exercise on Striatal Dopaminergic Indicators in Chronic MPD
We further evaluated the dopaminergic biomarkers in the neostriatum among the three groups of animals. The sedentary chronic MPD displayed moderate but significant losses in the level of striatal TH, DA, and DAT [P<0.03] suggesting terminal dysfunction of the dopaminergic neurons (Fig. 3A-C). The striatal levels of TH, DA and DAT were significantly elevated in the 18-week exercised chronic MPD when compared with the sedentary chronic MPD or when all groups were analyzed with one-way ANOVA [F (2, 14) = 9.015, P = 0.003 for TH; F (2, 11) = 18.42, P = 0.0003 for DA; F (2, 13) = 5.711, P = 0.017 for DAT] (Fig. 3A-C). When parallel studies were conducted in moderate chronic MPD with only 10 weeks of treadmill exercise, the number of TH-positive cells in SNpc, the levels of TH in SN and striatum, and the amount of striatal DA were not statistically different from that of their sedentary counterparts (Fig. S1). Our observations suggest that long-term endurance exercise has a neuroprotective potential for restoring the nigrostriatal DA function in the chronic MPD with moderate neurodegeneration, whereas neurorecovery was not evident with short-term exercise (Fig. S1) or in chronic MPD with severe neurodegeneration (Pothakos et al., 2009).
Figure 3. Neuroprotective effect of 18 weeks of exercise in the striatum of the chronic MPD.

The striatal TH level (A), DA content (B), and dopamine uptake transporter (DAT) expression (C) were significantly decreased in the sedentary chronic MPD when compared with the control group (*P=0.031, 0.003 and 0.0018, respectively), whereas their levels were significantly higher in the 18-week exercised chronic MPD than in the sedentary chronic MPD (*P=0.0001, 0.008 and 0.030, respectively). The striatal TH and DAT contents in the exercised chronic MPD were not statistically different from that in the control animals (A and C, **P=0.266 and 0.152, respectively), whereas the striatal DA level in the exercised chronic MPD was still significantly lower than the control group (B, *P=0.003). N=4–6 per each group. Statistical comparisons were performed with unpaired Student’s t-test.
Impact of 18-Week Exercise on Movement and Balance Performance in Chronic MPD
We also assessed the animal’s behavioral manifestations by monitoring their balance and motor coordination skills on a challenging beam at the end of 18-week study. The sedentary chronic MPD mice performed poorly by committing more foot-slips while traversing on the balancing beam [P=0.013] (Fig. 4A) and presented a delayed latency for returning to the home cage [P=0.018] (Fig. 4B) when compared with the control group of animals. The deficits in balance and motor performance were not detected in 18-week exercise-trained chronic MPD; they performed significantly better than the sedentary chronic MPD in both foot-slip and latency measures when all groups were analyzed with one-way ANOVA [F (2, 78) = 20.39, P < 0.0001; F (2, 78) = 4.416, P = 0.019, respectively] (Fig. 4A-B).
Figure 4. Convalescent effect of 18 weeks of exercise on impaired movement in the chronic MPD.

The ability of the chronic MPD in maintaining balance and motor coordination on a challenging beam was carried out 48 hours after the completion of 18 weeks of endurance exercise training. While traversing the challenging beam, the sedentary chronic MPD group of mice significantly made more limb slip errors (A, *P = 0.013) and took longer to complete the task (B, *P=0.018) than the control mice. Following 18 weeks of training on motorized treadmill, the exercised chronic MPD made considerably less foot slips on the challenging beam (A, *P<0.0001) and spent less time for completing the traversing task (B, *P=0.038) than the sedentary chronic MPD. The exercised chronic MPD even made significantly less foot slips (A, *P<0.0001) with no change of time latency for completing the task (B, **P=0.549), when compared with the control animals. N=25–31 per group. Statistical comparisons were performed with unpaired Student’s t-test.
Effects of 18-Week Exercise on Striatal Mitochondrial Indicators in Chronic MPD
Next, we addressed whether oxidative protein damage and mitochondrial dysfunction could be detected in the chronic MPD and whether exercise would be beneficial in averting these injuries. When neuronal cells undergo oxidative stress and death, damages typically occur to cellular DNA, proteins and lipids. In the present study, we demonstrated that striatal carbonylated proteins were significantly elevated [P=0.014] (Fig. 5A), whereas the mitochondrial antioxidant enzymes, Mn SOD and Cu-Zn SOD were considerably reduced [P<0.001] (Fig. 5B) in the sedentary chronic MPD. The state 3 and state 4 mitochondrial respiration in sedentary chronic MPD were substantially lower than that of the control animals [P<0.01] (Fig. 6A-B). Thus, the striatal mitochondrial respiratory function when that is expressed in terms of the respiratory control ratio (state 3/state 4 respiration, RCR) was regarded impaired in the chronic MPD (Fig. 6C). The striatal concentration of mitochondrial ATP was also significantly reduced in the sedentary chronic MPD [P=0.0002] (Fig. 6D). These results collectively substantiate that oxidative damage and mitochondrial dysfunction persist in the chronic MPD in association with the neurodegenerative condition.
Figure 5. Deterrent action of 18 weeks of exercise on striatal carbonylated proteins and mitochondrial antioxidant enzyme levels in the chronic MPD.
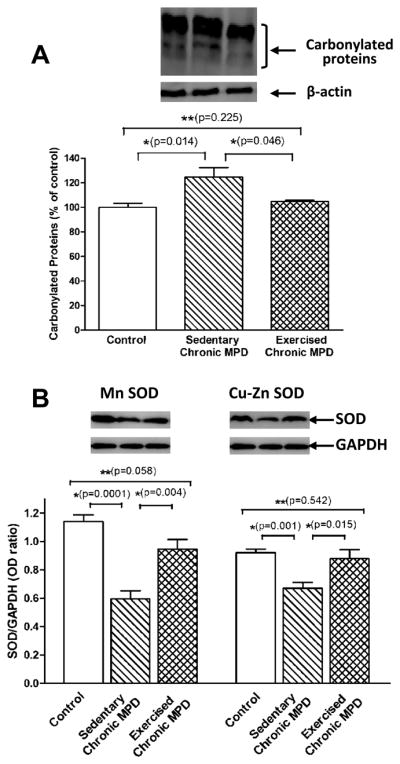
The total intensity of striatal carbonylated proteins between 43–94 Kd as an indicator of oxidative stress (A) and the level of striatal antioxidant enzymes, Mn SOD and Cu-Zn SOD (B) were measured by immunoblotting. In the sedentary chronic MPD, the striatal carbonylated protein level was significantly elevated (A, *P=0.014); the Mn SOD and Cu-Zn SOD levels were considerably reduced (B, *P=0.0001 and 0.001, respectively), when compared with those in the control animals. Following 18 weeks of treadmill exercise, the levels of carbonylated protein, Mn SOD, and Cu-Zn SOD in the striatum of chronic MPD were returned to normal; they are significantly different from the sedentary chronic MPD (*P=0.046, 0.004 and 0.015, respectively) and not statistically different from the control mice (**P=0.225, 0.058 and 0.542, respectively) (A-B). N=5–8 per group. Statistical comparisons were performed with unpaired Student’s t-test.
Figure 6. Protective effect of 18 weeks of exercise on mitochondrial functional indicators in the striatum of chronic MPD.

The rate of mitochondrial state 3 and state 4 respiration (A-B), and ATP content (D) in the striatal tissue were measured. The respiratory control ratio (rate of state 3/rate of state 4 respiration, RCR) was used to signify the mitochondrial respiratory function (C). Both striatal mitochondrial respiration (state 3, state 4, RCR) and ATP level were found markedly lower in the sedentary chronic MPD than in the control mice (*P<0.007), whereas their levels were significantly higher in the 18-week exercised chronic MPD than in the sedentary chronic MPD (*P<0.022). The mitochondrial state 3, state 4 respiration, RCR in the exercised chronic MPD were not statistically different from that in the control animals (A- C, **P=0.704, 0.685 and 0.119, respectively), whereas the mitochondrial ATP level in the exercised chronic MPD was still significantly lower than in the control group (D, *P=0.036). N=6 per group. Statistical comparisons were performed with unpaired Student’s t-test.
Strikingly, when the chronic MPD was exercise-trained for 18 weeks, we were able to demonstrate that their striatal carbonylated protein level was similar to the normal animals and was significantly lower than that in the sedentary chronic MPD when all groups were analyzed with one-way ANOVA [F (2, 14) = 6.713, P = 0.009] (Fig. 5A). The contents of striatal antioxidant enzymes, Mn SOD and Cu-Zn SOD were also considerably higher than that in the sedentary MPD when all groups were analyzed with one-way ANOVA [F (2, 13) = 20.98, P < 0.0001 for Mn SOD; F (2, 15) = 9.557, P <0.002 for Cu-Zn SOD] (Fig. 5B). The state 3 and state 4 mitochondrial respiration in the exercised chronic MPD were noticeably higher than that in the sedentary chronic MPD (Fig. 6A-B). These values hence resulted in an RCR that is greater than that detected in the sedentary chronic MPD and is not different from the control animal group MPD when all groups were analyzed with one-way ANOVA [F (2, 15) = 53.33, P < 0.0001] (Fig. 6C). Similarly, the striatal mitochondrial ATP level in the exercised chronic MPD was considerably higher than that in the sedentary chronic MPD when all groups were analyzed with one-way ANOVA [F (2, 14) = 13.98, P < 0.0005] (Fig. 6D). These data clearly illustrate that 18 weeks of exercise scheme prevented the loss of striatal mitochondrial integrity and function due to chronic MPD and suggest that long-term exercise is mitochondria-protective as well.
Effects of 18-Week Exercise on Nigrostriatal Neurotrophic Factors in Chronic MPD
Neurotrophic factors, such as the brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF) have been considered to play an important role in exercise-induced neuroprotection (Cotman & Berchtold, 2002; Hennigan et al., 2007). In the present study, we additionally investigated whether or not the endogenous BDNF and GDNF are altered in the chronic MPD and then elucidated whether or not long-term exercise would change the BDNF and GDNF levels. In the C57BL/6 mouse species, we found that the amount of endogenous BDNF was higher in the SN than in the striatum (18.04 ± 1.54 vs. 9.97 ± 0.64 pg/mg protein, respectively) [P=0.001] (Fig. 7A); whereas the endogenous GDNF content was not statistically different between the SN and the striatum (14.57 ± 1.88 vs. 19.51 ± 2.43 pg/mg protein, respectively) [P=0.147] (Fig. 7B). The endogenous BDNF or GDNF concentrations in both SN and striatum were not altered in the sedentary chronic MPD (Fig. 7A-B). However, 18 weeks of treadmill exercise in chronic MPD produced brain region specific effects: exercise significantly elevated BDNF level in the SN [F (2, 12) = 4.098, P = 0.047] but not in the striatum [F (2,12) = 1.114, P = 0.363] when all groups were analyzed with one-way ANOVA (Fig. 6A). Exercise drastically increased GDNF level in the striatum [F (2, 12) = 16.09, P = 0.0005] but not in the SN [F (2, 12) = 2.252, P = 0.151] when all groups were analyzed with one-way ANOVA, although the GDNF level in the SN of exercised chronic MPD group appeared to be significantly higher than that in the sedentary chronic MPD group when both groups were analyzed by unpaired Student’s t-test [P=0.035] (Fig. 7B). These data demonstrated that although baseline levels of BDNF and GDNF are not altered in the chronic MPD, neurotrophic factor levels are increased in response to long-term exercise, in a neurotrophin-specific and brain region-specific manner.
Figure 7. Elevation of endogenous nigrostriatal neurotrophic factors by 18 weeks of exercise training in the chronic MPD.

The endogenous contents of BDNF and GDNF in the SN and striatum were measured by immuno-specific ELISA. The basal level of endogenous BDNF was significantly higher in the SN than in the striatum (A, *P<0.002); whereas the basal content of endogenous GDNF in the SN and striatum were not statistically different (B, P=0.1466). The endogenous concentrations of BDNF (A) and GDNF (B) in the SN and striatum were not significantly altered in the sedentary chronic MPD (P>0.05). However, the BDNF level in the SN (A, **P=0.042) and the GDNF level in the striatum (B, **P=0.0005) were significantly elevated in the exercised chronic MPD when compared with the sedentary chronic MPD or the control group. N=5 per group. Statistical comparisons were performed with unpaired Student’s t-test.
Discussion
The purpose of this study was to investigate the impact of exercise on a chronic experimental model of PD with a moderate level of neurodegeneration and to explore possible mechanisms of exercise-induced neuroprotection. We found that when exercise was administered one week before, 5 weeks during and 12 weeks after the chronic MPD induction, the number and morphological integrity of the nigrostriatal neurons and the amount of striatal TH, DA and DAT were significantly less impaired than that found in the sedentary chronic MPD. The neuroprotective impact of exercise was confirmed by behavioral improvement as well; thus, the exercise-trained chronic MPD performed better than the sedentary chronic MPD on the balancing beam. We also found that long-term exercise protected against mitochondrial dysfunction that was detected in the chronic MPD. Exercise reduced the level of striatal carbonylated proteins possibly due to neurotoxin-induced oxidative stress. Exercise functionally restored mitochondrial respiration, ATP and antioxidant SOD levels in the striatum. Exercise was further shown to raise the level of endogenous BDNF and GDNF in the substantia nigra and striatum, correspondingly. We conclude that long-term exercise not only prevents neurobehavioral and mitochondrial deficits that are associated with chronic MPD, exercise also promotes neurotrophic activity in the nigrostriatal neurons in the chronic PD model.
After considering the published studies on exercise effects in animal models of PD (Table S1), we adopted several novel approaches in designing the present research. First, we used a chronically induced parkinsonian model with moderate neuronal deficit, which is in contrast to most published reports that utilize a rather severe, acutely or subacutely induced animal model. Second, our data collection and analyses were performed at least 48 hours after any experimental manipulation of the animals, including exercise, to avoid possible misinterpretation of results that might be associated with the residual effect of the last exercise training. Third, we evaluated exercise outcomes for a total of 18 weeks encompassing a period before, during and after the chronic MPD induction, whereas other studies measured effects shortly after the disease model induction or exercise training. Fourth, we applied a moderate exercise regimen that had been established and characterized by our laboratory to achieve desirable cardiovascular, respiratory and metabolic outcomes (Al-Jarrah et al., 2007). Under these experimental conditions, we have successfully demonstrated here that exercise is neuroprotective, which cannot be accomplished with shorter exercise training or in chronic MPD with severe neuronal deficit (Pothakos et al., 2009).
Gradual declines in the ability to carry out routine motor tasks and to recall events and memories are common phenomena and normal complaints among the aged population. Furthermore, aging itself is considered a risk factor for various neurodegenerative disorders. Inadequate physical activity or lack of exercise on a continuous basis can lead to an early sedentary lifestyle in young people and pose health risks later in life in normal elders or in individuals suffering chronic diseases, such as the neurodegenerative PD. Conventionally, exercise is thought to produce an overall benefit for physical fitness and mental stimulation, to slow down aging and defend against the onset of chronic diseases. In human and animal studies, the impact of exercise on promoting brain angiogenesis and neurogenesis in the dentate gyrus of the hippocampus has been well established supporting a notion that exercise can derail the slow decline of cognitive and memory functions during the course of normal aging (van Praag et al., 2005; Pereira et al., 2007). This research clearly demonstrates that long-term exercise attenuates neurological and motor declines in a chronic neurodegenerative PD model.
The neuroprotective effect of exercise in chronic MPD with moderate neuronal deficit may be explained by several plausible mechanisms underlying the current neurodegenerative hypotheses. Conceivably, neurons are extremely active cells that require a constant supply of energy in order to carry out highly specialized functions, such as regulating the activities of neuronal transmission, receptors, ion channels, transporters and synapses. Thus, mitochondria are vital for maintaining the homeostasis and integrity of neuronal functions. Neuronal oxidative stress and mitochondrial dysfunction have been implicated in aging and neurodegenerative processes (Calabrese et al., 2001; Betarbet et al., 2002; Beal, 2005). In human PD, reduced mitochondrial complex I activity is found in the SN and platelets (Beal, 2002), and mitochondrial DNA (mtDNA) mutations have been detected (Bender et al., 2006).
The link between mitochondrial dysfunction and PD-like neurodegeneration has also been demonstrated in vitro and in selected genetic animal models. For example, the accumulation of α-synuclein in cultured human DA neurons induces reactive oxygen species (ROS)-mediated apoptosis (Xu et al., 2002). Parkinsonian symptoms are replicated in genetically engineered mice that are deficient of mitochondrial respiratory chains (Ekstrand et al., 2007). Infusion of the mitochondrial complex I inhibitor rotenone or MPTP produces oxidative damage resulting in selective loss of SN DA neurons and buildup of cytoplamic α-synuclein-immunoreactive inclusions (Betarbet et al., 2000; Fornai et al., 2005). Parkinsonism is shown in genetic models with point mutations of mtDNA (Thyagarajan et al., 2000). Administration of MPTP to α-synuclein overexpressed mice produces pathological features in mitochondria (Song et al., 2004). Parkin is an ubiquitin E3 ligase that is important for the disposition of oxidatively damaged proteins. Parkin is associated with the outer mitochondrial membrane and protects against mitochondrial damage and the release of cytochrome c, which is triggered by various xenobiotic insults including ROS (Darios et al., 2003). By introducing mutations to the parkin molecule or in parkin-deficient mice, ROS levels are elevated; oxidative damage of the mitochondria and DA neuron apoptosis are exhibited (Palacino et al., 2004). We also demonstrated that inserting the mitochondrial NADH-quinone oxidoreductase (NDI1) gene into the SN rescued the DA neurons in the chronic MPD (Barber-Singh et al., 2009). Taken together, mitochondrial dysfunction can be considered as a contributing factor leading to PD-associated neuronal death.
ROS can be accumulated from external sources or produced by mitochondria as toxic byproducts of oxidative phosphorylation during their energy generating process. Exercise appears to lower the level of ROS and improve mitochondrial functions, neurological performance and survival (Conley et al., 2007; Navarro & Boveris, 2007). In the present study, oxidative protein damage, suppressed mitochondrial respiratory function, and depleted antioxidant enzyme and ATP levels in connection with neuronal and behavioral deficits were replicated in the moderate chronic MPD. In addition, we demonstrated that long-term exercise is neuron-protective and mitochondria-protective in tandem. This dual protective capability of exercise further strengthens the notion of a possible cause-and-effect relationship between mitochondrial dysfunction and neurodegeneration.
Several neurotrophic factors in the brain, particularly the BDNF and GDNF have been recognized for supporting the survival of adult neurons and for playing a role in exercise-induced neuroprotection (Cotman & Berchtold, 2002; Hennigan et al., 2007). BDNF or GDNF expression is significantly reduced in PD brains; thus it is inferred that PD neurodegeneration may be in part caused by defective synaptic plasticity associated with insufficient neurotrophic input (Parain et al., 1999; Howells et al., 2000; Chauhan et al., 2001). In animal studies, GDNF has been shown to protect DA cells in 6-OHDA and acute MPTP Parkinson’s models (Tang et al., 1998; Cheng et al., 1998). It is also shown that exercise-induced neuronal recovery in 6-OHDA-treated parkinsonian rats involves an apparent increase of the striatal BDNF and GDNF (Cohen et al., 2003; Tajiri et al., 2010).
While the impact of exercise on neurotrophic factors and neuronal plasticity in the hippocampus has been well documented, studies of exercise effects on BDNF and GDNF levels in nigrostriatal dopaminergic neurons are limited, especially with long-term exercise in chronic parkinsonism. In the case of chronic MPD as demonstrated in the current study, the endogenous BDNF and GDNF levels in the SN and striatum are not affected when compared with the normal control animals, suggesting that in the absence of neuroprotective intervention, the basal level of neurotrophic factors may not be sufficient for reviving the neurons that are undergoing degeneration. However, long-term exercise training is able to raise the endogenous BDNF and GDNF above their basal levels. Interestingly, we further discovered that exercise has produced a response that is trophic factor-selective and neuronal region-specific. These observations may provide insight into future pharmacological research on target specificity and neurotrophic factor selectivity in potential therapies for neurodegenerative diseases.
Finally, we have considered the possibility that when performed concomitantly with chronic MPD induction, exercise may enhance the elimination of MPTP and its metabolite, 1-methyl-4-phenylpyridinium (MPP+). Although measuring MPTP and MPP+ levels in the striatum would be technically challenging and was not carried out with the moderate chronic MPD, we believe that co-administration of probenecid would alleviate this concern. Furthermore, our findings that concomitant exercise with MPTP administration did not significantly reduce parkinsonism in 10-week exercised, moderate chronic MPD or in 18-week exercised, severe chronic MPD would suggest that exercise may not directly affect the removal of MPTP and MPP+ from the brain. On the other hand, this investigation indicates that concomitant and sustained long term exercise is beneficial even at a moderate activity level (450 m/day) in test animals with chronic exposure to low levels of neurotoxin. This finding might be considered relevant to PD in humans, who are potentially exposed to low levels of neurotoxic xenobiotics throughout life, according to the environmental hypothesis.
In summary, this research has demonstrated that the chronic MPD with moderate neurodegeneration is characterized by striatal protein oxidation and mitochondrial dysfunction, but no change in endogenous levels of BDNF or GDNF. Long-term treadmill exercise training in this chronic MPD effectively protected against the neurotoxin-induced protein oxidation, impaired mitochondrial function, loss of dopaminergic neurons and transmission, and elevated nigrostriatal neurotrophic factors. Endurance exercise may additionally produce neuroprotective benefits through other multi-faceted mechanisms. By modifying lifestyle and increasing physical activity, exercise would be an alternative, non-pharmacological neuroprotective approach for averting neuronal degenerative processes as demonstrated in the chronic MPD. Uncovering the mechanistic pathways using this endurance exercise-neuroprotective model and approach will allow the development of potential exercise-mimetic strategies for slowing down the progression of neurodegenerative disorders in individuals for which exercise may not be a feasible or acceptable option.
Supplementary Material
Acknowledgments
This work was supported by a grant from the US National Institute of Neurological Disorders and Stroke (NS 47920 to Y.S.L.). The technical assistance of Drs. Chao Li, Lesya Novikova, Konstantino Pothakos, and Ji-Hyuk Park is greatly acknowledged. Preliminary results of this study were presented in abstract form at the U.S. Society for Neuroscience Annual Meeting, October 17-21, 2009 in Chicago, IL, U.S.A.
Abbreviations
- ANOVA
analysis of variance
- BDNF
brain-derived neurotrophic factor
- DA
dopamine
- DAT
dopamine uptake transporter
- GAPDH
glyceraldehyde 3 phosphate dehydrogenase
- GDNF
glial cell line-derived neurotrophic factor
- MPD
mouse model of Parkinson’s disease
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA
6-hydroxydopamine
- PD
Parkinson’s disease
- RCR
respiratory control ratio
- ROS
reactive oxygen species
- SNpc
substantia nigra pars compacta
- SOD
superoxide dismutase
- TH
tyrosine hydroxylase
References
- Al-Jarrah M, Pothakos K, Novikova L, Smirnova IV, Kurz MJ, Stehno-Bittel L, Lau YS. Endurance exercise promotes cardiorespiratory rehabilitation without neurorestoration in the chronic mouse model of parkinsonism with severe neurodegeneration. Neuroscience. 2007;149:28–37. doi: 10.1016/j.neuroscience.2007.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baatile J, Langbein WE, Weaver F, Maloney C, Jost MB. Effect of exercise on perceived quality of life of individuals with Parkinson's disease. J Rehabil Res Dev. 2000;37:529–534. [PubMed] [Google Scholar]
- Barber-Singh J, Seo BB, Nakamaru-Ogiso E, Lau YS, Matsuno-Yagi A, Yagi T. Neuroprotective effect of long-term NDI1 gene expression in a chronic mouse model of Parkinson disorder. Rejuvenation Res. 2009;12:259–267. doi: 10.1089/rej.2009.0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Oxidatively modified proteins in aging and disease. Free Radic Biol Med. 2002;32:797–803. doi: 10.1016/s0891-5849(02)00780-3. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol. 2005;58 :495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, Di Monte DA, Greenamyre JT. Mechanistic approaches to Parkinson's disease pathogenesis. Brain Pathol. 2002;12:499–510. doi: 10.1111/j.1750-3639.2002.tb00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Scapagnini G, Giuffrida Stella AM, Bates TE, Clark JB. Mitochondrial involvement in brain function and dysfunction: relevance to aging, neurodegenerative disorders and longevity. Neurochem Res. 2001;26:739–764. doi: 10.1023/a:1010955807739. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ, Lee JM. Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson's disease brain. J Chem Neuroanat. 2001;21:277–288. doi: 10.1016/s0891-0618(01)00115-6. [DOI] [PubMed] [Google Scholar]
- Cheng FC, Ni DR, Wu MC, Kuo JS, Chia LG. Glial cell line-derived neurotrophic factor protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity in C57BL/6 mice. Neurosci Lett. 1998;252:87–90. doi: 10.1016/s0304-3940(98)00554-0. [DOI] [PubMed] [Google Scholar]
- Cohen AD, Tillerson JL, Smith AD, Schallert T, Zigmond MJ. Neuroprotective effects of prior limb use in 6-hydroxydopamine-treated rats: possible role of GDNF. J Neurochem. 2003;85:299–305. doi: 10.1046/j.1471-4159.2003.01657.x. [DOI] [PubMed] [Google Scholar]
- Comella CL, Stebbins GT, Brown-Toms N, Goetz CG. Physical therapy and Parkinson's disease: a controlled clinical trial. Neurology. 1994;44:376–378. doi: 10.1212/wnl.44.3_part_1.376. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA, Amara CE, Marcinek DJ. Mitochondrial dysfunction: impact on exercise performance and cellular aging. Exerc Sport Sci Rev. 2007;35:43–49. doi: 10.1249/JES.0b013e31803e88e9. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Drucker-Colin R, Garcia-Hernandez F. A new motor test sensitive to aging and dopaminergic function. J Neurosci Methods. 1991;39:153–161. doi: 10.1016/0165-0270(91)90081-a. [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc Natl Acad Sci U S A. 2007;104:1325–1330. doi: 10.1073/pnas.0605208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar R, Marincek C. Physical activity for elderly persons with neurological impairment: a review. Scand J Rehabil Med. 2000;32:99–103. [PubMed] [Google Scholar]
- Fisher BE, Petzinger GM, Nixon K, Hogg E, Bremmer S, Meshul CK, Jakowec MW. Exercise-induced behavioral recovery and neuroplasticity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse basal ganglia. J Neurosci Res. 2004;77:378–390. doi: 10.1002/jnr.20162. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haskell WL, Lee IM, Pate RR, Powell KE, Blair SN, Franklin BA, Macera CA, Heath GW, Thompson PD, Bauman A. Physical activity and public health: updated recommendation for adults from the American College of Sports Medicine and the American Heart Association. Circulation. 2007;116:1081–1093. doi: 10.1161/CIRCULATIONAHA.107.185649. [DOI] [PubMed] [Google Scholar]
- Hennigan A, O'Callaghan RM, Kelly AM. Neurotrophins and their receptors: roles in plasticity, neurodegeneration and neuroprotection. Biochem Soc Trans. 2007;35:424–427. doi: 10.1042/BST0350424. [DOI] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol. 2000;166:127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- Lau YS. Progressive neurodegeneration in the MPTP/probenecid model of Parkinson's disease. In: Ebadi M, Pfeiffer R, editors. Parkinson's Disease. CRC Press; Boca Raton, FL: 2005. pp. 109–115. [Google Scholar]
- Lau YS, Novikova L, Roels C. MPTP treatment in mice does not transmit and cause Parkinsonian neurotoxicity in non-treated cagemates through close contact. Neurosci Res. 2005;52 :371–378. doi: 10.1016/j.neures.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Lau YS, Trobough KL, Crampton JM, Wilson JA. Effects of probenecid on striatal dopamine depletion in acute and long-term 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. Gen Pharmacol. 1990;21:181–187. doi: 10.1016/0306-3623(90)90898-v. [DOI] [PubMed] [Google Scholar]
- Mabandla M, Kellaway L, St Clair Gibson A, Russell VA. Voluntary running provides neuroprotection in rats after 6-hydroxydopamine injection into the medial forebrain bundle. Metab Brain Dis. 2004;19:43–50. doi: 10.1023/b:mebr.0000027416.13070.c3. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Neuroprotective signaling and the aging brain: take away my food and let me run. Brain Res. 2000;886:47–53. doi: 10.1016/s0006-8993(00)02790-6. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Petroske E, Santa Cruz K, Callison RC, Jr, Lau YS. Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson's disease. Brain Res. 2002;956:156–165. doi: 10.1016/s0006-8993(02)03514-x. [DOI] [PubMed] [Google Scholar]
- Miyai I, Fujimoto Y, Ueda Y, Yamamoto H, Nozaki S, Saito T, Kang J. Treadmill training with body weight support: its effect on Parkinson's disease. Arch Phys Med Rehabil. 2000;81:849–852. doi: 10.1053/apmr.2000.4439. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Brain mitochondrial dysfunction in aging: conditions that improve survival, neurological performance and mitochondrial function. Front Biosci. 2007;12 :1154–1163. doi: 10.2741/2133. [DOI] [PubMed] [Google Scholar]
- Novikova L, Garris BL, Garris DR, Lau YS. Early signs of neuronal apoptosis in the substantia nigra pars compacta of the progressive neurodegenerative mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of Parkinson's disease. Neuroscience. 2006;140:67–76. doi: 10.1016/j.neuroscience.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R. Reduced expression of brain-derived neurotrophic factor protein in Parkinson's disease substantia nigra. Neuroreport. 1999;10:557–561. doi: 10.1097/00001756-199902250-00021. [DOI] [PubMed] [Google Scholar]
- Patki G, Che Y, Lau YS. Mitochondrial dysfunction in the striatum of aged chronic mouse model of Parkinson's disease. Front Aging Neurosci. 2009;1:3. doi: 10.3389/neuro.24.003.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AC, Huddleston DE, Brickman AM, Sosunov AA, Hen R, McKhann GM, Sloan R, Gage FH, Brown TR, Small SA. An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A. 2007;104:5638–5643. doi: 10.1073/pnas.0611721104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroske E, Meredith GE, Callen S, Totterdell S, Lau YS. Mouse model of Parkinsonism: a comparison between subacute MPTP and chronic MPTP/probenecid treatment. Neuroscience. 2001;106:589–601. doi: 10.1016/s0306-4522(01)00295-0. [DOI] [PubMed] [Google Scholar]
- Pothakos K, Kurz MJ, Lau YS. Restorative effect of endurance exercise on behavioral deficits in the chronic mouse model of Parkinson's disease with severe neurodegeneration. BMC Neurosci. 2009;10:6. doi: 10.1186/1471-2202-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulton NP, Muir GD. Treadmill training ameliorates dopamine loss but not behavioral deficits in hemi-parkinsonian rats. Exp Neurol. 2005;193:181–197. doi: 10.1016/j.expneurol.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Reuter I, Engelhardt M, Stecker K, Baas H. Therapeutic value of exercise training in Parkinson's disease. Med Sci Sports Exerc. 1999;31:1544–1549. doi: 10.1097/00005768-199911000-00008. [DOI] [PubMed] [Google Scholar]
- Schallert T, Woodlee MT, Fleming SM. Disentangling multiple types of recovery from brain injury. In: Krieglstein J, Klumpp S, editors. Pharmacology of Cerebral Ischemia. Medpharm Scientific Publishers; Stuttgart, Germany: 2002. pp. 201–216. [Google Scholar]
- Scott SA, Diaz NM, Ahmad SO. Stereologic analysis of cell number and size during postnatal development in the rat substantia nigra. Neurosci Lett. 2007;419:34–37. doi: 10.1016/j.neulet.2007.03.067. [DOI] [PubMed] [Google Scholar]
- Smith AD, Zigmond MJ. Can the brain be protected through exercise? Lessons from an animal model of parkinsonism. Exp Neurol. 2003;184:31–39. doi: 10.1016/j.expneurol.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–172. doi: 10.1016/S0014-4886(03)00342-X. [DOI] [PubMed] [Google Scholar]
- Tajiri N, Yasuhara T, Shingo T, Kondo A, Yuan W, Kadota T, Wang F, Baba T, Tayra JT, Morimoto T, Jing M, Kikuchi Y, Kuramoto S, Agari T, Miyoshi Y, Fujino H, Obata F, Takeda I, Furuta T, Date I. Exercise exerts neuroprotective effects on Parkinson's disease model of rats. Brain Res. 2010;1310:200–207. doi: 10.1016/j.brainres.2009.10.075. [DOI] [PubMed] [Google Scholar]
- Tang FI, Tien LT, Zhou FC, Hoffer BJ, Wang Y. Intranigral ventral mesencephalic grafts and nigrostriatal injections of glial cell line-derived neurotrophic factor restore dopamine release in the striatum of 6-hydroxydopamine-lesioned rats. Exp Brain Res. 1998;119:287–296. doi: 10.1007/s002210050344. [DOI] [PubMed] [Google Scholar]
- Thyagarajan D, Bressman S, Bruno C, Przedborski S, Shanske S, Lynch T, Fahn S, DiMauro S. A novel mitochondrial 12SrRNA point mutation in parkinsonism, deafness, and neuropathy. Ann Neurol. 2000;48:730–736. [PubMed] [Google Scholar]
- Tillerson JL, Caudle WM, Reveron ME, Miller GW. Exercise induces behavioral recovery and attenuates neurochemical deficits in rodent models of Parkinson's disease. Neuroscience. 2003;119:899–911. doi: 10.1016/s0306-4522(03)00096-4. [DOI] [PubMed] [Google Scholar]
- van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25:8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND, Fowler JS, Franceschi D, Logan J, Pappas NR, Wong CT, Netusil N. PET studies of the effects of aerobic exercise on human striatal dopamine release. J Nucl Med. 2000;41:1352–1356. [PubMed] [Google Scholar]
- Xu J, Kao SY, Lee FJ, Song W, Jin LW, Yankner BA. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med. 2002;8:600–606. doi: 10.1038/nm0602-600. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.