
Figure 1.
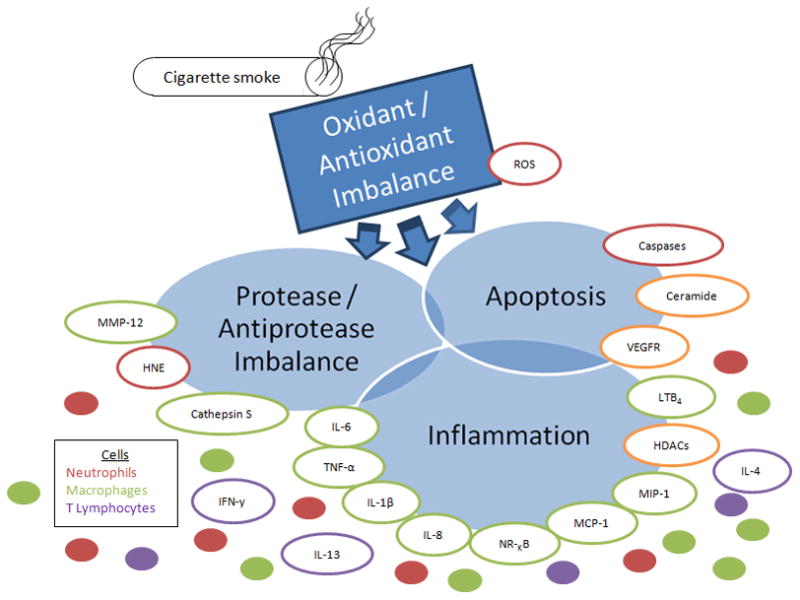
Confluence and interplay of major processes and mediators associated with COPD pathogenesis. Lung tissue damage resulting from the inhalation of free radicals in cigarette smoke results in oxidative stress and leads to the recruitment of neutrophils, macrophages and T lymphocytes to the lungs. Oxidative stress also leads to upregulation of ceramide, resulting in apoptosis of lung epithelial cells. Neutrophils release HNE while macrophages release MMP-12, and cathepsin S which collectively degrade lung elastin and exracellular matrix components. In addition, macrophages activate NF-κB and release various pro-inflammatory and chemotactic mediators which amplify inflammation while neutrophils release reactive oxygen species. MMP-12, oxidants in cigarette smoke, and reactive oxygen species inactivate alpha-1-proteinase inhibitor, the physiological inhibitor of HNE, resulting in a protease/anti-protease imbalance which allows the renegade proteases to degrade lung elastin. HNE inactivates TIMPs, the physiological inhibitor of MMP-12, augmenting elastin degradation. Elastin fragments are chemoattractants to macrophages and drive the progression of the disease. Alveolar wall homeostasis is eventually compromised, leading to enlargement of the airspaces and the COPD phenotype. Abbreviations: MMP-12, macrophage metalloelastase; HNE, human neutrophil elastase; TNF-α, tumor necrosis factor alpha; IL-1β, interleukin 1beta; IL-8, interleukin 8; MCP-1, monocyte chemotactic protein; MIP-1, monocyte inflammatory protein; HDACs, histone deacetylases; LTB4, leukotriene B4; VEGFR, vascular endothelium growth fibroblast factor; NF-κB, nuclear factor kappa B; IFN-γ, interferon gamma; ROS, reactive oxygen species.