

Abstract
Human thymidine phosphorylase (hTP) is responsible for thymidine (dT) homeostasis, promotes angiogenesis, and is involved in metabolic inactivation of antiproliferative agents that inhibit thymidylate synthase. Understanding its transition state structure is on the path to design transition state analogues. Arsenolysis of dT by hTP permits kinetic isotope effect (KIE) analysis of the reaction by forming thymine and the chemically unstable 2-deoxyribose 1-arsenate. The transition state for the arsenolytic reaction was characterized using multiple KIEs and computational analysis. Transition state analysis revealed a concerted bimolecular (ANDN) mechanism. A transition state constrained to match the intrinsic KIE values was found using density functional theory [B3LYP/6-31+G*]. An active site histidine is implicated as the catalytic base responsible for activation of the arsenate nucleophile and stabilization of the thymine leaving group during the isotopically sensitive step. At the transition state, the deoxyribose ring exhibits significant oxocarbenium ion character with bond-breaking (rC-N = 2.45 Å) nearly complete but no significant bond-making to the attacking nucleophile (rC-O = 2.95 Å). The transition state model predicts a deoxyribose conformation with a 2′-endo ring geometry. Transition state structure for the slow hydrolytic reaction of hTP involves a stepwise mechanism (Schwartz, P.A., Vetticatt, M.J., Schramm, V.L. (2010) J. Am. Chem. Soc. 132, 13425–13433), in contrast to the concerted mechanism described here for arsenolysis.
Human thymidine phosphorylase (hTP)1 catalyzes the reversible phosphorolytic depyrimidination of thymidine (dT) (1, 2):
Arsenate is a substrate analogue of phosphate and reduces the reverse reaction to permit transition state analysis by kinetic isotope effects (KIEs) (see commitments to catalysis). The hTP-catalyzed arsenolytic depyrimidination of dT forms an unstable 2-deoxy-α-D-ribose 1-arsenate which undergoes spontaneous hydrolysis (Fig. 1).
Figure 1.
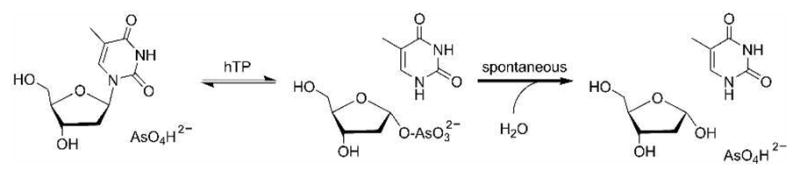
hTP-catalyzed arsenolytic depyrimidination of dT.
hTP is involved in dT homeostasis by participating in pyrimidine salvage (3, 4). The enzyme is also involved in promoting angiogenesis (5–7). hTP is linked to angiogenesis by the production of 2-deoxyribose (dRib) from dT, a chemoattractant that stimulates the endothelial cell migration for new capillary formation (8, 9). Angiogenesis is essential in the progression of solid tumors, as they fail to grow beyond a few mm3 without a capillary bed to meet demands for nutrients and oxygen (10). hTP inhibition is proposed to halt the angiogenesis required for tumor hyperproliferation (11). hTP also degrades antiproliferative agents including 5-trifluorothymidine (TFT) and 2′-deoxy-5-fluorouridine (5FdU), compounds designed to inhibit thymidylate synthase, an enzyme crucial in de novo DNA synthesis (12–14). Administration of an hTP inhibitor in combination with these drugs increases their efficacy, permitting reduced dose and decreased off-target toxicity (15).
Transition state analysis has been used to design transition state analogues in other N-ribosyltransferases (16). A common feature for N-ribosidic bond cleavage reactions of nucleosides and nucleotides is the appearance of an oxocarbenium ion intermediate (stepwise process) or a transition state exhibiting oxocarbenium ion character (concerted bimolecular process) (Fig. 2) (17). The majority of these reactions proceed through highly dissociative concerted ANDN mechanisms2 where leaving group departure is advanced before nucleophile approach (18–22). Stepwise mechanisms predominantly involve 2-deoxyribosides where all 2-deoxyriboside hydrolysis reactions are stepwise. Base excision repair enzymes uracil DNA glycosylase (23, 24) and MutY (25) proceed through stepwise DN‡*AN and DN*AN‡ mechanisms, respectively. Ricin A-chain catalyzes the depurination of small stem-loop DNA in a DN‡*AN reaction (26). Recent studies reveal that the hTP-catalyzed hydrolysis of dT involves a stepwise DN*AN‡ process (27).
Figure 2.

Generic mechanism for glycosidic bond cleavage of ribofuranosides. In the upper pathway, attack from the nucleophile (Nu) displaces the leaving group (LG) in a concerted bimolecular process (ANDN). In the lower pathway, departure of LG occurs in an independent step (DN) from attack of the 2-deoxyribosyl oxocarbenium ion by Nu (AN). In a DN‡*AN mechanism, the DN step is rate-limiting. In a DN*AN‡ mechanism, the AN step is rate-limiting, and the glycosidic bond cleaves and reforms many times before nucleophilic capture.
Here, the arsenolytic depyrimidination of dT by hTP is documented kinetically and by multiple KIEs. hTP catalyzes a concerted, dissociative ANDN mechanism reaction with ribooxocarbenium ion character at the transition state. Important hydrogen bond interactions between the nucleophile and the leaving group to active site His 116 facilitates catalysis.
EXPERIMENTAL PROCEDURES
Materials
3H- and 14C- labeled riboses and glucoses and [5′-3H]dT were purchased from American Radiolabeled chemicals. 15N-labeled thymine was a generous gifts from Industrial Research Limited (Lower Hutt, New Zealand). Tetrabutylammonium bisulfate (Fluka), and 2-deoxyribose (dRib, Acros) were purchased commercially. Ultima Gold scintillation fluid (Perkin-Elmer) was used for all scintillation counting. Acetonitrile, methanol, trifluoroacetic acid, and 14.6 cm glass Pasteur pipettes for charcoal columns were purchased from Fisher. Ribonucleotide-triphosphate reductase was a generous gift from Dr. Gary Gerfen (Albert Einstein College of Medicine) (28). Ribokinase (29), phospho-D-ribosyl-1-pyrophosphate synthase (29), and adenine phosphoribosyltransferase (30) were prepared as described previously. All other reagents and synthetic enzymes were from Sigma-Aldrich.
The gene encoding hTP was subcloned into a pTWIN1 expression vector (New England Biolabs) and overexpressed in the K BR2566 (T7 express) cell strain of Escherichia coli (New England Biolabs). hTP was purified as described elsewhere (27) and concentrated by ultrafiltration to ~ 20 mg/mL as determined by the calculated molar extinction coefficient of 23,490 M−1 cm−1 at 280 nm with a specific activity of 10 U/mg at 22 °C for the phosphorolysis of dT. This construct and associated after-expression processing generates the native amino acid sequence encoded by human mRNA for hTP. Stock enzyme was stored in 20 mM phosphate pH 7.4. Before use, stock enzyme was thawed on ice, inserted into a 0.5 mL slide-a-lyzer dialysis cassette, and dialyzed against argon-saturated 20 mM HEPES pH 7.4 buffer at 4 °C with 7 × 300 mL exchanges over 20 hours. A constant stream of argon was bubbled through the buffer during dialysis.
Analytical Methods
hTP activity was monitored by spectrophotometer (Cary 300) in a 1 cm−1 path length quartz cuvette containing 1 mL of 20 mM HEPES pH 7.4, 50 mM potassium phosphate (or 10 mM sodium arsenate), 1 mM dT, and ~ 50 nM hTP. The reaction progress was monitored by the decrease in absorbance of dT upon depyrimidination at 290 nm using an extinction coefficient of Δε290 = 1000 M−1 cm−1.
Steady-State Parameters for hTP Arsenolysis
A model SX-20 stopped-flow spectrometer (Applied Photophysics) outfitted with a mercury-xenon lamp was used to follow the arsenolytic depyrimidination of dT by hTP in order to capture the initial velocity period of the steady state at low substrate concentrations, while still maintaining enough hTP to accurately measure a rate. In the reaction chamber, 20 mM HEPES pH 7.4, 1 mM DTT, and varying concentrations of dT or sodium arsenate under saturating concentrations of the second substrate (2.5 mM sodium arsenate or 0.5 mM dT, respectively) were monitored for hTP-catalyzed arsenolysis using the parameters described above (see analytical methods). hTP concentration was maintained at 175 nM (22 °C) or 90 nM (37 °C) in the assay. Apparent Km’s and kcat were determined using the Michaelis-Menten equation.
Synthesis of radiolabeled dT
Except for the 5′-3H and 2′-3H labels, dTs were synthesized enzymatically in three steps via ATP and dATP based on an enzymatic synthesis described previously (31). The exact experimental procedure used for this enzymatic synthesis has been reported (27).
Glucose or ribose was converted into ATP through the action of adenine phosphoribosyltransferase, phospho-D-ribosyl-1-pyrophosphate synthase, pyruvate kinase, and myokinase. In reactions starting from glucose additional enzymes were added: hexokinase, glucose-6-phosphate dehydrogenase, phosphogluconic acid dehydrogenase, phosphoriboisomerase, and glutamate dehydrogenase. In reactions starting from ribose, ribokinase was present. Radiolabeled ATP was converted into dATP using class II ribonucleotide-triphosphate reductase. dATP was converted to dT in a two part procedure previously described using hexokinase, myokinase, adenosine deaminase, alkaline phosphatase, hTP, purine nucleoside phosphorylase, and xanthine oxidase (32).
2′-3H labels were synthesized enzymatically from specifically radiolabeled dRib with ribokinase, phosphoglucomutase, and hTP. 5′-3H labeled dT was commercially available. Labels and their starting material can be found in Table 1 and the original synthetic procedure is documented elsewhere (27).
Table 1.
Radiolabeled starting material used to synthesize isotopically enriched thymidines and remote labels used in KIE experiments.
| radiolabeled thymidine | starting material | remote label |
|---|---|---|
| [1′-14C]dT | [1-14C]ribose | [5′-3H]dT |
| [1-15N, 5′-14C]dT | [6-14C]glucosea | [5′-3H]dT |
| [1′-3H]dT | [1-3H]ribose | [5′-14C]dT |
| [2′R-3H]dT | [2R-3H]-2-deoxyribose | [5′-14C]dT |
| [2′S-3H]dT | [2S-3H]-2-deoxyribose | [5′-14C]dT |
| [5′-3H]dT | [6-3H]glucose | [5′-14C]dT |
| [5′-14C]dT | Commercially available | n.a.b |
1-15N labeled thymine was also used in the synthesis.
The KIE of [5′-14C]dT is assumed to be unity (see text).
Determination of KIEs by Scintillation Counting
Competitive KIEs for substrate isotopically enriched with either 3H or 14C at various positions were measured for the arsenolytic reaction catalyzed by hTP. The isotopic label of interest was mixed with the remote label listed in Table 1 at a CPM ratio of 2:1 3H:14C. KIEs were determined by measuring the difference in the 3H/14C ratio of products from a partially reacted sample and a reaction taken to completion (33). Two 200 μL reactions in 20 mM HEPES pH 7.4 were run in parallel, the partially reacted and the completely reacted samples, and augmented with 100 μM or 10 mM sodium arsenate, respectively. A master mix containing the appropriate pair of radiolabels (5 × 105 cpm of 14C) and cold carrier was used to make each sample 100 μM dT. The reactions were initiated with the addition of either 5 nM hTP to the partially reacted sample or 300 nM hTP to the completely reacted sample. After incubation for 1 h at 37 °C, 40 μL aliquots (6 from each reaction mixture) were loaded onto activated charcoal columns poured in 14.6 cm glass Pasteur pipettes plugged with glass wool. Charcoal columns contained ~1.3 mL of a 4:1 cellulose/charcoal resin poured from a slurry in 10 mM dRib wash buffer. After the sample entered the charcoal bed, the wall of the pipette was rinsed with 3 × 40 μL of wash buffer. One mL of wash buffer was added to the column and eluted. The wash step was followed by 3 × 1 mL elution steps using wash buffer augmented with 10% ethanol. All elution steps from one column were collected directly into one scintillation vial. Samples were dried on a speed-vac, resuspended in 200 μL of H2O, mixed with 10 mL of scintillation fluid, and measured by scintillation counting.
3H/14C ratios were determined by counting samples for 10 × 5 min in a dual channel scintillation detector (Wallac Winspectral model 1414) and using an appropriate 14C standard to relate channel cpm to total cpm from either radiolabel. Complete reaction in the appropriate sample was verified by the reversed-phase HPLC on a small aliquot quenched in 2 μL of TFA (27). The fractional extent of the reaction was determined both chromatographically, in a similar fashion to the completely reacted sample and radiometrically, by comparing the remote label in the partially and completely reacted samples. Observed KIEs were calculated according to eq 1.
| (1) |
Where f is the fractional extent of the reaction and Rp and R0 are the isotope ratios in the product at fractional and total reaction, respectively.
Competitive KIEs using a remote label are a function of the apparent KIEs for both labeled and remote labeled substrate. The measured KIE for 5′-14C was assumed to be unity as it is three bonds removed from the reaction center and 14C does not manifest significant isotope effects for geometric changes or binding (in contrast to remote 3H labels) (34, 35). The KIE for the position of interest was calculated by first determining the KIE for the remote 3H label and using that value to correct the observed KIE. Radiolabeled substrates and the remote labels used are listed in Table 1. The apparent KIE for the 3H label used a separate experiment with [5′-14C]dT as the remote label. Apparent KIEs for the positions at or near the reaction coordinate were calculated according to eq 2.
| (2) |
KIEapp is the apparent KIE for the position of interest, KIEobs is the experimentally measured KIE, and KIEremote is the observed KIE for the remote label.
Computational analysis
The arsenolytic reaction of thymidine catalyzed by hTP was studied using B3LYP method with a 6-31G* basis set as implemented in Gaussian 09. Reactant, intermediate, and product geometries were located as global minima and frequency calculations performed on these optimized geometries had no imaginary frequencies. Most transition state structures, located with and without geometric constraints, were found to have only one imaginary frequency, characteristic of true potential energy saddle points. The errors associated with isotope effect predictions for geometries with more than one imaginary frequency are discussed elsewhere (37). Isotope effects were calculated for each of these transition structures using ISOEFF 98 (38). For the final model, a one-dimensional infinite parabola correction was applied to account for tunneling contributions (39).
RESULTS
Steady-state Kinetic Parameters for hTP-Catalyzed Arsenolysis
Kinetic parameters for the arsenolytic depyrimidination of dT by hTP were measured using a stopped-flow spectrophotometer. Data followed Michaelis-Menten type kinetics and values for apparent KM, kcat, and the second-order rate constant (kcat/KM) are summarized in Table 2 along with the previously reported values (27) for the phosphorolytic reaction measured using the same assay.
Table 2.
Steady state kinetic parameters for the arsenolytic and phosphorolytic depyrimidination of dT by hTP.
| conditiona | kcat (s−1) | KM dTb, c (μM) | KM nucleophileb, c (μM) | kcat/KMd (M−1 s−1) |
|---|---|---|---|---|
| Phosphorolysis, 22 °Ce | 2 ± 0.1 | 31 ± 6 | 5 ± 0.6 | (6.7 ± 1.3) × 104 |
| Arsenolysis, 22 °C | 3 ± 0.1 | 60 ± 4 | 5 ± 0.9 | (5 ± 0.4) × 104 |
| Phosphorolysis, 37 °Ce | 7 ± 0.2 | 30 ± 4 | 11 ± 2 | (2.3 ± 0.3) × 105 |
| Arsenolysis, 37 °C | 8 ± 0.4 | 112 ± 15 | 29 ± 7 | (7.3 ± 1) × 104 |
hTP was incubated at 22 °C and 37 °C and steady state kinetic parameters determined as described.
Nucleophile is PO4 in phosphorolysis and AsO4 in arsenolysis.
Apparent KM was determined for varying concentrations of substrate at a saturating concentration of the other substrate.
kcat/KM value listed is for dT at saturating concentrations of the nucleophilic substrate.
Data from Ref. 32.
Values of kcat for the arsenolytic and phosphorolytic reactions were similar at 22 °C and 37 °C. The KM values for dT are similar at 22 °C, but at 37 °C, dT shows fourfold lower affinity with saturating arsenate than with phosphate. Dependence of affinity for dT on the nucleophile supports the report that phosphate binding causes conformational changes to form the active site in the structurally related E. coli thymidine phosphorylase (40). Differences in AsO4 and PO4 size and charge cause changes in the active site that translate into the differences in dT affinity.
Commitments to Catalysis
KIEs measured by the competitive label method give the apparent isotope effect on kcat/KM and reflect all steps from free reactants through the first irreversible step (41). These include contributions from non-chemical steps, steps that achieve equilibrium during the reaction, and are frequently manifesting as commitments to catalysis (42). Intrinsic KIEs can be approached by corrections for commitment factors.
Isotope effect studies on the N-ribosyltransferases are usually influenced by large commitments to catalysis. Arsenate is often useful to reduce commitments in these systems (19, 43, 44). The product of arsenolysis, ribose 1-arsenate, is unstable and rapidly hydrolyzes to ribose and arsenate (45). Hydrolysis prior to product release prevents internal reversibility, thus removing the reverse commitment. A decrease in the rate of the chemical step can shift the rate-determining step from a non-chemical one. A decrease in the affinity of enzyme for labeled substrate or product would lead to a reduction in the commitments.
KIE measurements for hTP arsenolysis were made at a variety of temperatures, and at 22 °C partial suppression of KIEs was observed (data not shown). Similar experiments were performed at 37 °C showing more significant expression of KIEs (Table 3). Traditionally, a forward commitment has been assessed with isotope trapping, but low affinity of hTP for dT in the absence of an inorganic ester and the presence of a an alternate hydrolytic activity make such measurements impossible (data not shown) (27). Computational analysis (see below) reveled that the experimental 1-15N KIE of 1.018 was within experimental error of the maximum theoretical KIE for this position (1.020). Thus, masking of the experimental KIEs by commitment factors at 37 °C must be small and the observed KIE approximates the intrinsic KIE.
Table 3.
Experimental competitive KIEs for the arsenolytic depyrimidination of dT by hTP.
| labeled dT | type of KIE | apparent KIEa, b |
|---|---|---|
| [1′-14C]dT | primary 14C | 1.025 ± 0.003 (2)c |
| [1-15N, 5′-14C]dT | primary 15N | 1.018 ± 0.002 (2)c |
| [1′-3H]dT | α-secondary 3H | 1.177 ± 0.002 (2) |
| [2′R-3H]dT | β-(R)-secondary 3H | 1.028 ± 0.001 (2) |
| [2′S-3H]dT | β-(S)-secondary 3H | 1.048 ± 0.003 (2) |
| [5′-3H]dT | δ-secondary 3H | 1.019 ± 0.002 (2) |
The number in parentheses is the number of independent KIE experiments.
The experimentally measured KIE for the position of interest (corrected for remote label where noted) is equal to the apparent KIE (see text).
KIEs were corrected for the remote 3H label according to the expression: KIEapp = KIEobs × KIEremote, where KIEapp is the apparent KIE for the position of interest, KIEobs is the experimentally measured KIE, and KIEremote is the observed KIE for the remote label.
KIEs for dT Arsenolysis
Intrinsic KIEs reflect the nature of the transition state at the kinetically significant (rate-determining) chemical step (41, 42). Measurement of multiple KIEs for substrates provides information related to transition state structure. KIE measurements were made on multiple labeled substrates for the hTP-catalyzed arsenolytic depyrimidination of dT (Table 3).
1′-14C KIE
Primary 14C KIEs for N-glycoside hydrolysis and transfer reactions report on the extent of oxocarbenium ion character at the transition state. Values on the order of 1.00 to 1.03 are expected for reactions involving well-developed oxocarbenium ion transition states and of 1.080 to 1.13 for associative concerted bimolecular reactions with neutrally charged pentavalent transition states (17). The value observed for hTP arsenolysis of 1.025 indicates significant oxocarbenium ion character at the chemical step.
1-15N KIE
The extent of C1′-N1 bond cleavage is related to the primary 15N KIE and lies between unity for early (substrate like) transition states and the equilibrium isotope effect (EIE) for late (product like) transition states (41). The value of 1.018 indicates considerable C1′-N1 bond cleavage and is in agreement in with the 1′-14C KIE which demonstrates significant oxocarbenium ion character during the reaction.
1′-3H KIE
Support for a well-developed oxocarbenium ion-character in the arsenolytic reaction is given by the α-secondary 3H KIE and is consistent with other enzyme-catalyzed N-glycoside bond cleavage reactions demonstrating this characteristic. A range of 1.15 to 1.34 for α-secondary 3H KIEs is typical in oxocarbenium ion-like transition states (22). Rehybridization at C1′ from sp3 in the reactant to sp2 in the transition state gives increased freedom to the out-of-plane bending mode of H1′ resulting in the large normal KIE (46, 47). The value of 1.177 is significantly lower than observed for hTP hydrolysis (1.325) (27). hTP hydrolysis was established to react via a DN*AN‡ mechanism. A steric effect from the leaving group or attacking AsO4 (absent in the TS for the hydrolytic reaction) may contribute to the observed decrease in the α-secondary KIE. This would support a dissociative ANDN mechanism for hTP arsenolysis.
2′-3H KIEs
The magnitudes of the β-secondary KIEs give geometric information on the ribose ring at the transition state. The oxocarbenium ion exhibits significant π-bonding character in the O4′-C1′ bond, favoring coplanarity between the C4′-O4′-C1′-C2′ atoms. Hyperconjugation between the C2′-H2′ σ-bond and the vacant p-orbital of C1′ can increase π-bonding character in the C1′-C2′ bond and weaken the C2′-H2′ σ-bond, resulting in a looser bonding environment for the H2′ hydrogen and an increasing normal KIE with optimization of orbital alignment (18, 48). This phenomenon stabilizes the positive charge on the anomeric carbon in the oxocarbenium ion. The low values observed for both 2′R -3H (1.028) and 2′S -3H (1.048) are unusual and indicate limited hyperconjugation at the TS. KIEs at this position for related N-glycosides with 2′-deoxyribose sugars are not widely documented, but show a trend of significant hyperconjugation to both 2′ hydrogens, with KIEs ranging from 1.08 to 1.15 (23, 25, 26). Other related reactions have indicated the absence of stabilizing hyperconjugative forces from the 2′ position at the transition state (43, 49, 50). Binding isotope effects or steric compression by active site residues at the transition state may diminish the apparent KIE, and these effects must also be considered.
The KIE measurements described here point to a transition state with extensive oxocarbenium ion character at the transition state. Computational matching of the experimental KIEs is required for a definitive mechanistic conclusion.
DISCUSSION
ANDN Mechanism and Predicted KIEs
This mechanism invokes simultaneous participation of the nucleophile (arsenate) and the leaving group (thymine) in the rate-limiting transition state. A previous theoretical study on a structurally related enzyme (TP from E. coli) proposed a key role for an active site histidine in the activation of phosphate and stabilization of a thymine anion by hydrogen bonding (51). Mutation of the analogous residue in hTP renders the enzyme catalytically inactive (52). We therefore incorporated a histidine mimic (imidazole) along with dT and arsenate in the computational analysis of the experimental KIEs.3
The KIEs of the atoms involved in the reaction coordinate (1′-14C, 1′-3H and 1′-15N) are primary indicators of transition state structure. A potential energy surface was generated by restraining the distances along the reaction coordinate, namely the breaking C-N bond (rC-N) and the forming C-O bond (rC-O).4 The best match between model and intrinsic KIEs corresponded to geometries where rC-N ranged from 2.5 – 2.9 Ǻ and rC-O from 3.0 – 2.5 Ǻ (Scheme 1) and when the sum of these distances was 5.4 Å. These geometries correspond to a dissociative transition state with significant oxocarbenium ion character. Several transition structures based on these select geometries were located by varying the orientation of arsenate and thymine with respect to the 2-deoxyribose ring. The predicted KIEs for these geometries led to a final transition state model within the framework of an ANDN mechanism (Fig. 3). It provided the best match of experimental and theoretical KIEs.
Scheme 1.

Figure 3.

Transition state structure determined for the hTP-catalyzed arsenolytic depyrimidination of dT. KIEs predicted by computational analysis are shown in blue.
The dissociative ANDN transition state exhibits significant oxocarbenium ion character, with a weak N-ribosidic bond (rC-N = 2.45 Å) and almost no significant bond order to the attacking nucleophile (rC-O = 2.95 Å) (Fig. 3). Favorable hydrogen bonds between the imidazole, the nucleophile and the leaving group determines their orientation with respect to the ribocation.5 The heavy atom KIE predictions (1′-14C and 1′-15N) are within the uncertainty of the experimental KIE measurements. The key 1′-3H KIE, which is sensitive to both reaction coordinate motion and steric effects enforced by the active site, is well predicted in this model. α-Secondary KIEs are often over-predicted since most computational models do not include contributions from the neighboring enzymatic groups. Including imidazole in this model provides an important step to improve the accurate modeling of this isotope effect.6
The KIEs that report on the ring-pucker of the 2′-deoxyribose ring, namely the 2′-3H(R) and 2′-3H(S) KIEs, are reasonably well predicted. Even though the calculations correctly predict a larger KIE for the (S) than the (R) hydrogen, the 2′-3H(S) KIE is over-predicted by 14% (1.188 versus the experimental KIE of 1.048). The most plausible explanation is that the KIEs for one of the two 2′-hydrogens is suppressed by factors that are not included in our theoretical model. Steric compression by an active-site residue is a reasonable hypothesis for this effect since: (1) hTP is selective for 2′-deoxy substrates indicating that the active site is crowded when a bulkier group is at this position (53), and (2) a similar phenomenon was observed in the prediction of KIEs for the hydrolytic reaction catalyzed by hTP where, at a geometry where all KIEs were consistent with experiment, the 2′-3H(S) KIE was over-predicted by nearly 5% (27). The observation that this effect is more pronounced in arsenolysis than in hydrolysis is also expected, as the binding of phosphate (and by analogy arsenate) causes a conformational change resulting in a domain closure around the active site (40). The low 2′-3H(R) KIE of 1.028 suggests that the transition state does not involve much overlap with this orbital and supports a mild 2′-endo transition structure (Fig. 3). The anticipated large 2′-3H(S) KIE is suppressed by interactions with the active site.
This transition state has low bond order to both the nucleophile and the leaving group (Fig. 3). We explored the effect of having only one of the two species participating at the rate limiting transition state, i.e. the possibility of a stepwise mechanism. The DN‡*AN (rate-limiting dissociation of thymine forming an oxocarbenium ion intermediate captured by arsenate at a subsequent step) or DN*AN‡ (reversible dissociation of thymine followed by rate-limiting capture of the oxocarbenium ion intermediate by arsenate) stepwise mechanisms were considered with and without the active site histidine (Schemes 2 and 3).
Scheme 2.

Scheme 3.

Stepwise Mechanisms and Predicted KIEs
For the DN‡*AN mechanism, dissociation of thymine was modeled by varying rC-N between 2.3 and 2.7 Ǻ at increments of 0.1 Ǻ. Each fixed distance geometry was optimized and frequency calculations performed. KIEs were calculated based on the single imaginary frequency corresponding to C-N bond cleavage (Table 4). Predicted KIEs for this mechanism are inconsistent with experiments. At longer C-N distances the 1′-14C KIEs are close to the experimental value of 1.025, but at these distances the 1′-3H KIE is larger due to the increased amplitude of the out-of-plane C-H bending mode. Including histidine (Scheme 2B) reduces this KIE, but the value is still 1.495, compared to the experimental value of 1.177. At short C-N distances the prediction for the 1′-3H KIE is in closer agreement (experimental value of 1.275), but the 1′-14C KIE of 1.048 is not well predicted.7 These observations do not support the DN‡*AN mechanism.8
Table 4.
Predicted KIEs along the reaction coordinate for a DN‡*AN mechanism.a
| KIE position |
rC-N (Å) |
||||
|---|---|---|---|---|---|
| 2.7 | 2.6 | 2.5 | 2.4 | 2.3 | |
| 1′-14C | 1.031 | 1.032 | 1.036 | 1.043 | 1.048 |
| 1′-14C + Hisb | 1.029 | 1.030 | 1.035 | 1.041 | 1.048 |
| 1-15N | 1.025 | 1.025 | 1.025 | 1.025 | 1.025 |
| 1-15N + Hisb | 1.023 | 1.023 | 1.024 | 1.024 | 1.024 |
| 1′-3H | 1.686 | 1.551 | 1.452 | 1.366 | 1.307 |
| 1′-3H + Hisb | 1.495 | 1.412 | 1.369 | 1.321 | 1.275 |
KIEs were computed using ISOEFF98 at 310.15 K. No correction for tunneling was incorporated.
KIEs of the model incorporating an active site histidine mimic.
A DN*AN‡ stepwise mechanism warrants scrutiny since the hTP-catalyzed hydrolytic reaction is characterized by this mechanism (27). Water is not the native nucleophile, resulting in an increased barrier for oxocarbenium ion capture. The hydrolytic transition state in best agreement with experimental KIEs involved reversible formation of thymine (lactam form) followed by capture of the oxocarbenium ion intermediate by an activated water nucleophile in the rate-limiting step.
A DN*AN‡ stepwise mechanism for arsenolysis predicts the KIE to be a product of the EIE for formation of the oxocarbenium ion and the intrinsic KIE for oxocarbenium ion capture by arsenate. The AN‡ step was modeled with and without the active site histidine mimic, by varying rC-O between 2.2 and 2.7 Å at fixed increments of 0.1 Å (Scheme 3; Table 5). These predicted KIEs show dependence of the 1′-14C KIEs on the nature of the nucleophile. At identical distances for the forming C-O bond, the 1′-14C KIEs are larger for histidine-activated arsenate than arsenate alone. When the predicted and experimental 1′-14C KIE are similar, the predicted 1′-3H KIE is significantly higher than experiment with or without the histidine mimic. The predicted KIEs for both stepwise mechanisms are inconsistent with the experimental measurements.
Table 5.
| KIE position |
rC-O (Å) |
|||||
|---|---|---|---|---|---|---|
| 2.2 | 2.3 | 2.4 | 2.5 | 2.6 | 2.7 | |
| 1′-14C | 1.040 | 1.031 | 1.026 | 1.019 | 1.013 | 1.012 |
| 1′-14C + Hisb | 1.059 | 1.054 | 1.048 | 1.044 | 1.040 | 1.035 |
| 1′-3H | 1.246 | 1.284 | 1.319 | 1.358 | 1.378 | 1.406 |
| 1′-3H + Hisb | 1.232 | 1.277 | 1.321 | 1.359 | 1.391 | 1.418 |
The KIEs shown in this table are a product of the EIE for the DN step and the intrinsic KIE for the AN step.
KIEs were computed using ISOEFF98 at 310.15 K. No correction for tunneling was incorporated.
KIEs of the model incorporating an active site histidine mimic.
Proposed Mechanism
Intrinsic KIEs establish that the arsenolytic depyrimidination of dT by hTP proceeds via a concerted ANDN mechanism with a dissociative transition state and significant oxocarbenium ion character. While common for N-riboside hydrolysis and transfer reactions, it is the first ANDN mechanism for a reaction with a 2′-deoxyribose. A DN*AN‡ mechanism is observed for the hydrolytic activity of hTP (27). A minimal kinetic mechanism and a qualitative free energy diagram can be proposed (Fig. 4).
Figure 4.

Chemical and kinetic mechanism of the hTP-catalyzed arsenolytic depyrimidination of dT. In the proposed mechanism the nucleophilic arsenate displaces the departing thymine in a concerted process. At the transition state the ribose has significant oxocarbenium character. (upper panel) The chemical species present at specific stages of catalysis. (middle panel) Free energy profile for enzyme catalyzed chemistry represented qualitatively. The proposed protonation step (see text) is represented by the dashed line (- - -). (lower panel) The minimal kinetic mechanism through release of the first product: E, enzyme free in solution; A or B, substrate free in solution; E•A, binary substrate complex; E•A•B, ternary substrate complex; E•P•Q, ternary product complex; E•Q, binary product complex; P or Q, product free in solution; kn, rate constant on step n. Catalysis is likely more complex, containing conformational changes after substrate binding and general acid/base chemistry after nucleophilic displacement.
A transition state with hydrogen bonding interactions between an active site histidine and both the departing thymine and attacking arsenate give the best match to the experimental KIEs. Stabilization of the leaving group and activation of the nucleophile result from these interactions. Similar interactions were predicted to facilitate catalysis in a molecular dynamics study of the structurally related E. coli thymidine phosphorylase (51). The conserved active site histidine serves a similar role in the human enzyme.
Protonation at N1 in the leaving group does not accurately predict the experimentally observed KIEs, suggesting N1 is likely not being protonated at the transition state. Hydrogen bonding to O2 by the active site histidine withdraws electron density and reproduces the KIEs. This raises the possibility that thymine leaves as a stabilized N1 anion. A related enzymatic reaction, uracil DNA glycosylase, catalyzes formation of a stabilized N1 anion of uracil in an analogous step (23). Protonation of O2 by the active site histidine to form the 1,2-lactim tautomer of thymine, cannot be ruled out. Leaving group protonation at the transition state occurs at N7 of adenine in the MutY-catalyzed hydrolytic depurination of DNA (25). In hTP hydrolysis, product thymine is rapidly protonated at N1 and this species equilibrates with the transition state, therefore occurring in a reversible step with oxocarbenium ion formation. Protonation at N1 explains the on-enzyme equilibrium between N1-protonated thymine and free reactant, which occurs before a rate limiting capture of the oxocarbenium ion by water. Similar chemistry is proposed to occur immediately after leaving group departure in arsenolysis. It is therefore proposed that after departure of the leaving group, hTP catalyzes the formation of N1-protonated thymine through general acid base chemistry (Figure 4). In the case of the N1 anion this would be direct protonation at N1, and in the case of the 1,2-lactim tautomer of thymine, an enzyme catalyzed tautomerization would occur.
Previous Arsenolytic Study
A previous study of the arsenolysis reaction catalyzed by hTP reported a near-symmetric ANDN nucleophilic transition state with bond orders near 0.5 to both the nucleophile and the leaving group (32). This conclusion was based on a large [1′-14C]dT KIE with smaller KIEs from the α- and β-3H effects. These isotope effects and conclusions on transition state structure differ substantially from the present experiments. The differences in the transition states is apparent from a More O’Ferrall-Jencks plot comparing the present DN*AN‡ transition state with that reported earlier (32) (Fig. 5). The transition state reported here for arsenolysis and that reported earlier for dT hydrolysis by hTP, using the same native hTP preparation, are related, as both proceed via ribocation formation (Fig. 5).
Figure 5.

Reaction space for N-riboside hydrolysis and transfer reactions. The axes are the Pauling bond order from the nucleophile (X-axis) or the leaving group (Y-axis) to the anomeric carbon, C1′. (X) Near-symmetric synchronous transition state for hTP arsenolysis as previously determined (32). (■) Transition state for hTP arsenolysis proposed from the current study. In this mechanism, the leaving group is nearly dissociated as the nucleophile begins to capture the anomeric carbon. (●) Recently identified transition state for the hTP-catalyzed hydrolytic depyrimidination of dT (27). In this mechanism, the nucleophile captures an oxocarbenium ion intermediate in the step after leaving group departure. Stepwise DN*AN‡ reactions lie on the axis at nLG = 0. The dashed line (- - -) represents the interface between associative and dissociative transition states of concerted processes, with synchronous mechanisms falling on the line. The solid line (—) encompasses the area that related enzyme-catalyzed reactions occupy (for which TS analysis has been performed). Graph adapted from elsewhere (22, 54).
The previous experiments were performed with hTP constructed and expressed with purification tags. More recent results linking protein dynamic architecture to transition state structure establish that mutations remote from the catalytic site are capable of altering transition state structure, providing one possible explanation for the departure from earlier work (55). In hTP, the effects may be more direct. Structures of hTP show the active sites near the dimer subunit interface. The N-terminal helices form this interface, thus the N-termini are near the catalytic sites (52). The sensitivity of transition state structure to structural and environmental changes argues for using the most physiological conditions available when transition state analysis is intended to guide inhibitor design.
Effects Contributing to Chemical Mechanism
Transition state analysis of the arsenolysis reaction provides an opportunity to compare hydrolysis (weak nucleophile) with a nucleophile (arsenate) more closely related to phosphate. The hydrolytic reaction is characterized by a stepwise DN*AN‡ mechanism with rate-limiting capture of the oxocarbenium ion intermediate by a water nucleophile hydrogen bonded to histidine (Fig. 2). With water, the AN step is the first irreversible step, and an equilibrium is established between the high energy oxocarbenium ion intermediate and free reactant. Conversely, arsenolysis proceeds by a concerted bimolecular mechanism, though hydrogen bonding interactions to the reacting nucleophile provides similarity for both catalytic activities. This shift in chemical mechanism is apparent in a More O’Ferrall-Jencks plot (Fig. 5).
The slightly larger volume of arsenate and altered pKa relative to phosphate raises the question of the effect of the nucleophile on transition state structure. At the transition state there is only weak bonding interactions between the attacking nucleophile and the ribocation, thus we propose these differences to be minimal. As the rates of arsenolysis and phosphorolysis are similar, despite differences in their chemical reactivity, we propose that the rate of nucleophile addition is governed by migration of the highly reactive deoxyribocation toward enzyme-bound nucleophile, a common mechanism in the N-ribosytransferases.
This fundamental shift in the chemical mechanism between hydrolysis and arsenolysis is caused by the relatively poor nucleophilicity of water when bound into a catalytic site designed to bind phosphate. Altered nucleophilicity in solution chemistry also promotes the shift from concerted to stepwise mechanisms.
CONCLUSIONS
hTP catalyzes the arsenolytic depyrimidination of dT, chemistry similar to the physiological phosphorolytic activity. The catalytic turnover numbers for arsenolysis and phosphorolysis reactions are similar. However the affinity of dT for hTP•arsenate is fourfold lower than for hTP•phosphate. hTP catalysis involves a transition state exhibiting significant ribooxocarbenium ion character. At the transition state, there is more bond order in the N-ribosidic bond (rC-N = 2.45 Å) than to the attacking nucleophile (rC-O = 2.95 Å). An active site histidine (His 116) is proposed to participate at the transition state to bridge the nucleophile and leaving group in a hydrogen bonding network. This catalytic site interaction activates the attacking arsenate and stabilizes the departing thymine. During the ANDN step, thymine leaves as an anionic species lacking a proton on N1 and general acid base chemistry is proposed to rapidly convert it to N1 protonated thymine.
Supplementary Material
Footnotes
Supported by NIH Research Grant GM41916
Abbreviations: dRib, 2-deoxyribose; dT, thymidine; EIE, equilibrium isotope effect; hTP, human thymidine phosphorylase; KIEs, kinetic isotope effects.
In IUPAC nomenclature, a reaction mechanism is symbolized by dividing the reaction into elementary steps. DN represents nucleofuge dissociation and AN represents nucleophile addition. When DN and AN occur in separate steps, and a discrete intermediate is formed (stepwise mechanism), the terms are separated by an asterisk (*) if the intermediate is too short-lived to diffusionally separate from the leaving group. Furthermore, the superscript “‡” denotes which step is the rate limiting chemical step. In a bimolecular reaction, when AN and DN occur in the same step (concerted mechanism), the terms are not separated (ANDN).
A similar model was presented in transition state analysis of the hydrolysis reaction catalyzed by hTP, where the imidazole was shown to activate the water nucleophile in the rate-determining transition state.
For a detailed description of the preliminary model and the predicted KIEs see Supporting Information.
The hydrogen bond between the nitrogen atom of the histidine and the –OH of the arsenate activates the nucleophile. The anionic leaving group thymine is stabilized by a second hydrogen bond between the protonated nitrogen of histidine and O2 of thymine. A similar model, with hydrogen bonding to N1 instead of O2 at the transition state, over-predicts the 15N KIE (1.026) supporting anionic leaving group stabilization by hydrogen bonding to O2 (Figure 3). Extending the argument, if thymine leaves as a neutral protonated species (as opposed to the anion at N1 stabilized by hydrogen bonding to O2), it is likely that the 1,2-lactim tautomer of thymine would be the initial product.
An identical transition state geometry without the histidine interaction gave a prediction of 30% for this KIE, almost double the experimental KIE of 17%.
Primary 14C KIEs are typically smaller than secondary 3H KIEs and can usually be predicted within ± 0.003 of the experimental value. Secondary 3H KIEs are sensitive to the environment around the hydron and a prediction within ± 0.05 of the experimental measurement is considered a good match.
The orientation of thymine with respect to the 2-deoxyribose ring was found to have an effect on the predicted 1′-14C and 1′-3H KIEs. For making a reasonable comparison with the ANDN model, the dihedral angle defined by C2-N2-1′C-2′C was fixed in the optimizations for the DN‡*AN model with the histidine mimic.
Supporting Information Available: Arsenolytic kinetic data and the complete calculation results are available in the supporting information available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Friedkin M, Roberts D. The enzymatic synthesis of nucleosides. I. Thymidine phosphorylase in mammalian tissue. J Biol Chem. 1954;207:245–256. [PubMed] [Google Scholar]
- 2.Krenitsky TA, Koszalka GW, Tuttle JV. Purine nucleoside synthesis, an efficient method employing nucleoside phosphorylases. Biochemistry. 1981;20:3615–3621. doi: 10.1021/bi00515a048. [DOI] [PubMed] [Google Scholar]
- 3.Krenitsky TA. Pentosyl transfer mechanisms of the mammalian nucleoside phosphorylases. J Biol Chem. 1968;243:2871–2875. [PubMed] [Google Scholar]
- 4.Zimmerman M. Deoxyribosyl Transfer: II. Nucleoside:pyrimidine deoxyribosyltransferase activity of three partially purified thymidine phosphorylases. J Biol Chem. 1964;239:2622–2627. [PubMed] [Google Scholar]
- 5.Miyazono K, Okabe T, Urabe A, Takaku F, Heldin CH. Purification and properties of an endothelial cell growth factor from human platelets. J Biol Chem. 1987;262:4098–4103. [PubMed] [Google Scholar]
- 6.Ishikawa F, Miyazono K, Hellman U, Drexler H, Wernstedt C, Hagiwara K, Usuki K, Takaku F, Risau W, Heldin CH. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature. 1989;338:557–562. doi: 10.1038/338557a0. [DOI] [PubMed] [Google Scholar]
- 7.Brown NS, Bicknell R. Thymidine phosphorylase, 2-deoxy-D-ribose and angiogenesis. Biochem J. 1998;334(Pt 1):1–8. doi: 10.1042/bj3340001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hotchkiss KA, Ashton AW, Schwartz EL. Thymidine phosphorylase and 2-deoxyribose stimulate human endothelial cell migration by specific activation of the integrins alpha 5 beta 1 and alpha V beta 3. J Biol Chem. 2003;278:19272–19279. doi: 10.1074/jbc.M212670200. [DOI] [PubMed] [Google Scholar]
- 9.de Bruin M, Smid K, Laan AC, Noordhuis P, Fukushima M, Hoekman K, Pinedo HM, Peters GJ. Rapid disappearance of deoxyribose-1-phosphate in platelet derived endothelial cell growth factor/thymidine phosphorylase overexpressing cells. Biochem Biophys Res Commun. 2003;301:675–679. doi: 10.1016/s0006-291x(03)00022-6. [DOI] [PubMed] [Google Scholar]
- 10.Takebayashi Y, Natsugoe S, Baba M, Akiba S, Fukumoto T, Miyadera K, Yamada Y, Takao S, Akiyama S, Aikou T. Thymidine phosphorylase in human esophageal squamous cell carcinoma. Cancer. 1999;85:282–289. doi: 10.1002/(sici)1097-0142(19990115)85:2<282::aid-cncr3>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 11.Focher F, Spadari S. Thymidine phosphorylase: a two-face Janus in anticancer chemotherapy. Curr Cancer Drug Targets. 2001;1:141–153. doi: 10.2174/1568009013334232. [DOI] [PubMed] [Google Scholar]
- 12.Foth H, Hellkamp J, Kunellis EM, Kahl GF. Pulmonary elimination and metabolism of 5-fluoro-2′-deoxyuridine in isolated perfused rat lung and lung slices. Drug Metab Dispos. 1990;18:1011–1017. [PubMed] [Google Scholar]
- 13.Cole C, Foster AJ, Freeman S, Jaffar M, Murray PE, Strafford IJ. The role of thymidine phosphorylase/PD-ECGF in cancer chemotherapy: a chemical perspective. Anticancer Drug Des. 1999;14:383–392. [PubMed] [Google Scholar]
- 14.Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, Yamada Y, Asao T. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2′-deoxyribonucleosides. Biochem Pharmacol. 2000;59:1227–1236. doi: 10.1016/s0006-2952(00)00253-7. [DOI] [PubMed] [Google Scholar]
- 15.de Bruin M, van Capel T, Van der Born K, Kruyt FA, Fukushima M, Hoekman K, Pinedo HM, Peters GJ. Role of platelet-derived endothelial cell growth factor/thymidine phosphorylase in fluoropyrimidine sensitivity. Br J Cancer. 2003;88:957–964. doi: 10.1038/sj.bjc.6600808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schramm VL. Enzymatic transition state poise and transition state analogues. Acc Chem Res. 2003;36:588–596. doi: 10.1021/ar0200495. [DOI] [PubMed] [Google Scholar]
- 17.Berti PJ, Tanaka KS. Transition state analysis using multiple kinetic isotope effects: mechanisms of enzymatic and non-enzymatic glycoside hydrolysis and transfer. Adv Phys Org Chem. 2002;37:239–314. [Google Scholar]
- 18.Parkin DW, Mentch F, Banks GA, Horenstein BA, Schramm VL. Transition-state analysis of a Vmax mutant of AMP nucleosidase by the application of heavy-atom kinetic isotope effects. Biochemistry. 1991;30:4586–4594. doi: 10.1021/bi00232a032. [DOI] [PubMed] [Google Scholar]
- 19.Kline PC, Schramm VL. Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction. Biochemistry. 1993;32:13212–13219. doi: 10.1021/bi00211a033. [DOI] [PubMed] [Google Scholar]
- 20.Berti PJ, Blanke SR, Schramm VL. Transition state structure for the hydrolysis of NAD catalyzed by diphtheria toxin. J Am Chem Soc. 1997;119:12079–12088. doi: 10.1021/ja971317a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bates C, Kendrick Z, McDonald N, Kline PC. Transition state analysis of adenosine nucleosidase from yellow lupin (Lupinus luteus) Phytochemistry. 2006;67:5–12. doi: 10.1016/j.phytochem.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Berti PJ, McCann JA. Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem Rev. 2006;106:506–555. doi: 10.1021/cr040461t. [DOI] [PubMed] [Google Scholar]
- 23.Werner RM, Stivers JT. Kinetic Isotope Effect Studies of the Reaction Catalyzed by Uracil DNA Glycosylase: Evidence for an Oxocarbenium Ion–Uracil Anion Intermediate. Biochemistry. 2000;39:14054–14064. doi: 10.1021/bi0018178. [DOI] [PubMed] [Google Scholar]
- 24.McCann JAB, Berti PJ. Transition State Analysis of Acid-Catalyzed dAMP Hydrolysis. J Am Chem Soc. 2007;129:7055–7064. doi: 10.1021/ja067371l. [DOI] [PubMed] [Google Scholar]
- 25.McCann JAB, Berti PJ. Transition-State Analysis of the DNA Repair Enzyme MutY. J Am Chem Soc. 2008;130:5789–5797. doi: 10.1021/ja711363s. [DOI] [PubMed] [Google Scholar]
- 26.Chen XY, Berti PJ, Schramm VL. Transition-State Analysis for Depurination of DNA by Ricin A-Chain. J Am Chem Soc. 2000;122:6527–6534. [Google Scholar]
- 27.Schwartz PA, Vetticatt MJ, Schramm VL. Transition State Analysis of Thymidine Hydrolysis by Human Thymidine Phosphorylase. J Am Chem Soc. 2010;132:13425–13433. doi: 10.1021/ja105041j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Booker S, Stubbe J. Cloning, sequencing, and expression of the adenosylcobalamin-dependent ribonucleotide reductase from Lactobacillus leichmannii. Proc Natl Acad Sci U S A. 1993;90:8352–8356. doi: 10.1073/pnas.90.18.8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merkler DJ, Kline PC, Weiss P, Schramm VL. Transition-state analysis of AMP deaminase. Biochemistry. 1993;32:12993–13001. doi: 10.1021/bi00211a007. [DOI] [PubMed] [Google Scholar]
- 30.Shi W, Tanaka KS, Crother TR, Taylor MW, Almo SC, Schramm VL. Structural analysis of adenine phosphoribosyltransferase from Saccharomyces cerevisiae. Biochemistry. 2001;40:10800–10809. doi: 10.1021/bi010465h. [DOI] [PubMed] [Google Scholar]
- 31.Parkin DW, Leung HB, Schramm VL. Synthesis of nucleotides with specific radiolabels in ribose. Primary 14C and secondary 3H kinetic isotope effects on acid-catalyzed glycosidic bond hydrolysis of AMP, dAMP, and inosine. J Biol Chem. 1984;259:9411–9417. [PubMed] [Google Scholar]
- 32.Birck MR, Schramm VL. Nucleophilic Participation in the Transition State for Human Thymidine Phosphorylase. J Am Chem Soc. 2004;126:2447–2453. doi: 10.1021/ja039260h. [DOI] [PubMed] [Google Scholar]
- 33.Parkin DW. In: Enzyme mechanism from isotope effects. Cook PF, editor. CRC Press, Inc; Boca Raton, FL: 1991. pp. 269–290. [Google Scholar]
- 34.Lewis BE, Schramm VL. Conformational equilibrium isotope effects in glucose by (13)C NMR spectroscopy and computational studies. J Am Chem Soc. 2001;123:1327–1336. doi: 10.1021/ja003291k. [DOI] [PubMed] [Google Scholar]
- 35.Lewis BE, Schramm VL. Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J Am Chem Soc. 2003;125:4785–4798. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 36.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 37.Hirschi JS, Takeya T, Hang C, Singleton DA. Transition-state geometry measurements from (13)C isotope effects. The experimental transition state for the epoxidation of alkenes with oxaziridines. J Am Chem Soc. 2009;131:2397–2403. doi: 10.1021/ja8088636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anisimov V, Paneth PJ. ISOEFF98. A program for studies of isotope effects using Hessian modifications. J Math Chem. 1999;26:75–86. [Google Scholar]
- 39.Bell RP. The Tunnel Effect in Chemistry. Chapman & Hall; London: 1980. [Google Scholar]
- 40.Pugmire MJ, Cook WJ, Jasanoff A, Walter MR, Ealick SE. Structural and theoretical studies suggest domain movement produces an active conformation of thymidine phosphorylase. J Mol Biol. 1998;281:285–299. doi: 10.1006/jmbi.1998.1941. [DOI] [PubMed] [Google Scholar]
- 41.Cook PF, Cleland WW. Enzyme Kinetics and Mechanism. Garland Science; New York: 2007. [Google Scholar]
- 42.Northrop DB. The expression of isotope effects on enzyme-catalyzed reactions. Annu Rev Biochem. 1981;50:103–131. doi: 10.1146/annurev.bi.50.070181.000535. [DOI] [PubMed] [Google Scholar]
- 43.Lewandowicz A, Schramm VL. Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry. 2004;43:1458–1468. doi: 10.1021/bi0359123. [DOI] [PubMed] [Google Scholar]
- 44.Singh V, Schramm VL. Transition-State Structure of Human 5′-Methylthioadenosine Phosphorylase. J Am Chem Soc. 2006;128:14691–14696. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parks RE, Jr, Agarwal RP. In: The Enzymes. Boyer PD, editor. Academic Press; New York: 1972. pp. 483–514. [Google Scholar]
- 46.Glad SS, Jensen F. Transition State Looseness and a-Secondary Kinetic Isotope Effects. J Am Chem Soc. 1997;119:227–232. [Google Scholar]
- 47.Matsson O, Westaway KC. Secondary Deuterium Kinetic Isotope Effects and Transition State Structure. In: Bethell D, editor. Adv Phys Org Chem. Academic Press; San Diego: 1998. pp. 143–248. [Google Scholar]
- 48.Sunko DE, Szele I, Hehre WJ. Hyperconjugation and the angular dependence of. beta-deuterium isotope effects. J Am Chem Soc. 1977;99:5000–5005. [Google Scholar]
- 49.Singh V, Lee JE, Nunez S, Howell PL, Schramm VL. Transition state structure of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry. 2005;44:11647–11659. doi: 10.1021/bi050863a. [DOI] [PubMed] [Google Scholar]
- 50.Chen XY, Berti PJ, Schramm VL. Ricin A-Chain: Kinetic Isotope Effects and Transition State Structure with Stem-Loop RNA. J Am Chem Soc. 2000;122:1609–1617. [Google Scholar]
- 51.Mendieta J, Martin-Santamaria S, Priego EM, Balzarini J, Camarasa MJ, Perez-Perez MJ, Gago F. Role of Histidine-85 in the Catalytic Mechanism of Thymidine Phosphorylase As Assessed by Targeted Molecular Dynamics Simulations and Quantum Mechanical Calculations†. Biochemistry. 2003;43:405–414. doi: 10.1021/bi034793o. [DOI] [PubMed] [Google Scholar]
- 52.Mitsiki E, Papageorgiou AC, Iyer S, Thiyagarajan N, Prior SH, Sleep D, Finnis C, Acharya KR. Structures of native human thymidine phosphorylase and in complex with 5-iodouracil. Biochem Biophys Res Commun. 2009;386:666–670. doi: 10.1016/j.bbrc.2009.06.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pugmire MJ, Ealick SE. Structural analyses reveal two distinct families of nucleoside phosphorylases. Biochem J. 2002;361:1–25. doi: 10.1042/0264-6021:3610001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schramm VL. Enzymatic transition state theory and transition state analogue design. J Biol Chem. 2007;282:28297–28300. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 55.Luo M, Li L, Schramm VL. Remote mutations alter transition-state structure of human purine nucleoside phosphorylase. Biochemistry. 2008;47:2565–2576. doi: 10.1021/bi702133x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.