

Abstract
The human p53 gene is a tumor suppressor mutated in half of colon cancers. Although p53 function appears important for proliferation arrest and apoptosis induced by cancer therapeutics, the prognostic significance of p53 mutations remains elusive. This suggests that p53 function is modulated at a posttranslational level and that dysfunctions affecting its modulators can have a prognostic impact. Among p53 modulators, homeodomain interacting protein kinase (HIPK) 2 emerges as a candidate “switch” governing p53 transition from a cytostatic to a proapoptotic function. Thus, we investigated the possible prognostic role of HIPK2 on a retrospective series of 80 colon cancer cases by setting up a multiplexed cytometric approach capable of exploring correlative protein expression at the single tumor cell level on TMA. Crossing the data with quantitative PCR and p53 gene sequencing and p53 functional assays, we observed the following: despite a strong impact on p21 transcription, the presence of disabling p53 mutations has no prognostic value, and the increased expression of the HIPK2 protein in tumor cells compared with paired normal tissue cells has a strong impact on survival. Unexpectedly, HIPK2 effect does not appear to be mediated by p53 function because it is also observed in p53-disabling mutated backgrounds. Thus, our results point to a prominent and p53-independent role of HIPK2 in colon cancer survival.
The human p53 gene acts as a tumor suppressor that plays a central role in protecting the genome against genotoxic stresses, such as oxidative stress, UV light, and ionizing radiation. In such conditions, p53 “senses” the DNA lesions and is activated, leading to the transactivation of target genes involved in cell cycle arrest and apoptosis. This either allows for DNA repair or, alternatively, when the damage is extensive, provokes cell self-elimination by apoptosis.1 Although tumor protein 53 (TP53) is mutated in approximately 50% of colon cancer cases,2 the prognostic significance of these mutations remains controversial. In a large cohort of colorectal carcinomas published by the TP53-CRC Collaborative Study Group, this prognostic impact seems intricate and depends on tumor site, type of mutation, adjuvant therapy, and stage of the disease.3,4
Posttranslational modifications of the p53 protein, such as ubiquitination, phosphorylation, and acetylation, contribute to p53 activity regulation, leading to protein stabilization, conformational changes, and modifications of its affinity for DNA and its interaction with transcriptional coactivator complexes.5 As a result, the molecular modulators of p53 could play a significant role by influencing the p53-dependent responses in tumor cells and altering the prognostic significance of the p53 genetic status of the tumor.
Among such modulators, homeodomain interacting protein kinase 2 (HIPK2)6 seems to be an important regulator of p53 functions acting through direct phosphorylation of p53 at Ser46 in response to severe DNA damage.7–9 The phosphorylation of p53 Ser46 may represent a sensor of DNA damage intensity that promotes a shift in p53 affinity from cell cycle arrest–related promoter genes to apoptosis genes.10–12 HIPK2 also forms a heterocomplex with p53, acetyltransferase Creb-binding protein, and promyelocytic leukemia protein in promyelocytic leukemia protein–nuclear bodies; and the phosphorylation of p53 at Ser46 by HIPK2 promotes the acetylation of p53 by acetyltransferase Creb-binding protein at lysines 373/382, playing a critical role in the induction of apoptosis.9,13,14
Moreover, the main mechanism of p53 expression-level regulation that is mediated by the ubiquitin ligase MDM2 is controlled by HIPK2. Indeed, HIPK2 inhibits the nuclear export and ubiquitination of p53 mediated by MDM2, thus neutralizing p53 degradation and promoting p53-dependent apoptosis.15 In addition, the phosphorylation of p53 at Ser46 by HIPK2 inhibits the MDM2 negative feedback loop by inhibiting p53-mediated MDM2 gene transcription. In addition, HIPK2 further inhibits MDM2 in a p53-independent and transcription-independent way: MDM2 can be phosphorylated by HIPK2, causing its cytoplasmic shuttling and provoking its proteasomal degradation.16
From a functional point of view, the knockdown of HIPK2 abolished repair after DNA damage in vitro.17 Moreover, in a recent study,18 a significant correlation between poor survival rates and low mRNA HIPK2 expression was reported in wild-type (WT) p53 colonic tumors.
HIPK2 can also modulate the apoptotic response in the absence of p53. Indeed, HIPK2 facilitates the phosphorylation of the antiapoptotic corepressor C-terminal binding protein at Ser422, directly and indirectly, through the c-Jun N-terminal kinase signaling pathway, resulting in C-terminal binding protein proteasomal degradation and the promotion of apoptosis.19–21
Furthermore, HIPK2 appears to be highly regulated at both the activity and expression levels.22 For instance, in response to severe DNA damage, caspase-6, a p53 target gene, removes an inhibitory C-terminal domain from HIPK2, resulting in a hyperactive kinase that potentiates p53 Ser46 phosphorylation.23,24
Regarding expression, MDM2 was reported to act as a negative regulator of HIPK2, mediating its ubiquitination and degradation on mild DNA damage.12 Recently, HIPK2 has undergone a rapid protein turnover in resting conditions orchestrated by the E3 ubiquitin ligase Siah-1, which, like MDM2, is another p53 target gene. In resting conditions, Siah-1 colocalizes and forms a complex with HIPK2 in nuclear bodies, whereas DNA damage triggers disruption of the complex, via the ATM/ATR pathway, resulting in HIPK2 stabilization and activation.25
Therefore, there is some evidence that HIPK2 is an important actor in DNA damage response, along with p53, which acts as a tumor suppressor gene.
Because DNA damage signaling is involved in both colon cancer progression and drug sensitivity, it is tempting to speculate the following: changes in HIPK2 expression could occur during cancer natural history, and HIPK2 expression could affect the drug sensitivity of tumor cells, modulating pharmacologically induced p53 signaling. Thus, it could potentially represent a prognostic marker.
To explore HIPK2 expression in colon carcinoma, we selected a collection of 80 pairs of carcinoma and the respective healthy mucosa.
By using immunofluorescence and TMA imaging, we developed and validated a novel semiautomatic method (which we coined “TMA cytometry”) for quantifying and analyzing multiple protein expression on archival paraffin-embedded tissue. By applying this technique to our series of colon carcinomas, we simultaneously studied HIPK2 and p53 expression at the cellular level, crossing these data with the mutational status of p53. Moreover, we compared the expression level between the tumor sample and the normal tissue of each patient to explore the possible variations of the protein level occurring during carcinogenesis.
Herein, we show that high HIPK2 overexpression is highly and positively correlated with prognosis, irrespective of the p53 mutational status and stage of the disease.
Materials and Methods
Clinical Data
Eighty patients with primary colorectal carcinoma, operated on and treated at the Institut Bergonié, Bordeaux, France, between January 1, 1999, and December 31, 2004, were included in the study. At surgery, fragments of tumor and normal mucosa (taken at a distance from the tumor) were frozen quickly in liquid nitrogen and stored at -140°C until extraction. The remaining material was fixed in Holland Bouin's solution and paraffin embedded for pathological evaluation. These frozen and fixed samples were used as paired specimens in the quantification of gene expression by real-time PCR and immunofluorescence analysis, respectively. The clinical characteristics of the series are described in Table 1. At analysis, 39 patients were considered alive without cancer, 8 were alive with cancer, and 4 were alive with another cancer. Twenty-nine patients were dead from cancer (n = 19), complications (n = 3), another cancer (n = 4), or other causes (n = 3).3 The 5–year overall survival (OS) was 63%, with a median follow–up of 54.6 months. A clear prognostic value of the stage was observed in this series (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Table 1.
Patient Characteristics
| Characteristics | Value |
|---|---|
| No. of patients | 80 |
| Age, median (range) (years) | 66 (31–88) |
| Male/female ratio | 1:10 |
| Primary tumor site | |
| Descending colon and sigma-rectum junction | 55 (68.8) |
| Ascending and transverse colon | 22 (27.5) |
| Rectum | 3 (3.8) |
| Tumor stage | |
| I and II | 29 |
| III | 20 |
| IV | 29 |
| Unknown | 2 |
| Adjuvant therapy | |
| None | 40 |
| Chemotherapy | 40 |
Data are given as number (percentage) of patients unless otherwise indicated.
TMA Construction
Tissue cores with a diameter of 0.6 mm were removed from fixed paraffin–embedded tissue blocks and arrayed on a recipient paraffin block using a tissue arrayer (Beecher Instruments Tissue Arrayer, Sun Prairie, WI). Each tumor sample was punched in triplicate, along with a core of nontumoral mucosal tissue punched far away from the tumor. Sections of the array were cut at 5 μm and placed on glass slides.
An independent TMA incorporating a series of 30 colorectal cancers with matched normal tissue for eight cases, together with patient survival data, was purchased from US Biomax (CO951, Rockville, MD).
Immunohistochemistry
IHC with p53 antibody (mouse monoclonal antibody DO–7 Ab, 1:100; Dako, Trappes, France) was performed on a 5–μm fixed paraffin–embedded TMA section mounted on a charged slide. The tissue section was deparaffinized in toluene. After heat-induced proteolytic epitope retrieval in citrate buffer (pH 6.0) using a microwave oven, the primary antibody was incubated for 1 hour at room temperature. Positive reactions were visualized using the avidin–biotin method. Staining was evaluated by visual examination using a semiquantitative approach, assessing the staining intensity with a four–point scale (from zero to three); the percentage of stained cells ranged from 0% to 100%.
Immunofluorescence
Double immunofluorescence with p53 (same antibody and conditions as IHC) and HIPK2 (rabbit polyclonal anti–HIPK2 28507 Ab, 1:300; Abcam, Cambridge, UK) was performed on a 5–μm fixed paraffin–embedded TMA section mounted on a charged slide. The tissue section was deparaffinized in toluene. After heat–induced proteolytic epitope retrieval in citrate buffer (pH 6.0) using a microwave oven, primary antibodies were incubated for 1 hour at room temperature. Secondary antibodies [ie, goat anti-rabbit (Alexa Fluor 594 and 532) and goat anti-mouse (Alexa Fluor 488); all from Molecular Probes, Eugene, OR] were diluted in antibody diluent (REAL; Dako) and incubated for 1 hour at room temperature in the dark. The section was washed with PBS three times and incubated with DAPI (1 μg/mL) for 15 minutes in the dark. After dehydration, the section was mounted with Fluoromount–G and stored at 4°C in the dark until analysis.
Immunofluorescence Image Acquisition and Quantification
Immunofluorescence signals were collected for each array core with a confocal microscope (LSM 510 META; Zeiss, Göttingen, Germany) using a ×20 objective lens. Images were quantified with the multiwavelength cell scoring application of Metamorph (Molecular Devices, Sunnyvale, CA). A second acquisition and analysis of both TMAs was performed with an automated laser scanning cytometer (iCys; Compucyte, Boston, MA) using phantom segmentation.
Quantification by Real-Time Quantitative RT–PCR Analysis
RNA samples were extracted from fresh–frozen fractions of tumor tissue and normal nontumoral mucosa using a kit (RNeasy Lipid Tissue Qiagen kit, 74804; Qiagen, Valencia, CA), following the manufacturer's instructions. Immediately after elution, RNA samples were cleaned with a DNase inactivation reagent kit (DNase Turbo DNA free; AMBION, Austin, TX), thus avoiding subsequent degradation (data not shown). The RNA samples were treated with DNase (DNase Turbo DNA free), and RNA quality was assessed on a bioanalyzer (Bioanalyseur 2100; Agilent, Santa Clara, CA). Only RNA of high quality [RNA integrity number (RIN) > 7] qualified for the study. First–strand cDNA was synthesized according to the manufacturer's instructions (Reverse Transcriptase Powerscript Clontech, Mountain View, CA). Forty–seven paired samples were available for further analysis.
Quantitative analysis was performed on DNA ENGINE OPTICON 2. For standardization, commercial kits for HIPK2 and the housekeeping gene have been used (Applied Biosystems, Carlsbad, CA). Among 10 housekeeping genes tested [ie, GAPDH (glyceraldehyde–3–phosphate dehydrogenase), B2M (β2 microglobulin), PGK1 (phosphorylated glycerate kinase 1), ACTB (actin β), TBP (TATA box binding protein), 18S (18S ribosomal RNA), GUSB (glycuronidase β), HPRT1 (hypoxanthine phosphoribosyl transferase), RPLPO (ribosomal protein large PO), and PPIA (cyclophilin A)], GUSB was the gene that showed less variability in our PCR conditions and was chosen as the housekeeping gene for subsequent quantitative analysis. Each sample was analyzed in triplicate. Paired samples (tumor and normal tissue) were plotted on the same plate. For each sample, fold change (FC) was calculated according to the 2–ΔΔCt method.
p53 Mutational Analyses
Sequencing
After evaluation of tumoral cellularity on a frozen H&E–stained section, DNA was extracted from fresh–frozen fractions of tumor tissues using the phenol chloroform method. Of 80 cases, 72 (cellularity >10%) were analyzed. Mutational analysis explored exons 2 to 11 of the p53 gene. Primer sequences (Table 2) were designed with software (Primer 3). PCR was performed in a thermocycler (GeneAmp PCR System 9700; Applied Biosystems). For exons 4 to 11, the PCR mix was composed of 1.5 mmol/L MgCl2, 3.5 μL 10× PCR Gold buffer (Applied Biosystems), dNTP (100 μmol/L each), primer (17.5 pmol each), 1.4 U of AmpliTaq Gold (Applied Biosystems), and 40 ng of DNA (QS, 35 μL). The PCR comprised 5 minutes at 94°C; 45 cycles of 30 seconds at 94°C, 45 seconds at 59°C, and 45 seconds at 72°C; and an elongation phase of 5 minutes at 72°C. For exons 2 to 3, MgCl2 was replaced by 1.5 mmol/L MgSO4, and AmpliTaq Gold was replaced by 1.4 U of Platinium TaqDNA polymerase (Invitrogen) in the PCR mix. The PCR comprised 2 minutes at 94°C; 35 cycles of 30 seconds at 94°C, 45 seconds at 61°C, and 45 seconds at 72°C; and an elongation phase of 5 minutes at 72°C. PCR product sizes were controlled by migration on 2% agarose electrophoresis gel and were purified with columns (GFX PCR DNA kit; Amersham Biosciences, Piscataway, NJ). Bidirectional direct sequencing was performed on the thermocycler (GeneAmp PCR System 9700; Applied Biosystems). Sequencing primers are the same as those of the PCR. Sequencing reaction mix comprises 3.5 μL of 5× sequencing buffer, 0.5 μL of Big Dye Terminator, 12.5 pmol of primer, and 4 μL of PCR products purified and diluted at 1:10 (QS, 20 μL). The sequencing cycle comprised 10 seconds at 94°C and 25 cycles of 5 seconds at 50°C and 4 minutes at 60°C. Purification of sequencing products was performed on a column (DyeEx; Qiagen). Sequence analyses were read on software (SeqScape).
Table 2.
Probe Sequences
| Exon | Primer |
Amplicon size (bp) | Annealing temp (°C) | |
|---|---|---|---|---|
| 1 | 2 | |||
| 2 and 3 | 5′-TCTCAGACACTGGCATGGTG-3′ | 5′-GGGGACAGCATCAAATCATC-3′ | 500 | 61 |
| 4 | 5′-CGTTCTGGTAAGGACAAGGG-3′ | 5′-GGAATCCCAAAGTTCCAAAC-3′ | 484 | 59 |
| 5 and 6 | 5′-GTTTGTTTCTTTGCTGCCG-3′ | 5′-TCATGGGGTTATAGGGAGGTC-3′ | 585 | 59 |
| 7 | 5′-CCTCCCCTGCTTGCCAC-3′ | 5′-GAGGTGGATGGGTAGTAGTATGG-3′ | 293 | 59 |
| 8 and 9 | 5′-TTGGGAGTAGATGGAGCCTG-3′ | 5′-AAACAGTCAAGAAGAAAACGGC-3′ | 473 | 59 |
| 10 | 5′-TCAAACAATTGTAACTTGAACCATC-3′ | 5′-GGCAGGATGAGAATGGAATC-3′ | 269 | 59 |
| 11 | 5′-GGGAAAAGGGGCACAGAC-3′ | 5′-GCAAGCAAGGGTTCAAAGAC-3′ | 244 | 59 |
Functional Analysis of Separated Alleles in Yeast Test
To detect inactivating mutations in the TP53 gene, a functional analysis of separated alleles in yeast (FASAY) assay was performed on tumoral RNA extracted for the quantitative RT–PCR analysis, as described by Flaman et al.26 The transcriptional activity of human p53 in tumor cells is assessed in Saccharomyces cerevisiae, where it activates a p53 target gene (Ade2). This reporter gene is under the control of a promoter that contains the p53 binding site. The mRNA specimens were reverse transcribed with random primers, and part of the TP53 open reading frame comprising exons 4 to 11 was amplified by PCR, leading to a PCR product of 1 kb. The reporter strains were grown and cotransformed with PCR amplicons and a linearized yeast expression vector carrying the 5′ and 3′ ends of the TP53 open reading frame. Activation of the reporter by WT p53 results in white colonies, whereas the mutant p53 produces smaller red colonies. The activity of the p53 mutant was determined by the color of at least 100 colonies per strain. Because of PCR-induced errors or alternatively spliced TP53 mRNA, the FASAY test result was considered negative (functional or WT p53) when <10% of red colonies were detected. The test result was considered doubtful when between 10% and 20% of red colonies were detected. The FASAY test result was considered positive (nonfunctional or mutated p53) for values higher than this cutoff. Positive and negative controls were included in each assay.
Statistical Analysis
Survival curves were calculated according to the Kaplan-Meier method; survival analysis was performed using the log-rank test. OS was calculated from the date of diagnosis until death or last follow-up. The Cox proportional hazards model was used for multivariate analysis, and calculation of the hazard ratios and confidence intervals was performed with the ascending step-by-step maximum likelihood method, including only variables significant at P < 0.05 in the univariate analysis.
Results
TMA Cytometry
To evaluate the expression of HIPK2 and p53 at the protein level in our tissue collection, we constructed a TMA containing one spot of healthy mucosa and three spots of tumors for each patient.
Double immunofluorescence with p53 and HIPK2 antibodies was performed on a 5-μm fixed paraffin-embedded TMA section mounted on a charged slide. The entire TMA section was analyzed by acquisition of p53, HIPK2, and DAPI signals by confocal microscopy (Figure 1, A and B). Each acquired image corresponding to tumoral or normal mucosa tissue core was reviewed for overall tissue content and quality assessment, and the best core for each tumor was chosen for subsequent quantification. Patients were excluded if all three cores were missing or not representative (eg, if they contained only stroma). A manual “virtual microdissection” of the images was performed by excluding the stromal component with a mask to keep only epithelial elements (Figure 1C). Based on DAPI labeling, we submitted each core to a segmentation process that determines the nuclear regions. Each segmented nucleus was quantified for p53 and HIPK2 expression, measuring the mean fluorescence intensity in each channel. Although the analysis routine allows us to automatically score the nuclei as positive or negative using a threshold-based procedure (Figure 1D), we preferred to analyze the distribution of fluorescence intensities of the overall cell populations of spots to avoid generating artifacts linked to the choice of an arbitrary threshold. The fluorescence intensity of each fluorochrome-labeled antibody was measured in individual nuclei, generating a data set for each sample. A mean of 767 cells was quantified in tumor samples (range, 231 to 1324 cells), and a mean of 309 cells was quantified in normal mucosa samples (range, 30 to 682 cells). The TMA from US Biomax contained 30 colorectal cancers (two cores per case), eight of which had matched normal tissue. It was acquired and analyzed with the same procedure as the home-constructed TMA.
Figure 1.
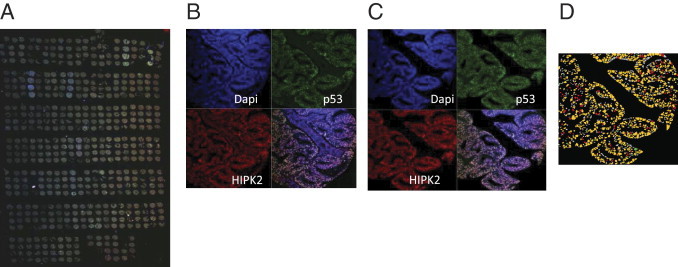
Acquisition and treatment of the TMA double labeled by immunofluorescence for p53 and HIPK2. A: Low-resolution acquisition of the overall TMA. B: Confocal microscopy acquisition of three fluorescent signals: DAPI (nuclear staining), p53 (green Alexa 488), and HIPK2 (red Alexa 594) of a single core. C: Operator-guided microdissection of images to eliminate stromal component. D: Segmentation and threshold-based cell scoring.
p53 Analysis
p53 Expression
First, we determined the distribution profiles of p53 nuclear intensities in normal mucosa samples (Figure 2A) and tumoral tissues (Figure 2B). The profiles of normal tissues are highly similar and display a homogeneous gaussian distribution. Tumor samples were more heterogeneous, with a wide range of fluorescence from low to high intensity. Some tumor profiles show the presence of two cell populations, one of them characterized by a high p53 expression level. Automated segmentation-based quantification of immunofluorescence provides a continuous range of values that reflects the real distribution of expression levels in the cell population of different tumors in contrast to the classic evaluation of intensity by discrete scoring given by pathologists. Thus, we compared our data with the pathologist scoring system performed on the IHC anti-p53 with the same antibody (Figure 2, C–E). This scoring is based on the determination of two parameters: the IHC intensity and the perceived percentage of positive cells.
Figure 2.
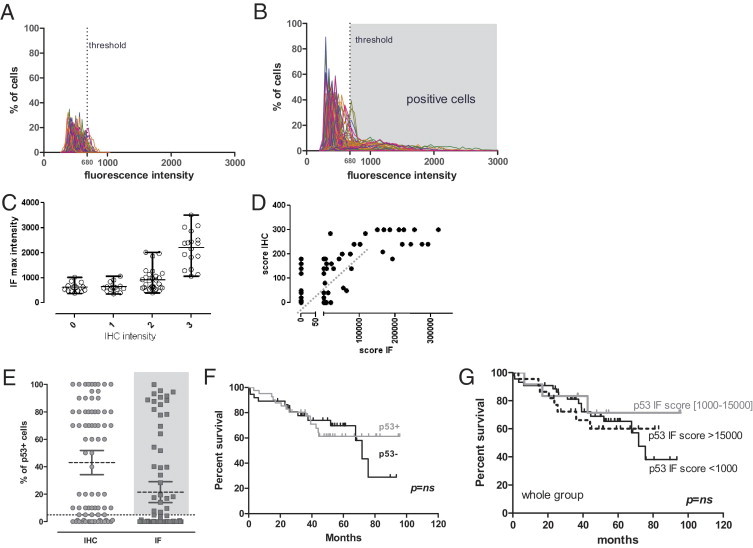
p53 Expression cytometry. A and B: Distribution of the p53 immunofluorescence nuclear intensities among normal mucosa (A) and tumoral (B) samples. Each curve represents a sample. The dotted line represents the threshold for p53 positivity. C: Correlation between the anatomopathologist discrete classification of intensity of IHC and the maximal (max) mean intensity of nuclei measured by Metamorph for immunofluorescence (IF). D: Correlation between the anatomopathologist index (percentage of positive cells × intensity) and the same index for IF. E: Percentage of p53-positive cells determined by the visual lecture of IHC by an anatomopathologist and the quantification of percentage of gated cells over the threshold using IF. Tumors with more than 5% of cells over the threshold were classified as p53 positive. F: Survival analysis of the p53+ group (highlighted by gray in E) and the p53- group. G: Survival analysis according to the IF score.
To compare the intensity values of immunofluorescence with the IHC pathologist scoring of intensity, we show the repartition of the maximal mean fluorescence intensity of cells in the tumors among the four IHC intensity groups defined by pathologist analysis (Figure 2D). We found a good correlation between the ranges of intensity in classes 2 and 3 of IHC; with this analysis, we did not detect a significant difference between group 0 and 1. This result could reflect immunofluorescence's lesser sensitivity in the low intensities or a certain level of arbitrariness of the classic procedure because of the forced classification in discrete groups.
When we considered the percentage of positive cells, 15 tumors were scored as p53 negative and 63 tumors were scored as p53 positive by IHC, with a range of tumor positivity from 1% to 100% of cells (two cases not available).
To determine p53 positivity for cells using immunofluorescence, we postulated that the normal protein p53 is present at low levels in normal tissues and calculated the 90th percentile of cell fluorescence for each normal tissue. Looking at the distribution of these values, we retained as the fluorescence threshold the 90th percentile of all these values (680 gray levels: minimum, 411; maximum, 923). At the end, 41 tumors presented a percentage of p53-positive cells ranging from 0.1% to 99.8%, whereas 37 tumors were completely negative (two cases not available). As seen in Figure 2C, the immunofluorescence quantification shows, as expected, a more gradual positivity for percentage of positive cells.
To quantify the real intensity of the positive cells, we measured the mean intensity of the nuclei considered positive on the basis of the chosen threshold. To obtain an integrated evaluation of the percentage of positivity and the intensity of expression, we considered the anatomopathologist score that combines the percentage of positive cells and the intensity score and compared it with the same index obtained for immunofluorescence by multiplying the percentage of positive cells (cells over the threshold) by their mean intensity. This representation shows a zone of good correlation between the two approaches for the intermediary cases; for the strong positive cases, the immunofluorescence quantification clearly shows a better resolution because all strong intensities are capped at intensity 3 using IHC.
Concerning the low-expressing tumors, the threshold-based analysis of immunofluorescence classifies this group of tumors as totally negative because the fluorescence intensities of nuclei are entirely lower than the threshold value. Among these cases, 30 are in good correlation with IHC analysis results, whereas five are more discordant (IHC score, 140 to 180).
We then analyzed the OS of the patients corresponding to the tumor series on the basis of p53 expression. We classified as p53 positive all of the tumors that presented more than 5% of cells over the threshold (gray zone in Figure 2E). The survival curves show that, in our group of patients, there was no difference in OS according to p53 expression measured by TMA cytometry (Figure 2F). We obtained the same results by grouping our samples on the basis of the immunofluorescence score, which better resolves the low- and high-expressing tumors (Figure 2G). The absence of prognostic value of p53 expression was also observed in the TMA from US Biomax (see Supplemental Figure S2 at http://ajp.amjpathol.org).
p53 Mutational Analysis
The increased expression of p53 in tumors is considered the signature of the presence of a p53 missense mutation. Thus, we explored the mutational status of tumors by direct sequencing and functional assay testing of the transcriptional activity of each tumor TP53 gene product in yeast; 21 (29%) of 72 cases analyzed by direct sequencing were WT. Figure 3A shows the distribution of the type of p53 mutations among the 51 mutated cases (71%). A total of 54 mutations were observed, with three cases showing two concomitant mutations. As expected, most mutations were hot-spot missense mutations (n = 36), followed by splice mutations (n = 7), nonsense mutations (n = 6), deletions (n = 4), and a silent mutation (n = 1). Details of sequencing results and correlation with FASAY testing are reported in Table 3. The FASAY test was performed in 46 cases. In six cases, the test result was considered noninterpretable because of insufficient number of colonies. One WT case was considered doubtful (12.4% of red colonies). In 28 cases, there was a perfect concordance between sequencing analysis and the FASAY test result (74%). On the other hand, we observed six cases classified as “mutated” by sequencing but FASAY test result negative and one case classified as WT by sequencing but FASAY test result positive, which could be because of sampling. In three cases, sequencing was considered nonrelevant (ie, no mutation detected but tumoral cellularity of the sample <10%); and the FASAY test result was negative in one case and positive in two cases. Finally, one case was not sequenced but was negative with the FASAY test. Combining sequencing and FASAY testing, 55 cases (69%) were considered mutated, 21 (26%) were considered WT, and 4 (5%) were considered undetermined. As expected, the mutated group of tumors and, more specifically, the subgroup presenting a missense mutation corresponded to the tumors presenting an elevated percentage of p53-positive cells (Figure 3B). Again, concerning survival, when we compared the OS of the WT p53 group with that of the mutated p53 group, we did not find any difference according to the p53 mutational status of tumors (Figure 3, C and D), confirming our previous result with p53 protein expression. By using quantitative PCR on RNA from fresh-frozen fractions of tissues included in the TMA, we sought to determine whether the presence of a mutated p53 had a measurable impact on the expression of two main transcriptional targets of p53: the proapoptotic BCL-2–associated X protein (Bax) and p21, which is responsible for p53-dependent cell growth arrest. For Bax, we found the same profile in the WT and mutated p53 group (ie, Bax expression appeared invariant or slightly increased; Figure 3E). However, for p21, we observed a decreased expression of p21 in the tumors with mutated p53 compared with the WT p53 group, showing a loss of transcriptional p53 function (Figure 3F).
Figure 3.
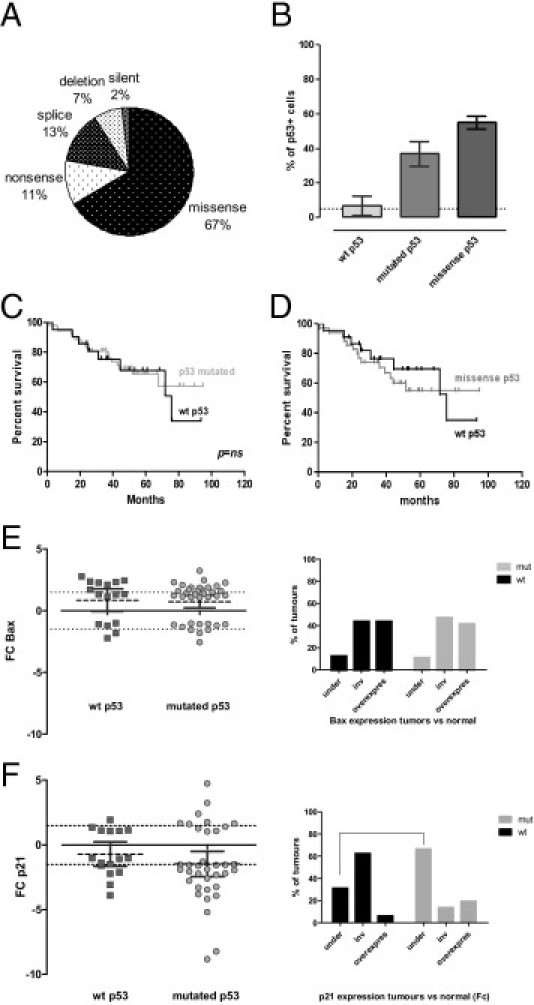
Effect of p53 mutations on p53 expression, survival, and expression of p53 targets. A: Repartition of the TP53 mutations among mutated cases. Fifty-four mutations were observed among 51 mutated cases (69%). Three cases showed two mutations: a missense mutation along with another missense mutation, a nonsense mutation, or a silent mutation. B: Percentage of p53-positive cells in the WT, mutated, and missense p53 tumor groups. C and D: Survival analysis according to p53 mutational status: survival of the WT p53 tumors versus the p53 mutated group (C) or the tumor group bearing a missense p53 mutation (D). E and F: Variation of the expression of Bax and p21 in the WT and mutated (mut) p53 groups of tumors, evaluated by quantitative RT-PCR: Bax FC and percentage of tumors that respectively have a decreased, invariant (inv), or increased expression of Bax (E) or p21 FC and percentage of tumors that respectively have a decreased, inv, or increased expression of p21 (F).
Table 3.
P53 Mutational Analysis (77 Samples)
| Tumor no. | AA or sequence change | Effect | FASAY test result | Final mutational status |
|---|---|---|---|---|
| 29 | p.C141T | Missense | ND | M |
| 50 | p.P151R | Missense | ND | M |
| 18 | p.Y163C | Missense | Nonfunctional | M |
| 47 | p.Y163S | Missense | Nonfunctional | M |
| 35 | p.V173L | Missense | Nonfunctional | M |
| 36 | p.V173L | Missense | ND | M |
| 16 | p.R175H | Missense | ND | M |
| 27 | p.R175H | Missense | Nonfunctional | M |
| 33 | p.R175H | Missense | ND | M |
| 70 | p.R175H | Missense | NC | M |
| 78 | p.R175H | Missense | Nonfunctional | M |
| 40 | p.C176F | Missense | Nonfunctional | M |
| 24 | p.P190L | Missense | Functional | M |
| 21 | p.H193Y | Missense | Nonfunctional | M |
| 63 | p.L194R | Missense | Nonfunctional | M |
| 44 | p.I195T | Missense | Nonfunctional | M |
| 5 | p.R213L | Missense | Nonfunctional | M |
| 6 | p.R213Q | Missense | NI | M |
| 37 | p.S215R | Missense | ND | M |
| 53 | p.Y236C | Missense | NC | M |
| 79 | p.G245D | Missense | ND | M |
| 39 | p.G245S/p.P36P | Missense/silent | Nonfunctional | M |
| 28 | p.R248Q | Missense | ND | M |
| 12 | p.R248W | Missense | ND | M |
| 14 | p.R273C | Missense | ND | M |
| 80 | p.R273C | Missense | Nonfunctional | M |
| 26 | p.R273H | Missense | NC | M |
| 51 | p.R273H | Missense | Nonfunctional | M |
| 75 | p.R273H | Missense | NC | M |
| 67 | p.C275Y | Missense | Functional | M |
| 43 | p.R282W | Missense | Nonfunctional | M |
| 55 | p.R282W | Missense | ND | M |
| 56 | p.R290P | Missense | NI | M |
| 72 | p.P152L/p.I255F | Missense/missense | Nonfunctional | M |
| 57 | p.C176F/p.R306X | Missense/nonsense | Nonfunctional | M |
| 11 | p.R196X | Nonsense | Nonfunctional | M |
| 13 | p.R306X | Nonsense | Nonfunctional | M |
| 23 | p.R342X | Nonsense | Functional | M |
| 31 | p.T146X | Nonsense | Functional | M |
| 58 | p.R213X | Nonsense | ND | M |
| 9 | c.375 + 2 T>C | Splice | ND | M |
| 52 | c.375 + 1 G>T | Splice | Nonfunctional | M |
| 77 | c.672 + 1 G>A | Splice | NC | M |
| 71 | c.673–1 G>C | Splice | Functional | M |
| 66 | c.782 + 1 G>T | Splice | Functional | M |
| 46 | c.919 + 1 G>A | Splice | ND | M |
| 34 | p.T125T | Splice | NC | M |
| 2 | c.743-750delGGAGGCC | Del | ND | M |
| 17 | c.764-766delTCA | Del | ND | M |
| 49 | c.1024delC | Del | ND | M |
| 25 | c.1146delA | Del | NC | M |
| 54 | NC | Nonfunctional | M | |
| 65 | NC | Nonfunctional | M | |
| 61 | WT | Nonfunctional | M | |
| 3 | WT | ND | WT | |
| 8 | WT | Doubtful | WT | |
| 42 | WT | Functional | WT | |
| 60 | WT | NI | WT | |
| 62 | WT | Functional | WT | |
| 59 | WT | ND | WT | |
| 15 | WT | ND | WT | |
| 73 | WT | Functional | WT | |
| 10 | WT | Functional | WT | |
| 64 | WT | NI | WT | |
| 22 | WT | ND | WT | |
| 48 | WT | NI | WT | |
| 74 | WT | NI | WT | |
| 32 | WT | Functional | WT | |
| 45 | WT | Functional | WT | |
| 20 | WT | ND | WT | |
| 4 | WT | Functional | WT | |
| 19 | WT | Functional | WT | |
| 41 | WT | Functional | WT | |
| 68 | WT | ND | WT | |
| 69 | ND | Functional | WT | |
| 76 | NC | ND | Unknown |
ND, not done; M, mutated; NC, non-contributive data; NI, non-interpretable data; Del, deletion; WT, wild type.
HIPK2 Analysis
HIPK2 Expression
HIPK2 expression analysis was analyzed from the TMA double labeled for p53 and HIPK2. After segmentation, the distribution profiles of HIPK2 immunofluorescence intensities in normal mucosa samples (Figure 4A) and tumoral tissues (Figure 4B) were built. These profiles show that both normal and tumoral tissues display a wide range of fluorescence intensities; however, each individual sample presented a normal cellular signal intensity distribution. In any case, we did not observe the appearance of a subpopulation of high-expressing cells in the tumors, as in the case of p53. Thus, it was not possible to determine a threshold of positivity for cells and a percentage of positive cells for a given tumor, as previously described for p53. Thus, to evaluate a possible impact of HIPK2 expression on survival, we considered two parameters: the absolute expression level of tumors and the variation of expression occurring in the transformation process.
Figure 4.
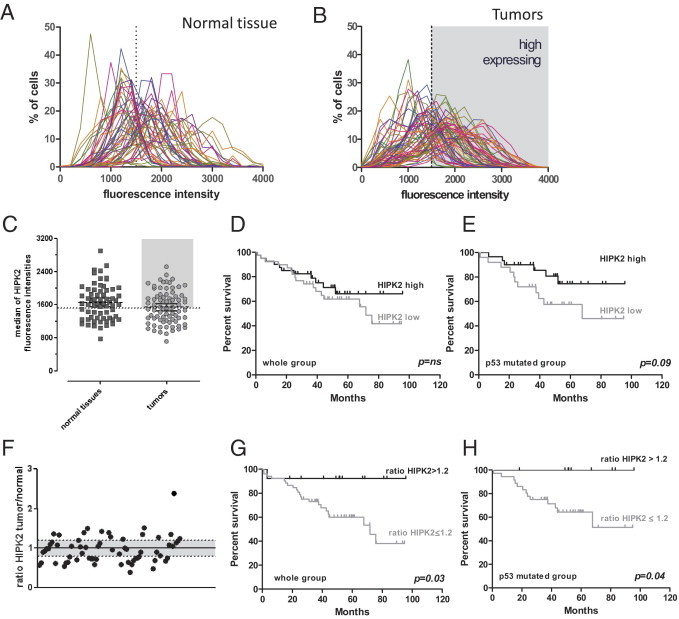
HIPK2 expression cytometry. A and B: Distribution profiles of nuclear HIPK2 fluorescence intensities among normal tissues (A) and tumors (B). Each curve represents a sample. C: Median fluorescence intensity for each sample. The dotted line in B and C represents the mean value of medians that corresponds to the threshold for HIPK2 high expression. D and E: Survival analysis according to HIPK2 expression (low and high, respectively) in the whole group (D) and in the p53 mutated group (E). F: Ratio of HIPK2 expression between tumor and normal tissue for each patient. G and H: Survival analysis comparing the HIPK2 overexpressing (ratio, ≥1.2) with the invariant or underexpressing tumors in the whole group (G) or the p53 mutated group (H).
We first analyzed the clinical data considering the absolute expression level of HIPK2 in the tumor samples. Each tumor sample was defined by its median of HIPK2 fluorescence intensity (Figure 4C). We compared the group with HIPK2 high expression (median of cell intensity expression is greater than all median fluorescence intensities) with the group with low expression (median of cell intensity expression is the median or less than all median fluorescence intensities). For OS in the whole group, the difference was not statistically different (Figure 4D); the same result was found in the p53 WT group (data not shown). In the p53 mutated group, even if the difference did not reach statistical significance, HIPK2 high expression seems to have a good prognostic impact (Figure 4E).
Consequently, we looked at the variations of expression between the normal mucosa and tumor in paired samples. To evaluate this parameter, we calculated the ratio of the medians of fluorescence intensity between tumor and normal tissue for each sample (Figure 4F). Sixty-six paired samples were available. We observed a ratio lower than 0.8 (underexpression) in 21 tumors, a ratio between 0.8 and 1.2 (no expression change) in 26 tumors, and a ratio >1.2 (overexpression) in 18 tumors. In all three groups, we observed WT p53 and mutated tumors (data not shown). There was no apparent correlation between these groups and the p53 mutational status of the tumors (ie, HIPK2 expression seems to be regulated independently of p53).
The survival analysis shows that samples that present an increased expression of HIPK2 in the tumor compared with the respective normal tissue, with a ratio >1.2, have a better outcome than tumors with a ratio of 1.2 or lower (Figure 4, G and H), irrespective of the p53 mutational status. More important, the absolute expression level of HIPK2 and its tumor/normal expression ratio did not correlate with stage, excluding a possible confounding effect of the latter. Moreover, the positive impact of the HIPK2 expression ratio on survival was also observed in the TMA from US Biomax (see Supplemental Figure S2 at http://ajp.amjpathol.org). By using multivariate Cox analysis, the independent prognostic value of the HIPK2 ratio in predicting survival in our series is of borderline significance (P = 0.08; 95% confidence interval, 0.8 to 44.9); stage IV is highly significant (P = 0.002; 95% confidence interval, 2.07 to 25.4), and stage 3 is not significant (P = 0.8; 95% confidence interval, 0.26 to 5.3).
To obtain indications on the level of control of HIPK2 expression (transcriptional or posttranscriptional), we also quantified HIPK2 mRNA level by quantitative RT-PCR.
Figure 5 shows the distribution of the mean Ct of HIPK2 and the housekeeping gene GUSB among normal mucosal tissues and tumoral tissues. The mean Ct value is similar between normal mucosa and tumor for the housekeeping gene and HIPK2, respectively. In the tumors, the range of HIPK2 expression distribution is slightly larger than in normal mucosa (Figure 5A).
Figure 5.
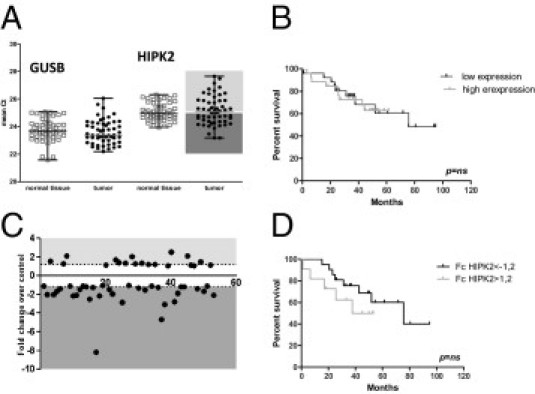
mRNA HIPK2 expression by quantitative RT-PCR. A: Distribution of mean Ct for HIPK2 and GUSB in normal tissues and tumors. Each tumor and normal tissue was studied in triplicate. The mean Ct represents the mean of the three values. Each point represents a different sample. B: Survival analysis according to HIPK2 mRNA expression (low and high that are less and greater, respectively, than the median Ct). C: FC between the tumor and normal sample. D: Survival analysis comparing the overexpressing with the underexpressing group.
Regarding mRNA variations among the tissue pairs of each patient, FCs vary from -8.18 to 2.57. Ten samples showed an FC lower than -2, with seven showing a slight variation between -2 and -3. Only four samples showed an FC >2 but <3, and 33 showed an FC between -2 and 2 (Figure 5C).
In 37 cases, we obtained both mRNA and protein HIPK2 expression results and did not find a correlation: among 11 cases with an IF ratio lower than 0.8, three had an FC lower than -2 and seven showed no variation; among five cases that have an IF ratio >1.2, only one showed an FC >2 (others showed no change). Among the 21 cases that have no IF ratio variation, five had an FC lower than -2, two had an FC >2, and 14 showed no significant FC variation. These results favored the hypothesis that the modification of HIPK2 protein levels between normal mucosa and tumors did not reside on a change in transcriptional control. This was confirmed by the survival data, in which absolute mRNA amount and FC of Ct between tumor and normal mucosa were not correlated with the prognosis of the tumors (Figure 5, B and D).
Biparametric Cytometry
The availability of paired samples makes the analysis of the evolution of these molecular markers in the transformation process possible. Moreover, the double-immunofluorescence technique associated with a quantitative analysis approach with a cellular resolution allows us to follow the simultaneous expression of the two proteins of interest at the individual cell level. To analyze if there exists a correlated variation of p53 and HIPK2 in tumor cells, we generated biparametric scatterplots of HIPK2 and p53 cellular intensities for each single cell of the tumors and of the corresponding paired normal samples (examples are shown in Figure 6 and Supplemental Figure S2E at http://ajp.amjpathol.org).
Figure 6.
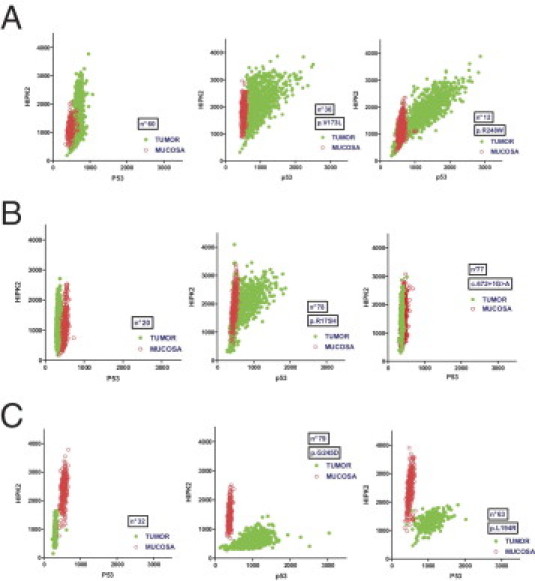
Biparametric cytometry for p53 and HIPK2 expression. Examples of biparametric scatterplots of paired samples showing p53 fluorescence (x axis) and HIPK2 fluorescence (y axis) nuclear intensities in tumors (green) and paired normal mucosa (red). A: Examples of tumors with increased HIPK2 expression compared with normal mucosa. B: Tumors with unmodified HIPK2 expression. C: Tumors with decreased HIPK2 expression. For A through C, the first scatterplot represents a p53 WT tumor, whereas the two others are p53 mutated.
Among WT p53 samples, most show a similar profile between normal mucosa and tumor for p53 and HIPK2 expression. Some samples show a population of tumoral cells with overexpression of HIPK2 and no change in p53 expression, whereas some show the opposite, a contingent of tumor cells with underexpression of HIPK2 with identical p53 expression. Among mutated p53 samples, different expression profiles have been observed. In deletion, nonsense, and splice mutations and, surprisingly, in some missense mutation samples, there is no evidence of p53 protein accumulation, as in WT p53 samples; HIPK2 could display either underexpression or overexpression compared with the corresponding normal mucosa. As expected in the missense mutation group, numerous samples exhibit p53 overexpression. Among these cases, some samples show concomitant HIPK2 overexpression. In the latter case, the expression level of p53 appears directly correlated with that of HIPK2 at the unicellular level. However, it is possible to identify a group of samples that shows a p53 accumulation associated with a loss of HIPK2 expression. As previously described herein, the group exhibiting HIPK2 overexpression compared with normal mucosa presents the better outcome for OS. As a mirror image, the group characterized by HIPK2 underexpression presents the worst survival rates.
Discussion
In this study, we analyzed the impact of HIPK2 expression on the survival of a series of patients with colorectal carcinoma and explored the possibility of a link with the p53 status of the tumors. The goal of this study was to analyze whether the functional role of HIPK2, which is recognized as an important modulator of the p53 response in in vitro cellular models, could be strong enough to have an impact on prognosis for patients. Thus, we constructed a TMA with 80 tumor-normal tissue pairs from patients with colorectal carcinoma and analyzed these samples by multiplexed tissue cytometry, simultaneously monitoring HIPK2 and p53 expression at the single cell level. In addition, we explored the p53 mutational status of each sample.
Concerning p53 mutations, we found a higher level of p53 mutations in our series compared with the large series published by the TP53-CRC Collaborative Study Group,4 which could be because of a higher percentage of stage III and IV disease; the percentage of p53 mutations tends to increase with stage. We did not find any prognostic impact of the p53 mutations in our series. This result is not surprising because, in most series, the prognostic impact of p53 mutations, if any, is always restricted to a subset of patients.3,4,27
Furthermore, the overexpression of p53, which could be observed in the group of tumors bearing a p53 missense mutation, is not associated with a significantly different prognosis, confirming that p53 is not a good marker for colorectal cancers. At the functional level, the only effect that we could measure as a direct consequence of the p53 mutations is the decrease of expression of p21 at the mRNA level in many mutated tumors, whereas the mRNA levels of the proapoptotic target Bax seem to not be affected. This p21 mRNA decrease could be compatible with a more actively proliferating status of the tumors; however, even if this holds true, this does not seem to have a strong clinical impact because the tumors concerned do not appear to be more aggressive.
For HIPK2, we did not observe a significant difference in the range of expression level in the tumors compared with the normal mucosa, in a subgroup of tumors, or in a subpopulation of cells in tumors.
However, the availability of the tumor-normal tissue pairs in our TMA allowed us to monitor the appearance of changes in the expression levels of HIPK2 between the normal and tumor tissue of each patient. Thus, we observed that an increase of HIPK2 protein level in the tumor compared with the corresponding normal tissue was associated with a better prognosis in univariate analysis. Interestingly, we did not find that the prognostic significance of HIPK2 overexpression was dependent on WT p53 status, as previously reported.18 Measuring HIPK2 at the protein level, we found a good prognostic impact of HIPK2 overexpression in tumors, irrespective of the presence of p53 mutations. Functionally, a possible explanation is that HIPK2 is able to modulate apoptosis in a p53-independent manner (eg, via the c-Jun N-terminal kinase pathway).19,20 Another hypothesis is that the increased expression of HIPK2 is able to activate residual p53 WT protein because of a WT residual allele or the p53 mutant protein conferring to these forms a transactivating activity that allows apoptosis. In this study, we developed and validated a method for quantifying and analyzing multiple protein expression at the single cell level on archival paraffin-embedded tissue using TMA technology and double immunofluorescence. Our data confirm the value of double-immunofluorescence labeling on histological material to evaluate, at the cellular level, the expression of several proteins implicated in the same pathway.28 Standard IHC and quantification by visual analysis provide semiquantitative values and a limited range, especially at a low or high expression level. By comparison, immunofluorescence coupled to image segmentation–based analysis allows the objective quantification of a wide range of expression levels in many cells. Moreover, the TMA approach, by the simultaneous analysis of hundreds of archival tumor samples, is becoming increasingly important for the validation of experimental data issues from transcriptomic analysis or functional approaches. Although “dedicated” imaging platforms have been developed to perform this kind of analysis at a high throughput, we show herein that this approach can be performed with any laser-scanning microscope associated with multipurpose segmentation-based image analysis software (ie, with common investigation tools available in most cell biology laboratories). More particularly, in this study, we show the interest of comparing, through quantitative analysis, the normal and tumor tissues in a TMA, thus highlighting differences of expression that would not be appreciated by standard IHC determined by visual analysis of colorimetric stains. Moreover, we compared our results with those obtained with a fully automated platform (see Supplemental Figure S3 at http://ajp.amjpathol.org). Both TMAs were automatically acquired and analyzed using a laser-scanning cytometer (iCys; Compucyte). Pseudocellular segmentation of the carcinoma (or epithelial) component of the cores was achieved by using random phantom segmentation. Automated exclusion of the stroma from analysis was obtained by gating out the phantoms that did not exhibit the DAPI staining feature combination (maximum pixel, integral, and mean intensity) that was distinctive of the carcinoma cells.
By performing this cross-platform verification, we confirmed our observations and highlighted some technical issues that could be critical for automated analysis.
We found similar and straightforward results with LSC and LSM acquisition for p53 that undergoes large variations of intensities and that could easily be expressed as a “cytometric percentage.”
HIPK2 analysis appeared more critical with the automated platform for several technical reasons: more important fluorescence background and less precise definition of the tumor epithelium using fixed parameters for the whole core series. The combination of both issues significantly affected the determination of the tumor/normal tissue expression ratio. Thus, even if we could find the same type of expression profiles (see Supplemental Figure S3, B and C, at http://ajp.amjpathol.org) and similar results for the small TMA from US Biomax, the fully automated analysis of the 80 cases of “homemade” TMA was less performant. We could conclude that, although the use of an automated platform is fully suitable for markers with a large range of signal variation, it should be used with care for more exploratory analyses. In this case, the operator-assisted LSM analysis, even with a low throughput, should be preferred in the first instance. Even if HIPK2 could not be considered a direct biomarker for colorectal cancer because its impact on prognosis resides more in the variations of expression than in the absolute expression level, this analysis could be the starting point to explore new molecular mechanisms that could determine the evolution of the pathological characteristics and/or the response to treatments for colon cancer.
Acknowledgments
We thank Dr. Pippa McKelvie-Sebileau for her help with the English version and Mrs. Dorothée Quincy (both from Institut Bergonié, Bordeaux, France) for manuscript editing.
Footnotes
Supported by the French National Institute for Medical Research (INSERM); Aquitaine Region, French Ministry of Research;Institut Bergonié; AMGEN France; ROCHE Laboratories; Association for Cancer Research; the Ligue Contre le Cancer; and Cancéropôle Grand Sud-Ouest and “Agir Cancer” Gironde.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi:10.1016/j.ajpath.2011.01.021.
Supplementary data
Stage-dependent survival of the colorectal cancer patient series used to build the TMA.
Analysis of an independent series of C-C chemokine receptors double labeled by immunofluorescence for p53 and HIPK2. This TMA contains 30 colorectal cancers, eight of which have matched normal adjacent tissue. A: Percentage of p53-positive cells in normal and tumor cores. B: Survival analysis of the p53+ group (tumors with more than five percent p53-positive cells) and the p53- group (tumors with less than five percent p53-positive cells). C: Expression of HIPK2 in normal and tumor cores. D: Correlation plot between the tumor/normal ratio of HIPK2 expression and the survival (in months) for the eight patients with matched tissues. E: An example of patients with an increased expression of HIPK2 (core images: overlay of DAPI, p53 green, and HIPK2 red channels) and of the cytometric quantification of the signal on the normal and matched tumor cores, respectively.
Comparison of LSM with automated LSC acquisition and analysis. A: Correlation of the percentage of p53-positive cells in tumor cores obtained by LSM (x axis) and LSC (y axis). Homemade TMA: black circles, TMA from US Biomax; gray squares, the border color of the scattegram displays. B: Examples of biparametric scatterplots of paired samples shown in Figure 6 (left) and the same scatterplots obtained by LSC analysis; each plot shows p53 (x axis) and HIPK2 (y axis) nuclear fluorescence intensities in tumors (green) and paired normal mucosa (red). C: Correlation plot between the medians of HIPK2 intensities obtained by LSM (x axis) and LSC (y axis) in the homemade TMA. D: Correlation plot between the tumor/normal ratio of HIPK2 expression calculated on the basis of the LSM (x axis) and LSC (y axis) acquisition and analyses, respectively.
References
- 1.Vousden K.H., Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 2.Iacopetta B. TP53 mutation in colorectal cancer. Hum Mutat. 2003;21:271–276. doi: 10.1002/humu.10175. [DOI] [PubMed] [Google Scholar]
- 3.Iacopetta B., Russo A., Bazan V., Dardanoni G., Gebbia N., Soussi T., Kerr D., Elsaleh H., Soong R., Kandioler D., Janschek E., Kappel S., Lung M., Leung C.S., Ko J.M., Yuen S., Ho J., Leung S.Y., Crapez E., Duffour J., Ychou M., Leahy D.T., O'Donoghue D.P., Agnese V., Cascio S., Di Fede G., Chieco-Bianchi L., Bertorelle R., Belluco C., Giaretti W., Castagnola P., Ricevuto E., Ficorella C., Bosari S., Arizzi C.D., Miyaki M., Onda M., Kampman E., Diergaarde B., Royds J., Lothe R.A., Diep C.B., Meling G.I., Ostrowski J., Trzeciak L., Guzinska-Ustymowicz K., Zalewski B., CapellÁ G.M., Moreno V., Peinado M.A., Lönnroth C., Lundholm K., Sun X.F., Jansson A., Bouzourene H., Hsieh L.L., Tang R., Smith D.R., Allen-Mersh T.G., Khan Z.A., Shorthouse A.J., Silverman M.L., Kato S., Ishioka C., TP53-CRC Collaborative Group Functional categories of TP53 mutation in colorectal cancer: results of an International Collaborative Study. Ann Oncol. 2006;17:842–847. doi: 10.1093/annonc/mdl035. [DOI] [PubMed] [Google Scholar]
- 4.Russo A., Bazan V., Iacopetta B., Kerr D., Soussi T., Gebbia N. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol. 2005;23:7518–7528. doi: 10.1200/JCO.2005.00.471. [DOI] [PubMed] [Google Scholar]
- 5.Kruse J.P., Gu W. Modes of p53 regulation. Cell. 2009;137:609–622. doi: 10.1016/j.cell.2009.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y.H., Choi C.Y., Lee S.J., Conti M.A., Kim Y. Homeodomain-interacting protein kinases, a novel family of co-repressors for homeodomain transcription factors. J Biol Chem. 1998;273:25875–25879. doi: 10.1074/jbc.273.40.25875. [DOI] [PubMed] [Google Scholar]
- 7.D'Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G., Piaggio G., Fanciulli M., Appella E., Soddu S. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol. 2002;4:11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 8.Dauth I., Kruger J., Hofmann T.G. Homeodomain-interacting protein kinase 2 is the ionizing radiation-activated p53 serine 46 kinase and is regulated by ATM. Cancer Res. 2007;67:2274–2279. doi: 10.1158/0008-5472.CAN-06-2884. [DOI] [PubMed] [Google Scholar]
- 9.Hofmann T.G., Möller A., Sirma H., Zentgraf H., Taya Y., Dröge W., Will H., Schmitz M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol. 2002;4:1–10. doi: 10.1038/ncb715. [DOI] [PubMed] [Google Scholar]
- 10.Mayo L.D., Seo Y.R., Jackson M.W., Smith M.L., Rivera Guzman J., Korgaonkar C.K., Donner D.B. Phosphorylation of human p53 at serine 46 determines promoter selection and whether apoptosis is attenuated or amplified. J Biol Chem. 2005;280:25953–25959. doi: 10.1074/jbc.M503026200. [DOI] [PubMed] [Google Scholar]
- 11.Oda K., Arakawa H., Tanaka T., Matsuda K., Tanikawa C., Mori T., Nishimori H., Tamai K., Tokino T., Nakamura Y., Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 12.Rinaldo C., Prodosmo A., Mancini F., Iacovelli S., Sacchi A., Moretti F., Soddu S. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol Cell. 2007;25:739–750. doi: 10.1016/j.molcel.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 13.Guo A., Salomoni P., Luo J., Shih A., Zhong S., Gu W., Pandolfi P.P. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- 14.Pearson M., Carbone R., Sebastiani C., Cioce M., Fagioli M., Saito S., Higashimoto Y., Appella E., Minucci S., Pandolfi P.P., Pelicci P.G. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 15.Di Stefano V., Blandino G., Sacchi A., Soddu S., D'Orazi G. HIPK2 neutralizes MDM2 inhibition rescuing p53 transcriptional activity and apoptotic function. Oncogene. 2004;23:5185–5192. doi: 10.1038/sj.onc.1207656. [DOI] [PubMed] [Google Scholar]
- 16.Di Stefano V., Mattiussi M., Sacchi A., D'Orazi G. HIPK2 inhibits both MDM2 gene and protein by, respectively, p53-dependent and independent regulations. FEBS Lett. 2005;579:5473–5480. doi: 10.1016/j.febslet.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Nardinocchi L., Puca R., Sacchi A., D'Orazi G. HIPK2 knock-down compromises tumor cell efficiency to repair damaged DNA. Biochem Biophys Res Commun. 2007;361:249–255. doi: 10.1016/j.bbrc.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 18.Puca R., Nardinocchi L., Gal H., Rechavi G., Amariglio N., Domany E., Notterman D.A., Scarsella M., Leonetti C., Sacchi A., Blandino G., Givol D., D'Orazi G. Reversible dysfunction of wild-type p53 following homeodomain-interacting protein kinase-2 knockdown. Cancer Res. 2008;68:3707–3714. doi: 10.1158/0008-5472.CAN-07-6776. [DOI] [PubMed] [Google Scholar]
- 19.Hofmann T.G., Stollberg N., Schmitz M.L., Will H. HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 2003;63:8271–8277. [PubMed] [Google Scholar]
- 20.Wang S.Y., Iordanov M., Zhang Q. c-Jun NH2-terminal kinase promotes apoptosis by down-regulating the transcriptional co-repressor CtBP. J Biol Chem. 2006;281:34810–34815. doi: 10.1074/jbc.M607484200. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Q., Nottke A., Goodman R.H. Homeodomain-interacting protein kinase-2 mediates CtBP phosphorylation and degradation in UV-triggered apoptosis. Proc Natl Acad Sci U S A. 2005;102:2802–2807. doi: 10.1073/pnas.0409373102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sombroek D., Hofmann T.G. How cells switch HIPK2 on and off. Cell Death Differ. 2009;16:187–194. doi: 10.1038/cdd.2008.154. [DOI] [PubMed] [Google Scholar]
- 23.Calzado M.A., Renner F., Roscic A., Schmitz M.L. HIPK2: a versatile switchboard regulating the transcription machinery and cell death. Cell Cycle. 2007;6:139–143. doi: 10.4161/cc.6.2.3788. [DOI] [PubMed] [Google Scholar]
- 24.Gresko E., Roscic A., Ritterhoff S., Vichalkovski A., del Sal G., Schmitz M.L. Autoregulatory control of the p53 response by caspase-mediated processing of HIPK2. EMBO J. 2006;25:1883–1894. doi: 10.1038/sj.emboj.7601077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Winter M., Sombroek D., Dauth I., Moehlenbrink J., Scheuermann K., Crone J., Hofmann T.G. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat Cell Biol. 2008;10:812–824. doi: 10.1038/ncb1743. [DOI] [PubMed] [Google Scholar]
- 26.Flaman J.M., Frebourg T., Moreau V., Charbonnier F., Martin C., Chappuis P., Sappino A.P., Limacher J.M., Bron L., Benhattar J., Tada M., van Meir E.G., Estreicher A., Iggo R.D. Simple p53 functional assay for screening cell lines, blood, and tumors. Proc Natl Acad Sci U S A. 1995;92:3963–3967. doi: 10.1073/pnas.92.9.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Westra J.L., Schaapveld M., Hollema H., de Boer J.P., Kraak M.M., de Jong D., ter Elst A., Mulder N.H., Buys C.H., Hofstra R.M., Plukker J.T. Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J Clin Oncol. 2005;23:5635–5643. doi: 10.1200/JCO.2005.04.096. [DOI] [PubMed] [Google Scholar]
- 28.Tellez C.S., Davis D.W., Prieto V.G., Gershenwald J.E., Johnson M.M., McCarty M.F., Bar Eli M. Quantitative analysis of melanocytic tissue array reveals inverse correlation between activator protein-2alpha and protease-activated receptor-1 expression during melanoma progression. J Invest Dermatol. 2007;127:387–393. doi: 10.1038/sj.jid.5700539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stage-dependent survival of the colorectal cancer patient series used to build the TMA.
Analysis of an independent series of C-C chemokine receptors double labeled by immunofluorescence for p53 and HIPK2. This TMA contains 30 colorectal cancers, eight of which have matched normal adjacent tissue. A: Percentage of p53-positive cells in normal and tumor cores. B: Survival analysis of the p53+ group (tumors with more than five percent p53-positive cells) and the p53- group (tumors with less than five percent p53-positive cells). C: Expression of HIPK2 in normal and tumor cores. D: Correlation plot between the tumor/normal ratio of HIPK2 expression and the survival (in months) for the eight patients with matched tissues. E: An example of patients with an increased expression of HIPK2 (core images: overlay of DAPI, p53 green, and HIPK2 red channels) and of the cytometric quantification of the signal on the normal and matched tumor cores, respectively.
Comparison of LSM with automated LSC acquisition and analysis. A: Correlation of the percentage of p53-positive cells in tumor cores obtained by LSM (x axis) and LSC (y axis). Homemade TMA: black circles, TMA from US Biomax; gray squares, the border color of the scattegram displays. B: Examples of biparametric scatterplots of paired samples shown in Figure 6 (left) and the same scatterplots obtained by LSC analysis; each plot shows p53 (x axis) and HIPK2 (y axis) nuclear fluorescence intensities in tumors (green) and paired normal mucosa (red). C: Correlation plot between the medians of HIPK2 intensities obtained by LSM (x axis) and LSC (y axis) in the homemade TMA. D: Correlation plot between the tumor/normal ratio of HIPK2 expression calculated on the basis of the LSM (x axis) and LSC (y axis) acquisition and analyses, respectively.