

Abstract
Lung cancer is the leading cause of cancer-related deaths in the United States. Approximately 85% of lung cancer is categorized as non–small cell lung cancer, and traditionally, non–small cell lung cancer has been treated with surgery, radiation, and chemotherapy. Targeted agents that inhibit the epidermal growth factor receptor pathway have been developed and integrated into the treatment regimens in non–small cell lung cancer. Currently, approved epidermal growth factor receptor inhibitors include the tyrosine kinase inhibitors erlotinib and gefitinib. Molecular determinants, such as epidermal growth factor receptor–activating mutations, have been associated with response to epidermal growth factor receptor tyrosine kinase inhibitors and may be used to guide treatment choices in patients with non–small cell lung cancer. Thus, treatment choice for patients with non–small cell lung cancer depends on molecular features of tumors; however, improved techniques are required to increase the specificity and efficiency of molecular profiling so that these methods can be incorporated into routine clinical practice. This review provides an overview of how genetic analysis is currently used to direct treatment choices in non–small cell lung cancer.
Lung cancer is the second most common malignancy (surpassed in incidence only by nonmelanoma skin cancer) and the leading cause of cancer-related death in the United States, with an estimated 219,440 new diagnoses and 159,390 deaths in 2009.1,2 Between 1996 and 2004, the 5-year survival rate was 15% for patients with lung cancer across all stages of disease—ranging from 2.8% for patients with distant metastases to nearly 50% for those presenting with local disease.1 Curative-intent surgery is the preferred treatment modality but has limited applicability, given the typical presentation at an unresectable locally advanced or metastatic stage, for which platinum-based chemotherapy has long been regarded as the standard of care (National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Non–Small Cell Lung Cancer. V. 2.2010; http://www.nccn.org/professionals/physician_gls/PDF/nscl.pdf, last accessed September 15, 2010).
Given the clinical burden of lung cancer, with non–small cell lung cancer (NSCLC) accounting for approximately 85% of cases, it has become a major platform for the clinical development of biological-targeting agents. To date, targeted agents that have been granted US Food and Drug Administration approval for treating advanced NSCLC are the anti-vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab (Avastin; Genentech, South San Francisco, CA), indicated as an adjunct to first-line chemotherapy with paclitaxel/carboplatin [Avastin (bevacizumab) package insert. Genentech, Inc., South San Francisco, CA], and the reversible epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) erlotinib and gefitinib in chemotherapy-pretreated patients (Figure 1) [Iressa (gefitinib tablets) package insert. AstraZeneca Pharmaceuticals LP, Wilmington, DE; Tarceva (erlotinib tablets) package insert. Genentech, Inc., South San Francisco, CA]. Erlotinib (Tarceva; Genentech) was initially approved by the US Food and Drug Administration in 2004 for patients with advanced NSCLC failing one chemotherapy regimen, a setting in which the objective response rate (ORR) was 9%, but overall survival (OS) was significantly improved by 2 months versus placebo [Tarceva (erlotinib tablets) package insert. Genentech, Inc., South San Francisco, CA].3 In April 2010, erlotinib received an additional indication in advanced NSCLC of maintenance use after four cycles of first-line, platinum-based chemotherapy, with a placebo-controlled trial showing significantly prolonged OS and progression-free survival (PFS) [Tarceva (erlotinib tablets) package insert. Genentech, Inc., South San Francisco, CA].4 Rapid and profound responses to gefitinib (Iressa; AstraZeneca, Wilmington, DE) in chemotherapy-pretreated patients, albeit only in select patients in the early trials (ORR was <20%, with additional symptomatic benefits),5,6 formed the basis of its regulatory approval in the absence of an OS benefit. In the United States, gefitinib is only indicated as monotherapy for advanced NSCLC after platinum-based and docetaxel chemotherapy in patients who are benefiting or have benefited from gefitinib therapy; however, according to the labeling, preference should be given to another EGFR TKI (ie, erlotinib) in light of the published survival gain with erlotinib [Iressa (gefitinib tablets) package insert. AstraZeneca Pharmaceuticals LP, Wilmington, DE].
Figure 1.
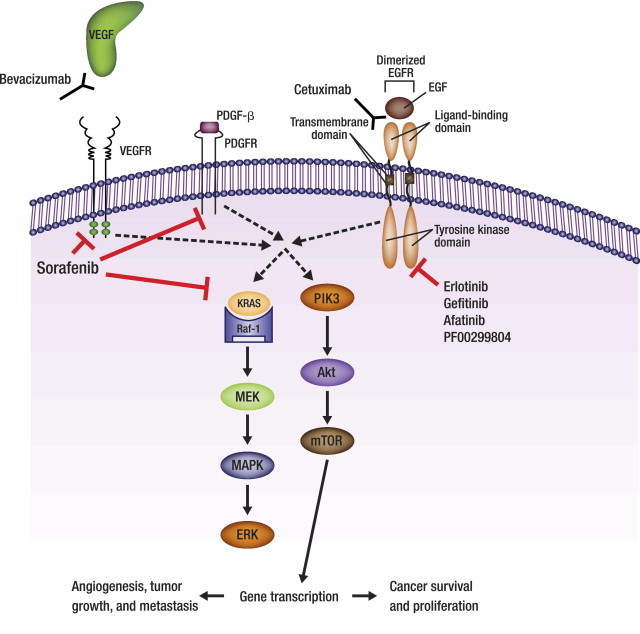
Targeting of NSCLC by VEGF inhibitors and EGFR TKIs. Bevacizumab, a monoclonal antibody, recognizes VEGF and prevents it from binding to the VEGFR which disrupts angiogenesis, halting tumor growth and metastasis. Sorafenib disrupts angiogenesis by inhibiting multiple receptors, including VEGFR, PDGFR-β, and RAF-1, as well as Flt3 and c-kit (not shown). Erlotinib and gefitinib reversibly bind to and inhibit the tyrosine kinase domain of the EGFR, cetuximab reversibly binds to the ligand-binding domain of the EGFR, and afatinib and PF00299804 irreversibly bind to and inhibit the tyrosine kinase domain of the EGFR to disrupt tumor proliferation and survival. Akt, protein kinase B; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; PDGF-β, platelet-derived growth factor-β; PDGFR, platelet-derived growth factor receptor; PIK3, phosphatidylinositol 3-kinase; Raf-1, v-raf 1 murine leukemia viral oncogene homolog 1; Ras, retrovirus-associated DNA sequences; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
In 2004, the highly anticipated phase III data for gefitinib or erlotinib as a component of first-line chemotherapy7–10 showed no benefit over placebo, tempering enthusiasm for EGFR-directed therapy for advanced NSCLC. That same year, however, a series of studies reported preferential activity for EGFR TKIs in patients with tumors exhibiting somatic EGFR mutations, revealing some of the unexplained favorable trends in certain subgroups and serving to refocus the clinical development of this class of agents for NSCLC.11–13 With the recognition that only a subset of patients with advanced NSCLC will benefit from erlotinib or gefitinib and that resistance invariably occurs, research efforts to further improve NSCLC-directed EGFR TKI therapy include the development of agents that irreversibly bind to their targets, inhibit several EGFR family receptors, and/or simultaneously inhibit EGFR and other oncogenic targets. Therapies targeting the EGFR pathway that are currently in the phase III stage of clinical development, including next-generation EGFR TKIs, are included in Table 1.
Table 1.
EGFR Inhibitors in Phase III Development for the Treatment of NSCLC
| Agent | Binding | Target(s) | Clinical setting | ClinicalTrials.gov identifier (status as of August 2010) |
|---|---|---|---|---|
| EGFR TKIs | ||||
| Afatinib (BIBW 2992) | Irreversible | EGFR, HER2 | Phase III trial of afatinib as first-line therapy versus pemetrexed/cisplatin in patients with confirmed EGFR-activating mutations (LUX-Lung 3) | NCT00949650 (recruiting) |
| Phase III trial of afatinib in combination with paclitaxel versus single-agent chemotherapy (chosen by the investigator) in patients with progressive disease after treatment with chemotherapy⁎ and erlotinib or gefitinib who subsequently received benefit from afatinib monotherapy (LUX-Lung 5) | NCT01085136 (recruiting) | |||
| Phase III trial of afatinib versus gemcitabine/cisplatin in patients with NSCLC harboring an EGFR mutation (LUX-Lung 6) | NCT01121393 (recruiting) | |||
| PF00299804 | Irreversible | EGFR, HER2, HER4 | Phase III trial of PF00299804 compared with placebo in patients with stage IIIB or IV NSCLC after failure of standard therapy for advanced or metastatic disease | NCT01000025 (recruiting) |
| EGFR monoclonal antibody | ||||
| Cetuximab | Reversible | EGFR | Phase III trial of cisplatin/vinorelbine with or without cetuximab as first-line treatment of patients with advanced NSCLC (FLEX) | NCT00148798 (active but not recruiting†) |
| Phase III trial of docetaxel or pemetrexed with or without cetuximab in patients with recurrent or progressive NSCLC | NCT00095199 (active but not recruiting) | |||
| Phase III trial of cetuximab as maintenance therapy after platinum-based chemotherapy in combination with cetuximab as first-line treatment of patients with advanced NSCLC (NEXT) | NCT00820755 (active but not recruiting) | |||
| Phase III trial of high-dose or standard-dose radiation therapy in combination with chemotherapy with or without cetuximab in patients with newly diagnosed, unresectable, stage III NSCLC | NCT00533949 (recruiting) | |||
| Phase III trial of carboplatin and paclitaxel with or without bevacizumab and/or cetuximab in patients with stage IV or recurrent NSCLC | NCT00946712 (recruiting) |
BSC, best supportive care; EGFR, epidermal growth factor receptor; HER, human epidermal growth factor receptor; NSCLC, non–small cell lung cancer; TKI, tyrosine kinase inhibitor.
Patients are exempt from prior chemotherapy if they have a confirmed EGFR mutation or had ≥6 months of benefit from erlotinib or gefitinib.
Favorable results have been published.14
Until recently, broad histologic classification (NSCLC versus small cell lung cancer), clinical or surgical staging, and performance status had been sufficient for guiding treatment decisions surrounding surgery, radiation, and/or cytotoxic chemotherapy in the lung cancer population. As part of the evolving treatment paradigm, the criticality of molecular profiling of tumor samples is now evident, as illustrated by the clinical trial experiences discussed herein.
Importance of Molecular Classification in NSCLC
EGFR Mutations as a Predictive Marker
The molecular features of NSCLC tumors have become an important consideration for predicting response to selected therapies. Point mutations, deletions, or insertions can occur in the catalytic tyrosine kinase domain (exons 18–21) of EGFR, located on chromosome 7.15 On the basis of a literature-based analysis of >2000 NSCLC samples containing 477 somatic mutations, in-frame deletions in exon 19 were the predominant form of all EGFR mutations, with an incidence of 44%.16 The second most common EGFR mutation was the single nucleotide substitution L858R in exon 21, accounting for 41% of all mutations. Rarer mutations included a single nucleotide substitution G719X (X can be A, C, or S) in exon 18, in-frame duplications/insertions in exon 20, and rare missense mutations in exons 18-21, accounting for 4%, 5%, and 6% of all EGFR mutations, respectively.
Efforts to elucidate the clinical implications of EGFR mutations have shown their variability across NSCLC histologies. As reviewed by Shigematsu and Gazdar,16 based on a collective assessment of published clinical studies and case reports, the incidence of EGFR mutations appears to vary greatly by histology and 3 other patient characteristics. There has been a significant predominance of EGFR mutations among patients with tumors of adenocarcinoma histology (30% versus 2% for other histologies), who were never smokers (45% versus 7% for ever smokers), and of female sex (38% versus 10% for men) or East Asian decent (33% versus 6% for non-Asian patients). A series of ongoing epidemiologic evaluations are expected to further elucidate the frequency, characteristics, and outcomes of patients with EGFR mutation-positive NSCLC (NCT00997230, NCT01081496, and NCT01100827).
EGFR Mutations Influencing Activity of EGFR TKI Monotherapy in Pretreated Patients
The high rate of tumor EGFR overexpression in patients with NSCLC, for which therapeutic gains had long remained elusive, provided a mechanistic rationale for studying EGFR TKI therapy in this population. Results of early phase II trials of the efficacy and safety of gefitinib monotherapy in pretreated advanced NSCLC showed modest activity when considering the study populations in their entirety; trends for greater benefit were also observed in certain subgroups, including women, never smokers, patients with adenocarcinoma histology, and patients of Japanese ethnicity.5,6 The tumor EGFR expression level, however, showed no correlation with outcome.5,6 As the potential for EGFR TKI treatment of advanced NSCLC appeared to be waning, reports by several research groups pointed to the same conclusion: the activity of gefitinib or erlotinib seen in a subset of patients was largely attributable to somatic mutations in the catalytic tyrosine kinase domain of EGFR.11–13 Across three reports published during 2004, the frequency of somatic EGFR mutation was 81% among the 31 patients achieving a partial response or experiencing notable improvement during EGFR TKI therapy compared with 0% among the 29 nonresponders.13 Since then, favorable efficacy results have been reported for EGFR TKI monotherapy, specifically in patients harboring EGFR mutations,17–19 with erlotinib listed within the National Comprehensive Cancer Network guidelines as a therapeutic option in these patients regardless of performance status level (National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Non–Small Cell Lung Cancer. V. 2.2010; http://www.nccn.org/professionals/physician_gls/PDF/nscl.pdf, last accessed September 15, 2010).
A prospective biomarker analysis of the phase III trial (INTEREST; IRESSA Non–small cell lung cancer Trial Evaluating REsponse and Survival against Taxotere) of gefitinib or docetaxel monotherapy in patients with platinum-pretreated advanced NSCLC, in which the primary endpoint of OS and other outcomes were similar between the two arms, was recently published.20 Biomarker evaluation was feasible in 453 patients, or approximately one-third of the 1466 patients in the randomized study population. Other than a significantly higher ORR with gefitinib versus docetaxel in patients with high EGFR copy number (13.0% versus 7.4%), no significant PFS or OS benefits for gefitinib versus docetaxel were observed based on EGFR protein expression or EGFR copy number. However, among 44 EGFR mutation-positive patients, gefitinib was significantly more effective than docetaxel in terms of a more prolonged PFS (7.0 versus 4.1 months) and a higher ORR (42.1% versus 21.1%). An OS benefit (14.2 versus 16.6 months) was not observed with a possible confounding influence of subsequent therapies. In addition to the between-treatment differences on the basis of EGFR mutation status, the more favorable survival outcomes among the EGFR mutation-positive subset relative to both the EGFR wild-type group (median survival, 6.4 and 6.0 months with gefitinib and docetaxel, respectively) and the overall study population (median survival, 7.6 and 8.0 months with gefitinib and docetaxel, respectively) are noteworthy.
A recent meta-analysis of clinical trials of erlotinib or gefitinib monotherapy for NSCLC,21 stratifying objective response data by EGFR mutation and/or copy number status, showed that EGFR mutations (1020 of 3101 patients) were good predictors of response. Sensitivity was 0.78 and specificity was 0.86, with positive and negative likelihood ratios of 5.6 and 0.26, respectively. Gain in the copy number of EGFR (542 of 1539 patients) was predictive of response to single-agent therapy with EGFR TKIs, but with less favorable sensitivity (0.61), specificity (0.71), and positive and negative likelihood ratios (2.1 and 0.55, respectively) relative to EGFR mutation status. For both mutation and gene copy number, diagnostic accuracy was better in white than in East Asian patients.
EGFR Mutations Influencing Activity of EGFR TKI Therapy in Previously Untreated Patients
Results of a retrospective biomarker analysis of the placebo-controlled phase III trial (TRIBUTE; Tarceva response in conjunction with paclitaxel and carboplatin) of erlotinib in advanced NSCLC (N = 1079), which had failed to show any benefit for adding the EGFR TKI to first-line paclitaxel/carboplatin chemotherapy, support the influence of EGFR mutation status in previously untreated patients undergoing chemotherapy with or without erlotinib.17 Of 228 samples with determinant EGFR status, 29 tumors (13%) had identifiable mutations. The ORR with erlotinib plus chemotherapy was 53% among the subset with EGFR mutation-positive tumors and was significantly higher than the 18% ORR for mutation-negative tumors. When combining the treatment arms, patients with EGFR mutation-positive versus mutation-negative tumors not only had a significantly higher ORR (38% versus 23%) but also experienced significantly prolonged time to progression (8 versus 5 months) and OS (not reached versus 10 months). Further subanalyses by treatment in the EGFR mutation-positive subset were limited by sample size, with erlotinib/chemotherapy associated with numerical but not statistically significant improvements in ORR (53% versus 21% for chemotherapy alone) and time to progression (12.5 versus 6.6 months for chemotherapy alone). With only four deaths occurring at the analysis cutoff, median OS for the EGFR mutation-positive subsets receiving erlotinib/chemotherapy versus chemotherapy alone could not be calculated; however, the Kaplan-Meier curves were similar between the arms.
Recently published results of the phase III open-label IPASS (IRESSA Pan-Asia Study) trial provide particularly compelling evidence for the influence of EGFR mutation status on response to EGFR TKI treatment in previously untreated patients.22 IPASS evaluated first-line gefitinib versus paclitaxel/carboplatin in an Asian advanced NSCLC population selected on the basis of clinical characteristics—specifically those with tumors of adenocarcinoma histology who were never smokers or former light smokers. Overall, across all patients, gefitinib significantly improved both PFS and ORR relative to paclitaxel/carboplatin. The EGFR mutation rate was high, approaching 60%, among the 437 patients evaluable for mutation status. Compared with paclitaxel/carboplatin, gefitinib conferred significantly longer PFS in patients with EGFR mutation-positive tumors and significantly shorter PFS in patients with EGFR mutation-negative tumors. The difference in gefitinib-associated ORR based on EGFR mutation status was striking: 71% for EGFR mutation-positive versus 1% for mutation-negative subsets, rates that in both cases were significantly different from those in the chemotherapy arm (47.3% and 23.5% for chemotherapy-treated EGFR mutation-positive and mutation-negative patients, respectively).22 The recent presentation of mature OS data of the trial indicated that there was no significant difference in OS between treatment groups in the overall population, EGFR mutation-positive subpopulation, and EGFR mutation-negative subpopulation.23
Similar results were recently reported in two trials in Japan, both of which evaluated first-line gefitinib in patients with NSCLC harboring an EGFR mutation with a primary endpoint of prolonged PFS.24,25 In the first trial, gefitinib was evaluated versus carboplatin/paclitaxel, and, at a planned interim analysis, PFS was significantly prolonged (median, 10.4 versus 5.5 months) and the ORR was significantly improved (73.7% versus 30.7%) with gefitinib; however, OS did not differ significantly between the two groups.24 In the second trial, gefitinib treatment resulted in significant improvements in PFS and ORR compared with cisplatin/docetaxel. OS data are not yet mature, and follow-up is ongoing.25
Additional Determinants of Response to EGFR TKIs
T790M Mutation
The T790M point mutation in exon 20 of EGFR is most often associated with acquired/secondary resistance to EGFR TKIs15 and has been detected in approximately 50% of NSCLC tumors that initially responded to erlotinib or gefitinib.15,26 Although T790M is the most common, there are several other secondary mutations of EGFR that have been linked to acquired resistance to reversible EGFR TKIs, including D761Y in exon 1927 and T854A mutation in exon 21.28
Data suggest that the T790M mutation may also contribute to primary resistance to EGFR TKI therapy. For example, in a small study, the T790M mutation was present in 10 of 26 NSCLC tumor samples (38%) before erlotinib or gefitinib therapy; patients whose tumors harbored the mutation had a significantly shorter PFS than patients who did not harbor the mutation (7.7 versus 16.5 months, respectively).29 It is probable that patients who initially respond to therapy may have T790M mutations in a small percentage of tumor cells, and then during treatment with erlotinib or gefitinib, clonal selection enables these T790M-expressing cells to populate more of the tumor mass over time.29,30
KRAS
Kirsten rat sarcoma viral oncogene homolog (KRAS, alias Ki-Ras) is a GTPase that transduces activated EGFR signaling, with roles in proliferation, differentiation, cell adhesion, apoptosis, and cell migration.31,32 Implicated in approximately one-third of NSCLC of adenocarcinoma histology,33 mutations in codon 12, 13, 59, or 61 of the KRAS gene (located on chromosome 12) are associated with impaired GTPase activity, which results in constitutive activation of the protein and may lead to signaling independent of EGFR activation.34,35 KRAS mutations have been described mostly in patients with a smoking history.36 In a prospective study of 106 patients with surgically resected primary lung adenocarcinoma during the late 1990s, KRAS mutations were identified in 43% of current/former smokers (90% of which were GC to TA transversions) compared with 0% of nonsmokers, but with no significant difference on the basis of extent of cigarette exposure. However, a recent mutational analysis of 482 tissue samples from patients with lung adenocarcinoma, in which the overall incidence of KRAS mutation was 21%, showed mutations in 15% (12 of 81) of never smokers, 22% (69 of 316) of former smokers, and 25% (21 of 85) of current smokers.37 Never smokers were significantly more likely than former or current smokers to have a transition mutation (G to A) compared with a transversion mutation (G to T or C), the latter of which is thought to be related to cigarette smoke exposure.
Although a recent meta-analysis concluded that KRAS mutations have no predictive value in patients undergoing traditional chemotherapy (whether for NSCLC, colorectal cancer, or other solid tumors),31,32 some evidence supports that determining KRAS mutation status is worthwhile, specifically in the context of EGFR TKI therapy. EGFR mutations and KRAS mutations are recognized as being mutually exclusive in the NSCLC population, with documentation of the latter suggesting a non-EGFR TKI therapeutic approach (National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology. Non–Small Cell Lung Cancer. V. 2.2010; http://www.nccn.org/professionals/physician_gls/PDF/nscl.pdf, last accessed September 15, 2010). An analysis at Memorial Sloan-Kettering Cancer Center, involving 60 gefitinib- or erlotinib-treated patients with lung adenocarcinomas, reported that KRAS mutations were present in more EGFR TKI-refractory patients [9 of 38 (24%)] than EGFR TKI-responsive patients (0 of 21).38 In a retrospective analysis of the aforementioned placebo-controlled phase III TRIBUTE trial, patients with KRAS mutations who received erlotinib plus paclitaxel/carboplatin instead of chemotherapy alone had a lower ORR (8% versus 23%) and significantly shorter median time to progression (3.4 versus 6 months) and OS (median, 4.4 versus 13.5 months).17 From a mechanistic standpoint, it is unclear as to why patients with KRAS mutations had a poorer outcome with erlotinib/chemotherapy than with chemotherapy alone, with the TRIBUTE investigators encouraging cautious interpretation of these findings in light of the limited sample size and other methodologic shortcomings inherent to retrospective subgroup analyses.
EML4-ALK
As some data begin to suggest the importance of KRAS screening in the clinic, preliminary evidence is also emerging to support a similar potential for echinoderm microtubule-associated protein-like 4 (EML4) fusion with anaplastic lymphoma kinase (ALK) screening. EML4-ALK, a fusion oncogene resulting from a small inversion in chromosome 2p, leads to the expression of the N-terminal half of EML4 fused with the intracellular kinase domain of ALK, thereby resulting in potent oncogenic activity.39 Although a generally low incidence of EML4-ALK has been reported among unselected patients (1.5% to 6.7%), the incidence was 13% in a study of primarily non-Asian (described as predominantly white) patients with metastatic NSCLC, increasing to 33% in the subset of EGFR mutation-negative never or light smokers.39 Results of this retrospective analysis also suggest that treatment responses tend to mirror those expected in EGFR mutation-negative patients, with no EML4-ALK–positive patients responding to erlotinib but a 25% partial response rate to platinum-based chemotherapy. Although there were notable differences between patients with EGFR mutation-positive and EML4-ALK–positive tumors [with the latter group having a greater tendency to be male, to be of younger age, and to have samples exhibiting a distinctive histologic pattern (adenocarcinomas predominantly of the signet ring cell subtype)], patient characteristics that also overlapped in both groups include never or light smokers and tumors of adenocarcinoma histology. In contrast, in another retrospective analysis, patients with EML4-ALK–positive tumors were primarily younger, female, and with acinar type adenocarcinoma.40 Regardless, clinical assessment would prove unreliable, requiring mutation testing for the identification of patients with EGFR mutation-positive versus EML4-ALK–positive disease.
Overall, mutation status is now regarded as the driving factor in the clinical decision making that surrounds EGFR TKI use in NSCLC and should be routinely performed particularly for EGFR mutations but also extending to other mutation types. Treatment with EGFR TKIs may be beneficial for patients with EGFR mutation-positive tumors but would probably be futile in patients harboring KRAS mutations, known to be associated with intrinsic resistance to EGFR TKIs but for which alternative therapies may be worthwhile. This point is underscored by the preliminary results of the BATTLE (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination) study, a novel prospective phase II trial conducted at the M.D. Anderson Cancer Center to assess potential predictive biomarkers with targeted therapy in patients with chemotherapy-pretreated NSCLC. All patients were first enrolled in an umbrella study during which tumor samples were screened for 11 biomarkers related to four NSCLC molecular pathways (EGFR, RAS/Raf, VEGF, and cyclin D1/retinoid X receptor). On the basis of these biopsy results, patients were “adaptively” randomly assigned to one of four treatment groups: erlotinib, sorafenib (multitargeted antiangiogenic agent), vandetanib (EGFR/VEGFR/RET inhibitor), or erlotinib/bexarotene (activates retinoid X receptors).41 Overall 8-week disease control (primary endpoint) was 46% (112 of 244), and this endpoint also predicted OS, with a significantly longer median OS in patients with disease control than in patients without (11.3 versus 7.5 months). Exploratory analyses indicated that, among the subset of patients whose tumors exhibited EGFR mutations, 8-week disease control rates were 71% (5 of 7) and 23% (3 of 13) with erlotinib and sorafenib, respectively. Conversely, sorafenib appeared to be highly effective in the KRAS mutation-positive subset, with an 8-week control rate of 61% (11 of 18) versus 22% (2 of 9) for erlotinib. Note, however, that the exploratory analyses of these patient subsets are based on small numbers of patients and of limited generalizability, thus requiring further validation in larger patient populations.
Predictive Biomarkers for Antiangiogenic Therapy
As discussed earlier, there are much data to support the clinical utility of EGFR mutation status for guiding personalized treatment of NSCLC. On the contrary, there are currently no validated tools for the selection of patients who may benefit from antiangiogenic treatment for NSCLC, and therapies are used by exclusion only. Candidate biomarkers, such as VEGF, have been investigated as predictors of response to antiangiogenic therapy, but research to date has been controversial. For example, results from a prospective biomarker study of the E4599 trial showed that patients with high plasma levels of VEGF had a significantly higher probability of response with chemotherapy plus bevacizumab than with chemotherapy alone, but VEGF levels did not predict OS.42 As previously described, the BATTLE program is assessing potential biomarkers for a variety of NSCLC treatment strategies, including the potential utility of KRAS for predicting treatment success with antiangiogenic agents (ie, sorafenib).41,43 Other potential biomarkers related to antiangiogenic therapy are being studied and include the soluble intracellular adhesion molecule.42
Perspective on Mutational Analysis
EGFR and KRAS are frequently analyzed by direct sequencing, a technique that requires large amounts of quality tissue, such as that derived from surgical specimens; however, this is relatively expensive, time consuming, and not feasible for unresectable tumors. Larger tumor samples are usually preferred for molecular analysis, but obtaining larger samples may at times be unrealistic or impractical.44 Current trends favor minimally invasive diagnostic procedures, including endobronchial biopsies, needle core biopsies, or cytology specimens. Although these procedures may require smaller tumor samples, current assay methods (eg, IHC) may require these samples to have a high ratio of tumor tissue to normal tissue content.44
Establishing new methods of mutational analyses is an active area of research that aims to reduce time and expense with acceptable sensitivity and use specimens other than resected tissue. For example, EGFR mutational testing with the use of Scorpion Amplification Refractory Mutation System technology (DxS) may be used to evaluate 29 of the most prevalent somatic EGFR mutations with the use of DNA from a variety of sample types (including fresh, frozen, or paraffin-embedded tumor tissue, as well as plasma or serum) [DxS EGFR Mutation Test Kit Instructions For Use. Version RU001b. Revised July 2009. DxS Diagnostic Innovations (currently, QIAGEN, Inc., Valencia, CA)]. This technology combines Scorpion probes with allele-specific amplification in real-time PCRs. A variety of other methods have also been developed for mutational analysis in NSCLC. Response Genetics, Inc., has developed technology that uses RT-PCR for detection of EGFR mutations and other molecular readouts, such as EML4-ALK expression, in patients with NSCLC.45 Another method that has been used for mutational analysis involves the collection of circulating tumor cells with the use of a microfluidic-based device called the circulating tumor cell chip.29 Finally, IHC with the use of antibodies to detect mutant EGFR is under development, but there are some issues of sensitivity with this method.46 In addition, when reflecting on the potential future of molecular testing in lung cancer, it is important to consider the association of the excision repair cross-complementation group 1 gene and high expression of the ribonucleotide reductase subunit 1 with platinum47,48 and gemcitabine resistance, respectively.49 Clearly, the ability to more easily determine mutation status in patients with advanced NSCLC would be invaluable for overcoming the barriers related to the relative or absolute lack of tumor tissue for direct sequencing.
Conclusions
The introduction of new options for treating advanced NSCLC has given way to a new era in which the molecular features of individual tumors are of unprecedented clinical relevance. Specifying mutational status in the pathology report is of clinical importance, because accurate characterization of molecular features is critical with the expanding role of targeted therapy in advanced NSCLC populations with various degrees of pretreatment and in the first-line setting. Although much effort may be required to identify additional pathways relevant to clinical outcomes in NSCLC, this will be essential to further advance our understanding of molecular determinants of response. Improvements in current techniques are necessary to decrease the time and expense of molecular characterization. Although mutation test kits may indeed prove valuable as diagnostic tools, controlled studies will be required to find the exact parameters for interpretation of results, and tissue analysis remains the “gold standard” for lung cancer diagnosis.
Acknowledgment
I thank Alyssa Tippens, PhD, (MedErgy, Yardley, PA) for writing and editorial assistance.
Footnotes
Supported by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI) for editorial assistance.
CME Disclosure: C.M. is supported by BIPI for editorial assistance, which is developing several compounds (including EGFR-based treatments) for non–small cell lung cancer; associated clinical trials are publicly disclosed at http://clinicaltrials.gov. C.M. asserts that he maintained full control and ownership of the scientific content of the article and that BIPI did not have a role in the content of the review article.
References
- 1.American Cancer Society . American Cancer Society, Inc.; Atlanta, GA: 2009. Cancer Facts & Figures, 2009. [Google Scholar]
- 2.Jemal A., Siegel R., Ward E., Hao Y., Xu J., Thun M.J. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd F.A., Rodrigues P.J., Ciuleanu T., Tan E.H., Hirsh V., Thongprasert S., Campos D., Maoleekoonpiroj S., Smylie M., Martins R., van K.M., Dediu M., Findlay B., Tu D., Johnston D., Bezjak A., Clark G., Santabarbara P., Seymour L. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 4.Cappuzzo F., Ciuleanu T., Stelmakh L., Cicenas S., Szczesna A., Juhasz E., Esteban E., Molinier O., Brugger W., Melezinek I., Klingelschmitt G., Klughammer B., Giaccone G. Erlotinib as maintenance treatment in advanced non-small-cell lung cancer: a multicentre, randomised, placebo-controlled phase 3 study. Lancet Oncol. 2010;11:521–529. doi: 10.1016/S1470-2045(10)70112-1. [DOI] [PubMed] [Google Scholar]
- 5.Fukuoka M., Yano S., Giaccone G., Tamura T., Nakagawa K., Douillard J.Y., Nishiwaki Y., Vansteenkiste J., Kudoh S., Rischin D., Eek R., Horai T., Noda K., Takata I., Smit E., Averbuch S., Macleod A., Feyereislova A., Dong R.P., Baselga J. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] J Clin Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 6.Kris M.G., Natale R.B., Herbst R.S., Lynch T.J., Jr, Prager D., Belani C.P., Schiller J.H., Kelly K., Spiridonidis H., Sandler A., Albain K.S., Cella D., Wolf M.K., Averbuch S.D., Ochs J.J., Kay A.C. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial. JAMA. 2003;290:2149–2158. doi: 10.1001/jama.290.16.2149. [DOI] [PubMed] [Google Scholar]
- 7.Gatzemeier U., Pluzanska A., Szczesna A., Kaukel E., Roubec J., De Rosa F., Milanowski J., Karnicka-Mlodkowski H., Pesek M., Serwatowski P., Ramlau R., Janaskova T., Vansteenkiste J., Strausz J., Manikhas G.M., Von Pawel J. Phase III study of erlotinib in combination with cisplatin and gemcitabine in advanced non-small-cell lung cancer: the Tarceva Lung Cancer Investigation Trial. J Clin Oncol. 2007;25:1545–1552. doi: 10.1200/JCO.2005.05.1474. [DOI] [PubMed] [Google Scholar]
- 8.Giaccone G., Herbst R.S., Manegold C., Scagliotti G., Rosell R., Miller V., Natale R.B., Schiller J.H., von P.J., Pluzanska A., Gatzemeier U., Grous J., Ochs J.S., Averbuch S.D., Wolf M.K., Rennie P., Fandi A., Johnson D.H. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 1. J Clin Oncol. 2004;22:777–784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 9.Herbst R.S., Giaccone G., Schiller J.H., Natale R.B., Miller V., Manegold C., Scagliotti G., Rosell R., Oliff I., Reeves J.A., Wolf M.K., Krebs A.D., Averbuch S.D., Ochs J.S., Grous J., Fandi A., Johnson D.H. Gefitinib in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: a phase III trial–INTACT 2. J Clin Oncol. 2004;22:785–794. doi: 10.1200/JCO.2004.07.215. [DOI] [PubMed] [Google Scholar]
- 10.Herbst R.S., Prager D., Hermann R., Fehrenbacher L., Johnson B.E., Sandler A., Kris M.G., Tran H.T., Klein P., Li X., Ramies D., Johnson D.H., Miller V.A. TRIBUTE: a phase III trial of erlotinib hydrochloride (OSI-774) combined with carboplatin and paclitaxel chemotherapy in advanced non-small-cell lung cancer. J Clin Oncol. 2005;23:5892–5899. doi: 10.1200/JCO.2005.02.840. [DOI] [PubMed] [Google Scholar]
- 11.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., Louis D.N., Christiani D.C., Settleman J., Haber D.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 12.Paez J.G., Janne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J., Naoki K., Sasaki H., Fujii Y., Eck M.J., Sellers W.R., Johnson B.E., Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 13.Pao W., Miller V., Zakowski M., Doherty J., Politi K., Sarkaria I., Singh B., Heelan R., Rusch V., Fulton L., Mardis E., Kupfer D., Wilson R., Kris M., Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirker R., Pereira J.R., Szczesna A., von Pawel J., Krzakowski M., Ramlau R., Vynnychenko I., Park K., Yu C.T., Ganul V., Roh J.K., Bajetta E., O'Byrne K., de Marinis F., Eberhardt W., Goddemeier T., Emig M., Gatzemeier U. Cetuximab plus chemotherapy in patients with advanced non-small-cell lung cancer (FLEX): an open-label randomised phase III trial. Lancet. 2009;373:1525–1531. doi: 10.1016/S0140-6736(09)60569-9. [DOI] [PubMed] [Google Scholar]
- 15.Sharma S.V., Bell D.W., Settleman J., Haber D.A. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–181. doi: 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- 16.Shigematsu H., Gazdar A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int J Cancer. 2006;118:257–262. doi: 10.1002/ijc.21496. [DOI] [PubMed] [Google Scholar]
- 17.Eberhard D.A., Johnson B.E., Amler L.C., Goddard A.D., Heldens S.L., Herbst R.S., Ince W.L., Janne P.A., Januario T., Johnson D.H., Klein P., Miller V.A., Ostland M.A., Ramies D.A., Sebisanovic D., Stinson J.A., Zhang Y.R., Seshagiri S., Hillan K.J. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–5909. doi: 10.1200/JCO.2005.02.857. [DOI] [PubMed] [Google Scholar]
- 18.Morita S., Okamoto I., Kobayashi K., Yamazaki K., Asahina H., Inoue A., Hagiwara K., Sunaga N., Yanagitani N., Hida T., Yoshida K., Hirashima T., Yasumoto K., Sugio K., Mitsudomi T., Fukuoka M., Nukiwa T. Combined survival analysis of prospective clinical trials of gefitinib for non-small cell lung cancer with EGFR mutations. Clin Cancer Res. 2009;15:4493–4498. doi: 10.1158/1078-0432.CCR-09-0391. [DOI] [PubMed] [Google Scholar]
- 19.Sequist L.V., Martins R.G., Spigel D., Grunberg S.M., Spira A., Janne P.A., Joshi V.A., McCollum D., Evans T.L., Muzikansky A., Kuhlmann G.L., Han M., Goldberg J.S., Settleman J., Iafrate A.J., Engelman J.A., Haber D.A., Johnson B.E., Lynch T.J. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol. 2008;26:2442–2449. doi: 10.1200/JCO.2007.14.8494. [DOI] [PubMed] [Google Scholar]
- 20.Douillard J.Y., Shepherd F.A., Hirsh V., Mok T., Socinski M.A., Gervais R., Liao M.L., Bischoff H., Reck M., Sellers M.V., Watkins C.L., Speake G., Armour A.A., Kim E.S. Molecular predictors of outcome with gefitinib and docetaxel in previously treated non-small-cell lung cancer: data from the randomized phase III INTEREST trial. J Clin Oncol. 2009;28:744–752. doi: 10.1200/JCO.2009.24.3030. [DOI] [PubMed] [Google Scholar]
- 21.Dahabreh I.J., Linardou H., Siannis F., Kosmidis P., Bafaloukos D., Murray S. Somatic EGFR mutation and gene copy gain as predictive biomarkers for response to tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2010;16:291–303. doi: 10.1158/1078-0432.CCR-09-1660. [DOI] [PubMed] [Google Scholar]
- 22.Mok T.S., Wu Y.L., Thongprasert S., Yang C.H., Chu D.T., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., Nishiwaki Y., Ohe Y., Yang J.J., Chewaskulyong B., Jiang H., Duffield E.L., Watkins C.L., Armour A.A., Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 23.Yang C.-H., Fukuoka M., Mok T.S., Wu Y.-L., Thongprasert S., Saijo N., Chu D.-T., Jiang H., Duffield E.L., Ichinose Y. Final overall survival (OS) results from a phase III, randomised, open-label, first-line study of gefitinib (G) V carboplatin/paclitaxel (C/P) in clinically selected patients with advanced non-small cell lung cancer (NSCLC) in Asia (iPASS) Ann Oncol. 2010;21:viii1–viii2. Abstract LBA2. [Google Scholar]
- 24.Maemondo M., Inoue A., Kobayashi K., Sugawara S., Oizumi S., Isobe H., Gemma A., Harada M., Yoshizawa H., Kinoshita I., Fujita Y., Okinaga S., Hirano H., Yoshimori K., Harada T., Ogura T., Ando M., Miyazawa H., Tanaka T., Saijo Y., Hagiwara K., Morita S., Nukiwa T. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 25.Mitsudomi T., Morita S., Yatabe Y., Negoro S., Okamoto I., Tsurutani J., Seto T., Satouchi M., Tada H., Hirashima T., Asami K., Katakami N., Takada M., Yoshioka H., Shibata K., Kudoh S., Shimizu E., Saito H., Toyooka S., Nakagawa K., Fukuoka M. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 26.Suda K., Onozato R., Yatabe Y., Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009;4:1–4. doi: 10.1097/JTO.0b013e3181913c9f. [DOI] [PubMed] [Google Scholar]
- 27.Balak M.N., Gong Y., Riely G.J., Somwar R., Li A.R., Zakowski M.F., Chiang A., Yang G., Ouerfelli O., Kris M.G., Ladanyi M., Miller V.A., Pao W. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–6501. doi: 10.1158/1078-0432.CCR-06-1570. [DOI] [PubMed] [Google Scholar]
- 28.Bean J., Riely G.J., Balak M., Marks J.L., Ladanyi M., Miller V.A., Pao W. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–7525. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maheswaran S., Sequist L.V., Nagrath S., Ulkus L., Brannigan B., Collura C.V., Inserra E., Diederichs S., Iafrate A.J., Bell D.W., Digumarthy S., Muzikansky A., Irimia D., Settleman J., Tompkins R.G., Lynch T.J., Toner M., Haber D.A. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–377. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inukai M., Toyooka S., Ito S., Asano H., Ichihara S., Soh J., Suehisa H., Ouchida M., Aoe K., Aoe M., Kiura K., Shimizu N., Date H. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 31.Fassina A., Gazziero A., Zardo D., Corradin M., Aldighieri E., Rossi G.P. Detection of EGFR and KRAS mutations on trans-thoracic needle aspiration of lung nodules by high resolution melting analysis. J Clin Pathol. 2009;62:1096–1102. doi: 10.1136/jcp.2009.067587. [DOI] [PubMed] [Google Scholar]
- 32.Loriot Y., Mordant P., Deutsch E., Olaussen K.A., Soria J.C. Are RAS mutations predictive markers of resistance to standard chemotherapy? Nat Rev Clin Oncol. 2009;6:528–534. doi: 10.1038/nrclinonc.2009.106. [DOI] [PubMed] [Google Scholar]
- 33.Bos J.L. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 34.Adjei A.A. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 35.Cepero V., Sierra J.R., Corso S., Ghiso E., Casorzo L., Perera T., Comoglio P.M., Giordano S. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70:7580–7590. doi: 10.1158/0008-5472.CAN-10-0436. [DOI] [PubMed] [Google Scholar]
- 36.Ahrendt S.A., Decker P.A., Alawi E.A., Zhu Yr Y.R., Sanchez-Cespedes M., Yang S.C., Haasler G.B., Kajdacsy-Balla A., Demeure M.J., Sidransky D. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 37.Riely G.J., Kris M.G., Rosenbaum D., Marks J., Li A., Chitale D.A., Nafa K., Riedel E.R., Hsu M., Pao W., Miller V.A., Ladanyi M. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14:5731–5734. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pao W., Wang T.Y., Riely G.J., Miller V.A., Pan Q., Ladanyi M., Zakowski M.F., Heelan R.T., Kris M.G., Varmus H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17. doi: 10.1371/journal.pmed.0020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaw A.T., Yeap B.Y., Mino-Kenudson M., Digumarthy S.R., Costa D.B., Heist R.S., Solomon B., Stubbs H., Admane S., McDermott U., Settleman J., Kobayashi S., Mark E.J., Rodig S.J., Chirieac L.R., Kwak E.L., Lynch T.J., Iafrate A.J. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitsudomi T., Suda K., Tomizawa K., Yatabe Y. Clinico-pathologic features of lung cancer with EML4-ALK translocation (abstract) J Clin Oncol. 2010;28 Abstract 10598. [Google Scholar]
- 41.Kim ES, Herbst RS, Lee JJ, Blumenschein GR Jr, Tsao A, Alden CM, Tang X, Liu S, Stewart DJ, Heymach JV, Tran HT, Hicks ME, Erasmus J Jr, Gupta S, Powis G, Lippman SM, Wistuba II, Hong WK: The BATTLE trial (Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination): personalizing therapy for lung cancer. Presented at the 101st Annual Meeting of the American Association for Cancer Research, 2010 April 17–21, Washington, DC
- 42.Dowlati A., Gray R., Sandler A.B., Schiller J.H., Johnson D.H. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab-an Eastern Cooperative Oncology Group Study. Clin Cancer Res. 2008;14:1407–1412. doi: 10.1158/1078-0432.CCR-07-1154. [DOI] [PubMed] [Google Scholar]
- 43.Herbst R.S., Blumenschein G.R., Jr, Kim E.S., Lee J., Tsao A.S., Alden C.M., Liu S., Stewart D.J., Wistuba I.I., Hong W.K. Sorafenib treatment efficacy and KRAS biomarker status in the Biomarker-Integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial (abstract) J Clin Oncol. 2010;28:7609. [Google Scholar]
- 44.Eberhard D.A., Giaccone G., Johnson B.E. Biomarkers of response to epidermal growth factor receptor inhibitors in Non-Small-Cell Lung Cancer Working Group: standardization for use in the clinical trial setting. J Clin Oncol. 2008;26:983–994. doi: 10.1200/JCO.2007.12.9858. [DOI] [PubMed] [Google Scholar]
- 45.Danenberg P.V., Stephens J., Cooc J., Gandara D.R., Mack P.C., Grimminger P.P., Danenberg K.D. A novel RT-PCR approach to detecting EML4-ALK fusion genes in archival NSCLC tissue (abstract) J Clin Oncol. 2010;28 Abstract 10535. [Google Scholar]
- 46.Kitamura A., Hosoda W., Sasaki E., Mitsudomi T., Yatabe Y. Immunohistochemical detection of EGFR mutation using mutation-specific antibodies in lung cancer. Clin Cancer Res. 2010;16:3349–3355. doi: 10.1158/1078-0432.CCR-10-0129. [DOI] [PubMed] [Google Scholar]
- 47.Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C., Declercq C., Pawling J., Moons L., Collen D., Risau W., Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 48.Rosell R., Lord R.V., Taron M., Reguart N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer. 2002;38:217–227. doi: 10.1016/s0169-5002(02)00224-6. [DOI] [PubMed] [Google Scholar]
- 49.Davidson J.D., Ma L., Flagella M., Geeganage S., Gelbert L.M., Slapak C.A. An increase in the expression of ribonucleotide reductase large subunit 1 is associated with gemcitabine resistance in non-small cell lung cancer cell lines. Cancer Res. 2004;64:3761–3766. doi: 10.1158/0008-5472.CAN-03-3363. [DOI] [PubMed] [Google Scholar]