

Abstract
In diabetic retinopathy (DR), abnormalities in vascular and neuronal function are closely related to the local production of inflammatory mediators whose potential source is microglia. A2A adenosine receptor (A2AAR) has been shown to possess anti-inflammatory properties that have not been studied in DR. Here, we evaluate the role of A2AAR and its underlying signaling in retinal complications associated with diabetes. Initial studies in wild-type mice revealed that the treatment with the A2AAR agonist resulted in marked decreases in hyperglycemia-induced retinal cell death and tumor necrosis factor (TNF)-α release. To further assess the role of A2AAR in DR, we studied the effects of A2AAR ablation on diabetes-induced retinal abnormalities. Diabetic A2AAR−/− mice had significantly more terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling-positive cells, TNF-α release, and intercellular adhesion molecule-1 expression compared with diabetic wild-type mice. To explore a potential mechanism by which A2AAR signaling regulates inflammation in DR, we performed additional studies using microglial cells treated with Amadori-glycated albumin, a risk factor in diabetic disorders. The results showed that activation of A2AAR attenuated Amadori-glycated albumin-induced TNF-α release in a cAMP/exchange protein directly activated by cAMP-dependent mechanism and significantly repressed the inflammatory cascade, C-Raf/extracellular signal-regulated kinase (ERK), in activated microglia. Collectively, this work provides pharmacological and genetic evidence for A2AAR signaling as a control point of cell death in DR and suggests that the retinal protective effect of A2AAR is mediated by abrogating the inflammatory response that occurs in microglia via interaction with C-Raf/ERK pathway.
Diabetic retinopathy (DR) is the leading cause of blindness in the Western world and the most common complication of diabetes.1,2 DR has been categorized as a vascular-neuroinflammatory disease. Early features of DR include signs of retinal inflammatory reactions, breakdown of the blood-retinal barrier function and loss of retinal neurons.3–5 Under these conditions, normally quiescent microglial cells become activated.6,7 Activated microglia release glutamate, reactive oxygen species, and proinflammatory mediators, such as tumor necrosis factor (TNF)-α.8–10 The retinal expression of TNF-α has been reported to be associated with neuronal and endothelial cell death, hallmark features of the disease,5,11 and inhibition of TNF-α has demonstrated beneficial effects in the prevention of early DR.12 Thus, research on retinal microglia activation may provide insights into the pathogenesis of DR.13
Adenosine has been proposed to modulate a variety of physiological responses.14 Under stress conditions, the local levels of extracellular adenosine are increased due to the increased need for energy supplied by ATP15 and the increased degradation of released ATP.16 The increased adenosine at inflamed sites can protect against cellular damage by activating A2A adenosine receptor (A2AAR), a Gs-coupled receptor.17 Treatment with an A2AAR agonist blocked the inflammation and functional changes associated with diabetic nephropathy in wild-type diabetic mice, but not in the diabetic A2AAR−/− mice.18
Recent efforts to understand how neurotoxic inflammatory cytokines are produced have shown that Ras/Raf/MEK/extracellular signal-regulated kinase (ERK) cascade is one of the attractive targets for intervention in human inflammatory-associated diseases, such as diabetes. However, this cascade does not operate alone, but rather interacts with other signaling systems, such as Gs-coupled receptor transducing pathway. Activation of this pathway results in accumulation of cAMP that interacts with the Ras/Raf/MEK/ERK kinase signaling to regulate cell functions.19
Previously we demonstrated the anti-inflammatory effect of A2AAR in acute retinal inflammation.20 Currently, we seek to determine the contribution of A2AAR in retinal protection against diabetes-induced retinal inflammation and injury. Moreover, we pursue to gain insight into the underlying signaling involved therein. Here, we report evidence that activation of A2AAR plays a protective role in diabetes-induced retinal cell death by enhancing the anti-inflammatory signaling, including the interaction with C-Raf/ERK cascade. These findings suggest that A2AAR agonists may be promising innovative agents in the treatment of diabetic retinopathy.
Materials and Methods
Animal Preparation and Experimental Design
All procedures with animals were performed in accordance with the Public Health Service Guide for the Care and Use of Laboratory Animals (Department of Health, Education, and Welfare, National Institutes of Health Publication No. 80-23) and the Medical College of Georgia guidelines. The 8-week-old male A2AAR−/− and corresponding littermate controls, wild-type (WT) mice in C57BL/6 background, were matched according to sex, age, and weight. Animals were given i.p. injections of vehicle or freshly prepared streptozotocin in 0.01 mol/L sodium citrate buffer, pH 4.5 (45 mg/kg) after a 4-hour fast each day for 5 consecutive days. Diabetes was confirmed by fasting blood glucose levels >250 mg/dl. In pharmacological studies, age-, weight- and sex-matched C57BL/6 mice were rendered diabetic and then injected i.p. with vehicle dimethyl sulfoxide, or CGS21680 (25 μg/kg) every other day for the duration of the study (n = 4/group to 6/group). The dose of CGS21680 was chosen according to what was previously reported.21 Four weeks after the establishment of diabetes, pooled retina and vitreous samples were used for TNF-α analysis by enzyme-linked immunosorbent assay (ELISA) and frozen eye sections were prepared to examine retinal cell death by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) or microglial activation and intercellular adhesion molecule 1 (ICAM-1) expression by immunofluorescence.
ELISA for TNF-α
TNF-α levels in pooled retina and vitreous samples or in the supernatants of culture media were estimated with ELISA kits (R & D, Minneapolis, MN) per the manufacturer's instructions. Standards and samples were added and bound by the immobilized antibody. After washing, an enzyme-linked polyclonal antibody specific for the cytokine was added to the wells followed by a substrate solution yielding a colored product. The intensity of the color was measured at 450 nm. The sample levels were calculated from the standard curve and corrected for protein concentration.
Retinal and vitreous lysates from mouse eyes were prepared for TNF-α ELISA according to our previous method with minor modifications.20 Briefly, pooled retina and vitreous from each eye were placed in 100 μL of lysis buffer [20 mmol/L imidazole HCl, 100 mmol/L KCl, 1 mmol/L MgCl2, 1% Triton X-100, 10 mmol/L NaF, 1 mmol/L Na3VO4, 1 mmol/L EGTA, and 1 mmol/L EDTA (pH 6.8)] supplemented with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) followed by homogenization with Ottawa sand using a Mini-Bead Beater (BiosSpec Products, Inc., Bartlesville, OK). The lysate was cleared of debris by centrifugation at 10,000 × g for 15 minutes (4°C), and 50 μL of the supernatant was used for ELISA directly without further dilution. The sample TNF-α levels were calculated from the standard curve and corrected for protein concentration.
Immunofluorescence
Immunofluorescence analysis was performed using frozen retinal sections. Briefly, cryostat sections (15 μm) were fixed in 4% paraformaldehyde, blocked with 10% normal goat serum and then incubated overnight at 4°C with primary antibodies: rabbit anti-Iba-1 (Wako Pure Chemical, Wako, TX), or mouse anti-ICAM-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA) together with Texas Red-labeled Isolectin B4 (Invitrogen, Carlsbad, CA). Thereafter, sections were briefly washed with PBS and incubated with appropriate secondary antibodies. Slides were examined by confocal microscopy (LSM 510, Carl Zeiss Inc.). Specificity of the reaction was confirmed by omitting the primary antibody. Data (10 fields/retina; n = 4 to 6 in each group) were analyzed using fluorescence microscopy and Ultra-View morphometric software to quantify the intensity of immunostaining.
TUNEL
TUNEL was performed in frozen sections using the TACS-2 TdT Fluorescein in situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD) counterstained with propidium iodide, according to the manufacturer's suggestions. Briefly, sections were hydrated with alcohol 100%, 95%, and 70%, then fixed in 3.7% paraformaldehyde. After washing, the slides were incubated in a mixture of TdT, Mn + 2, and TdT dNTP for 1 hour at 370C. The reaction was stopped with TdT Stop Buffer (Trevigen) for 5 minutes. After washing with deionized water, the slides were incubated with streptavidin-Fluorescein isothiocyanate (diluted 1:200) solution for 20 minutes at room temperature. Slides were counterstained, mounted, covered with coverslips, and visualized by confocal microscopy (LSM 510, Carl Zeiss, Inc). Apoptotic cells were identified as doubly labeled with TdT fluorescein and propidium iodide, and only the nuclei clearly labeled yellow were scored. After TUNEL assay, immunofluorescence was performed with the previously described method using the antibody against neuron-specific nuclear protein (NeuN; Chemicon, Temecula, CA).
Primary Retinal Microglia Culture
Microglial cells were isolated from retinas of newborn Sprague Dawley rats according to a previous procedure22 with minor modifications. Briefly, retinas were dissected from newborn Sprague Dawley rat pups. Tissues were collected into 0.01 M PBS and washed with ice-cold 0.01 M PBS, digested with 0.125% trypsin for 3 to 5 minutes and mixed with Dulbecco's modified Eagle's medium or Dulbecco's minimal essential medium/F12 (1:1) (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Atlanta Biologicals, Atlanta, GA) and 1% penicillin/streptomycin (Mediatech, Herndon, VA). Retina pieces were triturated by passing them through a disposable pipette several times until cells were dispersed. Cells were then filtered through a mesh (100 μm), collected by centrifugation, re-suspended in the culture medium, and plated onto T150 cell culture flasks (Corning, Corning, NY) at a density of 2 × 105 cells/cm2. All cultures were maintained in a humidified CO2 incubator at 37°C and 5% CO2, and were fed on the third day, and then once every 4 days. After 2 weeks, microglial cells were harvested by shaking the flasks at 100 rpm for 1 hour. The cell suspension was centrifuged and detached cells were re-plated in Dulbecco's modified Eagle's medium or Dulbecco's minimal essential medium/F12 (1:1) + 10% fetal bovine serum overnight, and then in serum-free/low protein media (Cellgro Complete containing 0.1% bovine serum albumin; Mediatech, Manassas, VA) at designated densities for various experiments. The purity of microglial cultures was 98%, as determined by immunocytochemical staining analysis for Iba1, a microglial marker. The morphology of microglia in culture was carefully examined by phase-contrast and fluorescent microscopy (data not shown).
Drug Treatment Effects on Cultured Microglial Cells
Microglial cells were seeded at a density of 5 × 105 cells/well in a 24-well tissue culture plate, or 1 × 105 cells/well in a 96-well plate. One day after seeding, the cultured wells were washed with Cellgro Complete (Mediatech, Manassas, VA) and incubated in the same media with various treatments. Cells were pretreated with adenosine receptor agonists (all from Sigma, St. Louis, MO), adenosine receptor antagonists [all from Sigma, except: 4-{2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-ylamino]ethyl}fphenol (ZM 241385), which was from Tocris, Ellisville, MO], 8-pCPT-cAMP, PKI 14–22 amide, or Rp-Adenosine 3′,5′-cyclic monophosphorothioate (all from Sigma) at the indicated concentrations or vehicle dimethyl sulfoxide for 30 minutes at 37°C before activation of microglia. For microglia activation, Amadori-glycated albumin (AGA; Sigma) with undetectable endotoxin (<0.125 units/mL, 10 EU = 1 ng lipopolysaccharide; Lonza, Basel, Switzerland) was added to each well at a final concentration of 500 μg/mL. At indicated time points, cells were homogenized for Western blot analysis, and culture media were taken and analyzed for TNF-α by ELISA.
cAMP Determination
Microglial cells in 96-well plates were pretreated with the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (500 μmol/L) for 10 minutes at 37°C followed by CGS21680 for 30 minutes. Cells were then treated with AGA for 4 hours. At the end of the incubation period, the cells were lysed (using 0.1 N HCl) and then scraped. cAMP content of the cell lysates was determined using a cAMP assay kit (Assay Design, Ann Arbor, MI) according to the manufacturer's instructions.
PKA Activity Assay
The protein kinase A (PKA) activity assay is based on a solid-phase ELISA that uses a pre-coated synthetic peptide as a substrate for PKA, and a polyclonal antibody that recognizes the phosphorylated form of the substrate. PKA activity was estimated with a nonradioactive PKA activity assay kit (Assay Design) according to the manufacturer's instructions. Briefly, PKA control or lysates of treated microglial cells were added, followed by the addition of ATP to initiate the reaction. The kinase reaction was terminated and a phospho-specific substrate antibody was added to the wells that bind specifically to the phosphorylated peptide substrate. The phospho-specific antibody was subsequently bound by a peroxidase-conjugated secondary antibody. The assay was developed with tetramethylbenzidine substrate (TMB) and a color developed in proportion to PKA phosphotransferase activity. The color development was stopped with acid stop solution and the intensity of the color was measured in a microplate reader at 450 nm.
Protein Extraction and Western Blot Analysis
Washed cultured cells were lysed in modified radioimmunoprecipitation assay buffer (Upstate, Lake Placid, NY) containing 50 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Nonidet P-40, 0.25% deoxycholate, supplemented with 40 mmol/L NaF, 2 mmol/L Na3VO4, 0.5 mmol/L phenylmethylsulfonyl fluoride, and 1:100 (v/v) of proteinase inhibitor cocktail (Sigma). Insoluble material was removed by centrifugation at 12,000 × g at 4°C for 30 minutes. Protein was determined by DC Protein Assay (Bio-Rad, Hercules, CA) and 50 μg was boiled in Laemmli sample buffer, separated by SDS-PAGE on a gradient gel (4 to 20%) (Pierce, Rockford, IL), transferred to nitrocellulose membrane and incubated with specific antibodies. Antibodies for β-actin (Sigma), phospho-C-Raf (Ser338), phospho-B-Raf, C-Raf, B-Raf, phospho-ERK, and ERK mitogen-activated protein kinase (Cell Signaling Technology, Beverly, MA) were detected with a horseradish peroxidase-conjugated antibody and enhanced chemiluminescence (ECL) (Amersham BioSciences, Buckinghamshire, UK). Intensity of immunoreactivity was measured by densitometry.
Data Analysis
The results are expressed as mean ± SD differences among experimental groups that were evaluated by analysis of variance, and the significance of differences between groups was assessed by the post hoc test (Fisher's Protected Least Significant Difference) when indicated. Significance was defined as P < 0.05.
Results
Role of A2AAR in Diabetes-Induced Retinal Cell Death and Inflammation
Significant death of retinal neural cells has been reported in experimental diabetes as early as 4 weeks.23 Because activation of the A2AAR has been implicated in the anti-inflammatory actions of adenosine in other tissues, we hypothesized that this receptor may also be effective in protecting retinal neurons from diabetes-induced inflammation and neurotoxicity. To test this hypothesis, we treated diabetic and nondiabetic mice with or without the A2AAR-selective agonist, CGS21680, and determined the effects on diabetes-induced retinal cell death and TNF-α production. Quantitative analysis of TUNEL-Fluorescein isothiocyanate and propidium iodide double-labeled cells in retinal tissue showed a significant increase in the frequency of retinal cell death mostly in the ganglion cell layer (in particular, neuronal cells) after 4 weeks of diabetes (Figure 1, A–C). The CGS21680 treatment significantly reduced the number of TUNEL-positive cells in the diabetic mice.
Figure 1.

Effect of A2AAR activation on diabetes-induced retinal cell death and inflammation. A2A adenosine receptor (A2AAR) activation inhibits retinal cell death and tumor necrosis factor (TNF)-α release in diabetes. Retinal cell death, TNF-α release, and microglial cells were determined or observed in 8-week-old C57BL mice after 4 weeks of streptozotocin-induced diabetes. Mice were treated with or without A2AAR agonist CGS21680 (25 μg/kg) every other day for 4 weeks. (A) Retinal distribution of terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)+ cells in normal, vehicle-treated diabetic, and CGS21680-treated diabetic mice. Sections were counterstained with propidium iodide (PI). For clearer TUNEL identification, the retinal area of the vehicle-treated diabetic animal was displayed with separated and merged TUNEL and PI staining. (B) Retinal quantitation of TUNEL+ cells. TUNEL+ cells were counted in 10 adjacent locations along the vertical meridian within 4 mm of the optic disk (10 fields/retina section). (C) Representative photomicrographs of apoptotic retinal neuronal cells that were immune-positive for TUNEL (green) and NeuN (red) in the vehicle-treated diabetic retinas. (D) Enzyme-linked immunosorbent assay measurement of TNF-α release in the pooled retina and vitreous samples. Data shown are the mean + SD (n = 4 to 6). (E) Morphological configuration of retinal microglial cells as determined by IHC for Iba1 and examined by confocal microscopy at ×20 and ×63 magnification. DMSO, dimethyl sulfoxide; GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
Next, we determined the treatment effect of CGS21680 on the levels of proinflammatory cytokine in the pooled retina and vitreous samples. Compared with age-matched nondiabetic animals, diabetic animals demonstrated a significant increase in TNF-α level (Figure 1D; P < 0.01) and the treatment with CGS21680 resulted in a marked reduction of diabetes-associated TNF-α release (Figure 1D; P < 0.02). Following this further, we have addressed an interesting feature (ie, acquisition of reactive microglial phenotype) that could be an important determinant for understanding the mechanisms by which CGS21680 affects TNF-α release. In this regard, we noted that when microglia encountered diabetic milieu, they transformed from their ramified resting state into an amoeboid shape, the activated and cytokines-releasing state (Figure 1E). Although no gross difference in cell number was observed between diabetic and nondiabetic conditions, treatment of diabetic mice with CGS21680 attenuated the morphological transformation of ramified microglia into an activated amoeboid microglia (Figure 1E).
To further assess the role of A2AAR in DR, we studied the effects of A2AAR ablation on diabetes-induced retinal cell death and inflammation. Initially, immunofluorescence revealed that A2AAR is localized in the ganglion cell layer and inner and outer plexiform layers (Figure 2A). Second, as shown in Figure 2, B and C, TUNEL+ cells in the retinas of diabetic A2AAR−/− mice were greatly increased (∼1.5-fold) compared with their age-matched diabetic WT littermates. Moreover, comparative analysis of TNF-α levels in the pooled retina and vitreous samples revealed that TNF-α release in the diabetic A2AAR−/− mice was increased almost twofold compared with the diabetic WT littermates (Figure 2D; P < 0.05). To further characterize the role of A2AAR in the retinal inflammation in diabetes, we determined the expression of ICAM-1 in the retinas of A2AAR−/− and WT littermates with and without diabetes. As shown in Figure 2, E and F, ICAM-1 expression in diabetic A2AAR−/− mice was increased significantly more than their age-matched diabetic WT mice. Finally, to determine whether the knockout of A2AAR would alter microglia phenotype in unison with TUNEL and cytokine expression profiles during diabetes, we examined the morphological features of microglia in WT versus knockout animals. As shown in Figure 2G, an amoeboid phenotypic configuration indicating active form of microglia was observed in the diabetic WT mouse retina at 4 weeks, and this phenotypic became more obvious in A2AAR−/− diabetic mice. Taken together, these results suggest that A2AAR plays a crucial role in limiting retinal inflammation and neuronal cell injury associated with diabetes.
Figure 2.
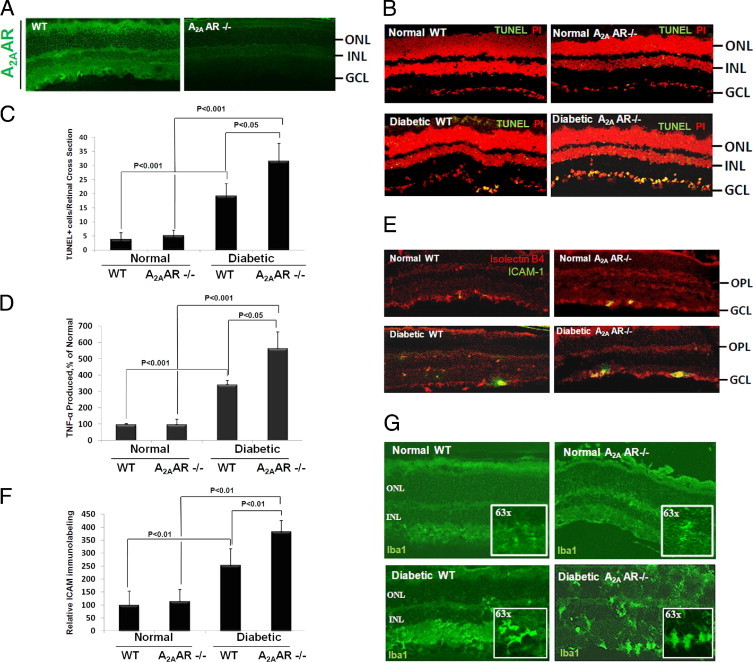
Effect of A2A adenosine receptor (A2AAR) ablation on diabetes-induced retinal cell death and inflammation. A2AAR ablation leads to a significant retinal cell death and vascular inflammation in diabetes. Eight-week-old A2AAR−/− mice in C57BL background and age-matched wild-type (WT) littermates were induced to diabetes with streptozotocin, and the retina was analyzed after 4 weeks of diabetes. (A) Retinal distribution of A2AAR. Retinal sections from A2AAR−/− and WT littermates were immunostained for A2AAR. (B) Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL)+ cell distribution in the retinal sections from A2AAR−/− and WT littermates with and without diabetes. (C) Retinal quantitation of TUNEL+ cells. TUNEL+ cells were counted in 10 adjacent locations along the vertical meridian within 4 mm of the optic disk (10 fields/retina section). (D) Enzyme-linked immunosorbent assay measurement of tumor necrosis factor (TNF)-α release in the pooled retina and vitreous samples from A2AAR−/− and WT littermates with and without diabetes. Data shown are the mean + SD (n = 4 to 6). (E) Intercellular adhesion molecule 1 (ICAM-1) distribution in the retinal sections from A2AAR−/− and WT littermates with and without diabetes. Sections were stained for ICAM-1 and Iso-lectin B4. (F) ICAM-1/Iso-lectin B4 immunofluoresence were quantified in 10 adjacent locations along the vertical meridian within 4 mm of the optic disk (10 fields/retina section). (G) Configuration of Iba1-stained retinal microglia in sections from A2AAR−/− and WT littermates with and without diabetes examined by confocal microscopy at ×20 and ×63 magnification. DMSO, dimethyl sulfoxide; GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; OPL, outer plexiform layer.
Activation of A2AAR Attenuates AGA-Induced TNF-α Release in Retinal Microglial Cells
After having shown that diabetic A2AAR−/− mice exhibit profound retinal cell death and inflammation. Next we sought to explore a potential mechanism by which A2AAR signaling regulates inflammation in DR with additional studies using microglial cells treated with AGA were performed. Treatment of microglia with AGA has been shown to simulate inflammation.22,24,25 As shown in Figure 3A, treatment of retinal microglial cells with AGA, but not with nonglycated albumin, triggered a prominent increase in TNF-α release, which was potently inhibited (30% to 40%) by pre-incubation with 1 μmol/L nonselective AR agonist (adenosine-5′-N-ethylcarboxamide). Wdhen the cells were pretreated with the A1AR antagonist (8-cyclopentyl-1,3-dipropylxanthine; 0.1 μmol/L), the A2BAR antagonist [8-[4-[((4-cyanophenyl) arbamoylmethyl) oxy]phenyl]-1,3-di(n-propyl) xanthine hydrate (MRS 1754); 1 μmol/L], or the A3AR antagonist [3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate; 2-chloro-N6-cyclopentyladenosine, 2-chloro-N6-cyclopentyladenosine (MRS 1523); 10 μmol/L], the inhibitory effect of adenosine-5′-N-ethylcarboxamide on TNF-α release was not affected. However, this effect was successfully blocked by the A2AAR antagonist (ZM241385; 0.1 μmol/L). To confirm the role of the A2AAR in regulating TNF-α release, we examined the effect of the selective A2AAR agonist, CGS21680, in AGA-induced TNF-α release. As shown in Figure 3B, activation of A2AAR with CGS21680 inhibited TNF-α release dose dependently. Together, these findings identify a signaling through A2AAR as a critical control point for TNF-α release in AGA-treated retinal microglial cells.
Figure 3.

Identification of A2A adenosine receptor (A2AAR) anti-inflammation signaling in retinal microglial cells. (A) Functional identification of AR subtypes in retinal microglial cells. Tumor necrosis factor (TNF)-α production was determined in retinal microglial cells treated with Amadori-glycated albumin (AGA) (500 μg/mL, 4 hours), in the presence or absence of the nonselective AR agonist adenosine-5′-N-ethylcarboxamide (1 μmol/L) and subtype-selective AR antagonists for A1AR (8-cyclopentyl-1,3-dipropylxanthine, 0.1 μmol/L), A2AAR (ZM 241385, 0.1 μmol/L), A2BAR (MRS 1754, 1 μmol/L), and A3AR (MRS 1523, 10 μmol/L). TNF-α levels were determined by enzyme-linked immunosorbent assay (ELISA). As a control for AGA, cells were also treated with nonglycated albumin (500 μg/mL, 4 hours). Data shown are the mean + SD of three experiments. (B) Effect of A2AAR activation on AGA-induced TNF-α release. Retinal microglial cells were treated with AGA (500 μg/mL, 4 hours) in the presence of different doses of the A2A-selective agonist, CGS21680. TNF-α levels were determined by ELISA. Data shown are the mean + SD of at least three different experiments. (C) Effect of CGS21680 on cAMP accumulation in 500 μg/mL AGA-treated microglia for 4 hours. Retinal microglial cells were pretreated with CGS21680, treated with AGA in the presence of the phosphodiesterase inhibitor, 3-isobutyl-1-methylxanthine, and cAMP was determined with enzyme immunometric assay. Data shown are the mean + SD of three experiments. (D) Effect of cAMP on AGA-induced TNF-α release. Retinal microglial cells were treated with AGA (500 μg/mL, 4 hours) in the presence of different doses of cAMP. TNF-α levels were determined by ELISA. Data shown are the mean + SD of three experiments. CCPA, 2-chloro-N6-cyclopentyladenosine; CGS 21680, 2-p-[2-Carboxyethyl]phenethylamino-5′-N-ethylcarboxamidoadenosine; CPX, 8-cyclopentyl-1,3-dipropylxanthine; ZM 241385, 4-{2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-ylamino]ethyl}fphenol; DMSO, dimethyl sulfoxide; MRS 1754, 8-[4-[((4-cyanophenyl) arbamoylmethyl) oxy]phenyl]-1,3-di(n-propyl) xanthine hydrate; MRS 1523, 3-propyl-6-ethyl-5[(ethylthio)carbonyl]-2-phenyl-4-propyl-3-pyridine-carboxylate; NECA, adenosine-5′-N-ethylcarboxamide; N.S., not significant; ZM, ZM 241385, 4-{2-[7-amino-2-(2-furyl)[1,2,4]triazolo-[2,3-a][1,3,5]triazin-5-ylamino]ethyl}phenol.
Next, we sought to gain insights into the molecular mechanisms mediating the anti-inflammatory effects of A2AAR signaling. A2AAR is known as a G-protein coupled receptor that activates Adenylyl cyclase, thereby increasing intracellular cAMP. To verify that the inhibitory effect of A2AAR activation on TNF-α release from activated microglia was mediated by elevating cAMP, we initially determined the effect of 2-p-[2-carboxyethyl]phenethylamino-5′-N-ethylcarboxamidoadenosine (CGS 21680) on cAMP production in AGA-stimulated microglia. In AGA-treated cells, pretreatment with CGS21680 resulted in approximately a twofold increase in cAMP level compared with a pretreated vehicle (Figure 3C). To assess the involvement of cAMP in TNF-α inhibition, retinal microglial cells were treated with AGA in the presence of a cell-permeable cAMP, 8-pCPT-cAMP (10 - 100 μmol/L), and TNF-α release was measured. As shown in Figure 3D, cAMP inhibited TNF-α release dose dependently. These results clearly demonstrate that inhibition of TNF-α release from activated microglial cells via the A2AAR signaling pathway is cAMP-dependent.
A2AAR-cAMP Mediated Inhibition of TNF-α Release in Activated Microglia Involves Activity of EPAC
Recent studies have shown that protein kinase A (PKA) and the exchange protein directly activated by cAMP (EPAC) are downstream targets of cAMP. Therefore, we sought to determine whether both PKA and EPAC were involved in A2AAR/cAMP-mediated anti-inflammatory signaling in AGA-treated microglia. As shown in Figure 4A, both CGS21680 and 8-pCPT-cAMP reduced TNF-α release significantly and this inhibition was not reversed by either Rp-cAMP or PKI 14–22, two different PKA inhibitors. To confirm this result, the effect of the cAMP-enhancing effect of CGS21680 on PKA kinase activity in the AGA-treated microglia was determined. Although PKA kinase activity was fully detectable in the positive control, this activity was almost undetectable in AGA-treated cells, and was also negligible following treatment with CGS21680 (Figure 4B). Next, we examine the role of EPAC in the cAMP-mediated anti-inflammatory signaling. As shown in Figure 4C, pretreating microglial cells with 20 and 200 μmol/L of the EPAC-selective agonist, 8′-CPT-cAMP-O-Me, dose-dependently inhibited AGA-induced TNF-α production. Taken together, these results demonstrate that the A2AAR-cAMP signaling regulating AGA-induced TNF-α release from microglial cells involves the activity of EPAC, but not PKA.
Figure 4.
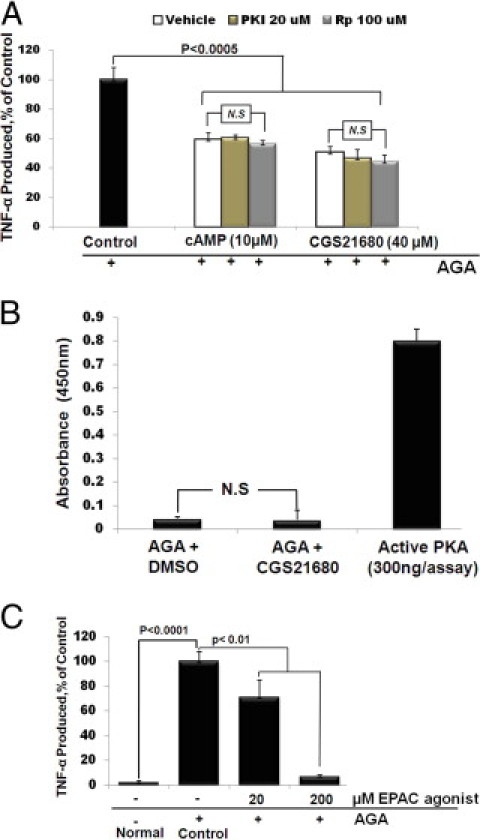
A2A adenosine receptor (A2AAR)-cAMP mediated inhibition of tumor necrosis factor (TNF)-α release in activated microglia involves activity of exchange protein directly activated by cAMP (EPAC). (A) Effect of protein kinase A (PKA) inhibitors on Amadori-glycated albumin (AGA)-induced TNF-α release from microglial cells. Cells were pretreated with CGS21680 or 8-pCPT-cAMP in the presence of PKI 14–22 amide or Rp-adenosine 3′,5′-cyclic monophosphorothioate (Rp-cAMP), then treated with AGA (500 μg/mL, 4 hours), and TNF-α release was determined by enzyme-linked immunosorbent assay (ELISA). Data shown are the mean + SD of three experiments. (B) Effect of CGS21680 on PKA kinase activity in the AGA-treated microglial cells. Cells were pretreated with CGS21680, and then treated with AGA (500 μg/mL), and PKA kinase activity was determined by ELISA 4 hours after AGA treatment. (C) Effect of EPAC agonist on AGA-induced TNF-α release from microglial cells. Cells were pretreated with 8′-CPT-cAMP-O-Me (20 and 200 μmol/L), and then treated with AGA (500 μg/mL, 4 hours), and TNF-α release was determined by ELISA. Data shown are the mean + SD of three experiments. DMSO, dimethyl sulfoxide.
A2AAR Signaling Mediates the Anti-Inflammatory Effect via Interaction with AGA-Activated C-Raf/ERK Pathway in Retinal Microglial Cells
Glycated albumin has been shown as a potent activator of Raf/ERK pathway in the course of TNF-α production (data not shown). Whether or not these kinases are modulated by the A2AAR signaling was also examined here. As shown in Figure 5A, CGS21680 dose-dependently inhibited AGA-induced ERK activation. To further delineate the interaction between A2AAR and mitogen-activated protein kinase signaling, we determined the effect of CGS21680 or 8-pCPT-cAMP on the activation of B-Raf or C-Raf, the upstream kinase of ERK, in AGA-treated retinal microglial cells. The results showed that phosphorylation of C-Raf occurred transiently at 15 minutes, whereas the phosphorylation of B-Raf was only slightly changed over the 60-minute experimental period. Both CGS21680 and 8-pCPT-cAMP inhibited AGA-induced activation of C-Raf, but not B-Raf (Figure 5, B and C). Collectively, these results suggest that the A2AAR-signaling cross talks with C-Raf/ERK pathway at a site upstream of C-Raf and that this cross talk modulates AGA-induced TNF-α expression in retinal microglial cells.
Figure 5.
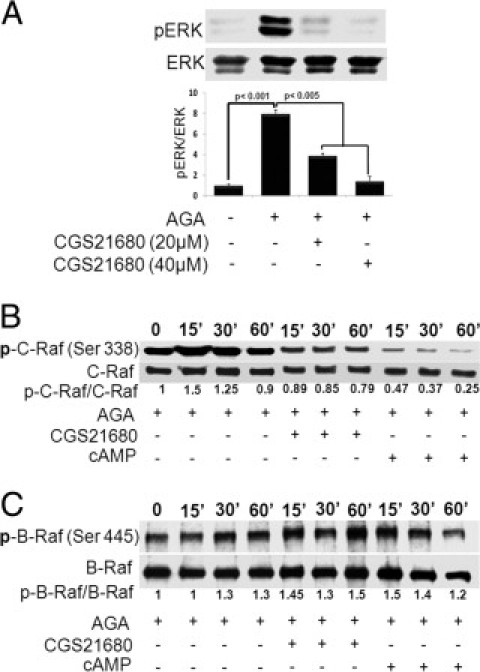
Role of A2A adenosine receptor (A2AAR)-cAMP signaling in the mitogen-activated protein (MAP) kinase pathway. (A) Effect of A2AAR activation on Amadori-glycated albumin (AGA)-induced extracellular signal-regulated kinase (ERK) MAP kinase activation. Retinal microglial cells were treated with AGA (500 μg/mL) in the presence or absence of CGS21680 (20 and 40 μmol/L) for 4 hours. Phospho-ERK (p-ERK) and total ERK MAP kinase in cell lysates were determined using Western blot analysis. Data shown are the mean + SD of three experiments. (B, C) Effect of A2AAR activation on AGA-induced C-Raf (B) and B-Raf (C) activation. Retinal microglial cells were treated with AGA (500 μg/mL) in the presence or absence of CGS21680 (20 μmol/L) or 8-pCPT-cAMP (250 μmol/L) for the indicated times (0 to 60 minutes). Phospho-C-Raf (Ser338), phospho-B-Raf, total C-Raf, and B-Raf in the cell lysates were determined using Western blot analysis.
Discussion
The early signs of DR include retinal inflammatory reactions, breakdown of the blood-retinal barrier function, and loss of retinal neurons.3–5 As the disease progresses, the retina may be damaged by oxidative stress induced by hyperglycemia, AGA, or advanced glycated proteins.26–28 This stress damages vascular and neuronal tissues of the retina, and activates microglial cells. Activated microglia further exacerbate the damage by releasing proinflammatory cytokines and cytotoxic molecules in response to oxidative stress.29 Thus, whereas the retina has traditionally been viewed as an immune privileged tissue, evidence is accumulating to support a role for local inflammation in the pathogenesis of DR.3,11,30,31 This suggests that pharmacological interventions that reduce inflammation may be effective neuroprotectants for DR.12,32
Adenosine has many diverse functions, all of which are dependent on its interaction with specific receptor subtypes.14 A2AAR has been mostly studied for its potent anti-inflammatory effects.33,34 High levels of A2AARs are found in macrophages and microglial cells that are poised, on activation, to abrogate the immune response.35 We have recently demonstrated that the activation of A2AAR, the dominant AR isoform in retinal microglial cells, blocks endotoxin-induced inflammation in these cells and in the retina.20 Here, pharmacological experiments with AR agonists, as well as AR antagonists have shown that A2AAR is the most likely candidate for mediating the adenosine effect on TNF-α suppression in activated microglial cells. Treatment with the A2AAR-selective agonist evokes the anti-inflammatory activity in AGA-activated retinal microglial cells via a cross talk with the Ras/C-Raf/MEK/ERK signaling. On activation of Gs-coupled receptor to regulate cell functions, cAMP increases to interact with the Ras/C-Raf/MEK/ERK signaling at a site upstream of C-Raf.36 Activation of C-Raf involves phosphorylation at multiple activating sites, including Ser338, Tyr341, Thr491, Ser494, Ser497, and Ser499.37 The cross talk between cAMP and the Ras/Raf/MEK/ERK signaling may lead to activation or inhibition of ERK activity, depending on the cell type. Evidence suggests that the expression of another Raf isoform, B-Raf, determines whether cAMP inhibits or activates ERK.36 In cells that express B-Raf, cAMP activates B-Raf and ERK, whereas in cells that express C-Raf, cAMP inhibits C-Raf activity, thereby suppressing ERK phosphorylation. Based on our observation that the activation of A2AAR-cAMP signaling inhibits ERK in retinal microglia, C-Raf is likely the major and functional Raf isoform in these cells. Moreover, the observed specificity in the cAMP signaling that is PKA-independent and EPAC-dependent suggests a novel mode in controlling the inflammatory events associated with microglia activation. A similar regulation of steroidogenesis has been observed for progesterone synthesis in ovarian granulosa cells.38 Figure 6 illustrates the pathways of AGA, a diabetic inflammatory trigger, which lead to the TNF-α release in stimulated microglia, as well as the mechanism by which A2AAR agonists block these processes.
Figure 6.
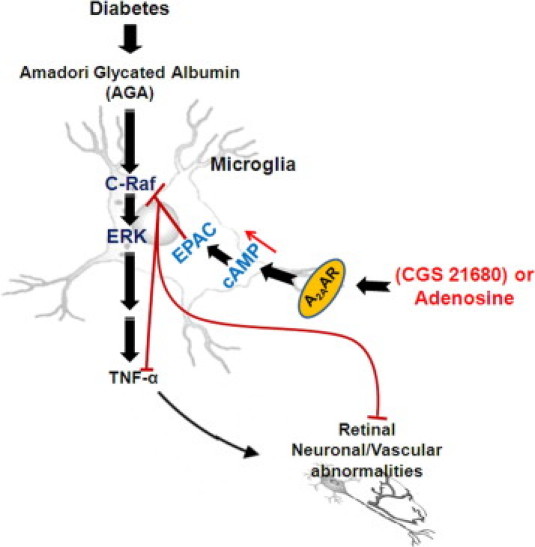
Proposed pathway of the Amadori-glycated albumin (AGA)-induced tumor necrosis factor (TNF)-α release, and A2A adenosine receptor (A2AAR) agonists mediated anti-inflammation in the retinal microglial cells during diabetes. EPAC, exchange protein directly activated by cAMP; ERK, extracellular signal-regulated kinase.
The present study further showed that diabetic mice that lack A2AAR exhibit significantly more retinal ICAM-1 expression, activated microglial cells, TNF-α release, and retinal cell death compared to diabetic mice that express A2AAR. Increases in the expression of ICAM-1 have been causally related to the increased retinal vascular inflammation and permeability in diabetes.3 Under stress conditions, the local levels of extracellular adenosine are elevated due to the increased need for energy supplied by ATP15 and the degradation of released ATP.16 This increased adenosine can protect against excessive cellular damage in a negative feedback manner.17 However, a recent study that determined the ability of rat cardiac fibroblasts to release adenosine under conditions of ATP depletion suggested that protection by adenosine may be insufficient. Although decreases in cellular ATP level were accompanied by increased extracellular adenosine, extracellular adenosine released by cells cultured in the absence of insulin was significantly less compared to cells cultured in the presence of insulin.39 Therefore, the apparently impaired protection in the WT diabetic mice with normal levels A2AAR could reflect decreases in the release of adenosine due to the insulin deficiency.
Another important point of this work is the demonstration of the in vivo efficacy of CGS21680 in reducing retinal inflammation and cell death associated with diabetes. This evaluation of the in vivo effect of CGS21680 is essential with the aim to develop a receptor-based therapy for diabetic retinopathy devoid of the invasive side effects associated with the standard therapy, laser photocoagulation. Future challenges include the development of compounds with high and selective-binding affinity to A2AAR, approaches to deliver A2AAR agonists to retina, and the definition of pharmacological strategies to safely use A2AAR agonists in diabetic patients. Ongoing studies by our group are in progress to further elucidate the role of ATL370 and ATL313, two potent and selective A2AR agonists, for their anti-inflammatory effects in diabetic retinopathy.
In summary, the current study provides preclinical evidence that the signaling through A2AAR is a critical pathway for attenuating retinal cell death associated with diabetes. Furthermore, this study suggests that the function of A2AAR agonists to inhibit C-Raf/ERK-mediated inflammatory cytokine release in activated microglia represents a novel therapeutic approach to treat ophthalmic complications associated with diabetes. Thus, with the discovery of new receptor-based therapies for diabetic retinopathy, A2AAR holds significant potential as a therapeutic target.
Footnotes
Supported by grants from the Egyptian Cultural and Educational Bureau (A.S.I.); the Juvenile Diabetes Research Foundation10-2009-575 (W.Z.); the Knights Templar Educational Foundation (G.I.L.); the Vision Discovery Institute (G.I.L.); the National Institutes of Health (NIH R01 EY04618 and NIH EY11766 (R.B.C.); and a VA Merit Review Award R.B.C.
References
- 1.Klein R., Klein B.E., Moss S.E., Davis M.D., DeMets D.L. The Wisconsin epidemiologic study of diabetic retinopathy: II Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch Ophthalmol. 1984;102:520–526. doi: 10.1001/archopht.1984.01040030398010. [DOI] [PubMed] [Google Scholar]
- 2.Sjolie A.K., Stephenson J., Aldington S., Kohner E., Janka H., Stevens L., Fuller J. Retinopathy and vision loss in insulin-dependent diabetes in Europe: The EURODIAB IDDM Complications Study. Ophthalmology. 1997;104:252–260. doi: 10.1016/s0161-6420(97)30327-3. [DOI] [PubMed] [Google Scholar]
- 3.Joussen A.M., Poulaki V., Le M.L., Koizumi K., Esser C., Janicki H., Schraermeyer U., Kociok N., Fauser S., Kirchhof B., Kern T.S., Adamis A.P. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452. doi: 10.1096/fj.03-1476fje. [DOI] [PubMed] [Google Scholar]
- 4.Barber A.J., Lieth E., Khin S.A., Antonetti D.A., Buchanan A.G., Gardner T.W. Neural apoptosis in the retina during experimental and human diabetes: Early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Remessy A.B., Al-Shabrawey M., Khalifa Y., Tsai N.T., Caldwell R.B., Liou G.I. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–244. doi: 10.2353/ajpath.2006.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeng X.X., Ng Y.K., Ling E.A. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. 2000;17:463–471. doi: 10.1017/s0952523800173122. [DOI] [PubMed] [Google Scholar]
- 7.Rungger-Brandle E., Dosso A.A., Leuenberger P.M. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1971–1980. [PubMed] [Google Scholar]
- 8.Sayyah M., Javad-Pour M., Ghazi-Khansari M. The bacterial endotoxin lipopolysaccharide enhances seizure susceptibility in mice: involvement of proinflammatory factors: nitric oxide and prostaglandins. Neuroscience. 2003;122:1073–1080. doi: 10.1016/j.neuroscience.2003.08.043. [DOI] [PubMed] [Google Scholar]
- 9.Yang L.P., Sun H.L., Wu L.M., Guo X.J., Dou H.L., Tso M.O., Zhao L., Li S.M. Baicalein reduces inflammatory process in a rodent model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2009;50:2319–2327. doi: 10.1167/iovs.08-2642. [DOI] [PubMed] [Google Scholar]
- 10.Liu W., Xu G.Z., Jiang C.H., Da C.D. Expression of macrophage colony-stimulating factor (M-CSF) and its receptor in streptozotocin-induced diabetic rats. Curr Eye Res. 2009;34:123–133. doi: 10.1080/02713680802650369. [DOI] [PubMed] [Google Scholar]
- 11.Joussen A.M., Doehmen S., Le M.L., Koizumi K., Radetzky S., Krohne T.U., Poulaki V., Semkova I., Kociok N. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428. [PMC free article] [PubMed] [Google Scholar]
- 12.Joussen A.M., Poulaki V., Mitsiades N., Kirchhof B., Koizumi K., Dohmen S., Adamis A.P. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J. 2002;16:438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 13.Gardner T.W., Antonetti D.A., Barber A.J., LaNoue K.F., Levison S.W. Diabetic retinopathy: more than meets the eye. Surv Ophthalmol. 2002;47(Suppl 2):S253–S262. doi: 10.1016/s0039-6257(02)00387-9. [DOI] [PubMed] [Google Scholar]
- 14.Collis M.G., Hourani S.M. Adenosine receptor subtypes. Trends Pharmacol Sci. 1993;14:360–366. doi: 10.1016/0165-6147(93)90094-z. [DOI] [PubMed] [Google Scholar]
- 15.Johnson S.M., Patel S., Bruckner F.E., Collins D.A. 5′-nucleotidase as a marker of both general and local inflammation in rheumatoid arthritis patients. Rheumatology (Oxford) 1999;38:391–396. doi: 10.1093/rheumatology/38.5.391. [DOI] [PubMed] [Google Scholar]
- 16.Wurm A., Iandiev I., Hollborn M., Wiedemann P., Reichenbach A., Zimmermann H., Bringmann A., Pannicke T. Purinergic receptor activation inhibits osmotic glial cell swelling in the diabetic rat retina. Exp Eye Res. 2008;87:385–393. doi: 10.1016/j.exer.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Ralevic V., Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 18.Awad A.S., Huang L., Ye H., Duong E.T., Bolton W.K., Linden J., Okusa M.D. Adenosine A2A receptor activation attenuates inflammation and injury in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290:F828–F837. doi: 10.1152/ajprenal.00310.2005. [DOI] [PubMed] [Google Scholar]
- 19.Gerits N., Kostenko S., Shiryaev A., Johannessen M., Moens U. Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways: comradeship and hostility. Cell Signal. 2008;20:1592–1607. doi: 10.1016/j.cellsig.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 20.Liou G.I., Auchampach J.A., Hillard C.J., Zhu G., Yousufzai B., Mian S., Khan S., Khalifa Y. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest Ophthalmol Vis Sci. 2008;49:5526–5531. doi: 10.1167/iovs.08-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese T., Melani A., Esposito E., Mazzon E., Di Paola R., Bramanti P., Pedata F., Cuzzocrea S. The selective adenosine A2A receptor agonist CGS 21680 reduces JNK MAPK activation in oligodendrocytes in injured spinal cord. Shock. 2009;32:578–585. doi: 10.1097/SHK.0b013e3181a20792. [DOI] [PubMed] [Google Scholar]
- 22.Wang A.L., Yu A.C., Lau L.T., Lee C., Wu le M., Zhu X., Tso M.O. Minocycline inhibits LPS-induced retinal microglia activation. Neurochem Int. 2005;47:152–158. doi: 10.1016/j.neuint.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Martin P.M., Roon P., Van Ells T.K., Ganapathy V., Smith S.B. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004;45:3330–3336. doi: 10.1167/iovs.04-0247. [DOI] [PubMed] [Google Scholar]
- 24.Wang A.L., Yu A.C., He Q.H., Zhu X., Tso M.O. AGEs mediated expression and secretion of TNF alpha in rat retinal microglia. Exp Eye Res. 2007;84:905–913. doi: 10.1016/j.exer.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Quan Y., Du J., Wang X. High glucose stimulates GRO secretion from rat microglia via ROS. PKC, and NF-kappaB pathways. J Neurosci Res. 2007;85:3150–3159. doi: 10.1002/jnr.21421. [DOI] [PubMed] [Google Scholar]
- 26.Al-Shabrawey M., Rojas M., Sanders T., Behzadian A., El-Remessy A., Bartoli M., Parpia A.K., Liou G., Caldwell R.B. Role of NADPH oxidase in retinal vascular inflammation. Invest Ophthalmol Vis Sci. 2008;49:3239–3244. doi: 10.1167/iovs.08-1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clements R.S., Jr., Robison W.G., Jr., Cohen M.P. Anti-glycated albumin therapy ameliorates early retinal microvascular pathology in db/db mice. J Diabetes Complications. 1998;12:28–33. doi: 10.1016/s1056-8727(97)00051-2. [DOI] [PubMed] [Google Scholar]
- 28.Stitt A.W., Bhaduri T., McMullen C.B., Gardiner T.A., Archer D.B. Advanced glycation end products induce blood-retinal barrier dysfunction in normoglycemic rats. Mol Cell Biol Res Commun. 2000;3:380–388. doi: 10.1006/mcbr.2000.0243. [DOI] [PubMed] [Google Scholar]
- 29.Kreutzberg G.W. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 30.Kern T.S. Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res. 2007;2007:95103. doi: 10.1155/2007/95103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adamis A.P., Berman A.J. Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol. 2008;30:65–84. doi: 10.1007/s00281-008-0111-x. [DOI] [PubMed] [Google Scholar]
- 32.Kim S.J., Flach A.J., Jampol L.M. Nonsteroidal anti-inflammatory drugs in ophthalmology. Surv Ophthalmol. 2010;55:108–133. doi: 10.1016/j.survophthal.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Bong G.W., Rosengren S., Firestein G.S. Spinal cord adenosine receptor stimulation in rats inhibits peripheral neutrophil accumulation: The role of N-methyl-D-aspartate receptors. J Clin Invest. 1996;98:2779–2785. doi: 10.1172/JCI119104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdmann A.A., Gao Z.G., Jung U., Foley J., Borenstein T., Jacobson K.A., Fowler D.H. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707–4714. doi: 10.1182/blood-2004-04-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trincavelli M.L., Melani A., Guidi S., Cuboni S., Cipriani S., Pedata F., Martini C. Regulation of A(2A) adenosine receptor expression and functioning following permanent focal ischemia in rat brain. J Neurochem. 2008;104:479–490. doi: 10.1111/j.1471-4159.2007.04990.x. [DOI] [PubMed] [Google Scholar]
- 36.Stork P.J., Schmitt J.M. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 37.Chong H., Lee J., Guan K.L. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001;20:3716–3727. doi: 10.1093/emboj/20.14.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chin E.C., Abayasekara D.R. Progesterone secretion by luteinizing human granulosa cells: a possible cAMP-dependent but PKA-independent mechanism involved in its regulation. J Endocrinol. 2004;183:51–60. doi: 10.1677/joe.1.05550. [DOI] [PubMed] [Google Scholar]
- 39.Podgorska M., Kocbuch K., Grden M., Szutowicz A., Pawelczyk T. Reduced ability to release adenosine by diabetic rat cardiac fibroblasts due to altered expression of nucleoside transporters. J Physiol. 2006;576:179–189. doi: 10.1113/jphysiol.2006.111278. [DOI] [PMC free article] [PubMed] [Google Scholar]