

Abstract
CD46 was discovered in 1986 during a search for novel C3b-binding proteins. CD46 is expressed ubiquitously and functions as a co-factor in the factor I-mediated proteolytic cleavage of C3b and C4b. Its vital role in preventing complement deposition on host tissue is underpinned by the fact that deficiency of CD46 is a predisposing factor for numerous disease conditions arising from complement-mediated ‘self-attack’. However, in the last 10 years, it has become apparent that CD46 is also heavily involved in a new and somewhat surprising functional aspect of the complement system: the down-modulation of adaptive T helper type 1 (Th1) immune responses by regulating the production of interferon (IFN)-γversus interleukin (IL)-10 within these cells. Specifically, this latter function of CD46 is a tantalizing discovery – it may not only have delivered the explanation as to why so many pathogens use and abuse CD46 as cell entry receptor but clearly has important clinical implications for the better understanding of Th1-mediated disease states and novel therapeutic approaches for their amelioration. Here, we summarize and discuss the current knowledge about CD46 and its expanding roles in the immune system.
Keywords: autoimmune disease, CD46, complement, IL-10, T cell regulation
Introduction
Complement activation initiates a defensive immune response targeted at infecting agents that breach the host epithelial barriers. Opsonization of microbes for phagocytic uptake by scavenger cells or for direct lysis and the induction of the inflammatory reaction are the effector functions that conventionally identified complement as the key component of the innate immune system. Research in recent years, however, has changed the perspective on how we look at complement today. Our knowledge has extended from a view of the complement cascade as an effector of humoral immune mechanisms to a network involved actively in initiating, shaping and also down-modulating adaptive immune responses [1].
The complement system, which consists of over 30 plasma and membrane-bound proteins, can be activated via three different pathways: the classical pathway (activated by antibodies and immune complexes), the lectin pathway (triggered by pathogen-specific lectins) and the alternative pathway (initiated directly on microbial surfaces or triggered by properdin binding to targets) (Fig. 1). All pathways converge to generate protein complexes (C3 convertases) that cleave and activate the central complement component C3 into the effector molecule C3b [2]. Deposition of C3b onto targets then induces the formation of the membrane attack complex (MAC) and subsequent lysis and/or the uptake of C3b-opsonized pathogens by macrophages and neutrophils. C3b generation also leads to production of the anaphylatoxins (C3a and C5a), potent chemoattractants and mediators of the inflammatory reaction [3].
Fig. 1.

Activation and regulation of the complement system. Complement can be activated through three pathways: the classical, lectin and alternative pathways. Note that properdin binding to a target [via specific glycosaminoglycans (GAGs)] and direct cleavage of C3 (or C5) through proteases belonging to the clotting cascade such as kallikrein and thrombin can also induce complement activation. Further, the initial deposition of C3b on a target surface initiates the feedback amplification loop. All activation conditions culminate in the formation of the C3-converting convertases and generation of C3b (C4b) and the anaphylatoxins C3a and C5a. These effectors mediate then direct lysis [membrane attack complex (MAC) formation] or immune adherence and phagocytosis of the pathogen (C3b and C4b) or function as chemokines, chemoattractants and activators of immunocompetent cells (C3a and C5a). Host tissue or unsuitable targets are protected from unwanted complement deposition by fluid-phase and cell-bound regulators (depicted in red), which either (1) degrade C3b and C4b deposited on tissue proteolytically, (2) accelerate the decay of already formed convertases or (3) inhibit MAC formation. C1 inhibitor prevents the activation and target-binding of C1.
Because complement activation is amplified quickly upon pathogen encounter, such a potent system needs to differentiate securely between self and non-self to avoid unwanted acute or chronic activation and consequential tissue damage. In humans, a set of soluble and membrane-bound complement inhibitory molecules and regulators provide this vital control and prevent the deposition of complement activation fragments on host cells [4]. Factor H (fH), C4b-binding protein (C4bp), vitronectin (S protein), clusterin (apolipoprotein J) and C1 inhibitor are soluble regulators found in blood and lymph and inhibit complement activation and cascade progression in the fluid phase [5,6]. CD46 (membrane co-factor protein, MCP), CD55 (decay accelerating factor, DAF), CD59 (protectin), CD35 (complement receptor 1, CR1) and CRIg (complement receptor of the immunoglobulin superfamily) [7], are membrane-bound regulators of the complement system [5,6]. These molecules are either co-factors (fH, C4bp, CD46 and CD35) in the factor I-mediated cleavage of C3b (and/or C4b) or can accelerate the decay (CD35 and CD55) of already formed C3 convertases, while CD59, clusterin and S protein prevent the complement formation of the MAC [2,3,6,8], although C1 inhibitor functions by binding to and inhibiting C1r and C1s activation within the C1 complex [6].
Although complement control by these molecules is clearly vital in the prevention of tissue damage (see below), it is now understood that the membrane-bound regulators specifically have a role beyond sole regulation on the cell surface. Upon ligand binding, these molecules also function as sensors for the degree of environmental complement activation and transmit intracellular signals which impact heavily not only intrinsically upon single cell function but upon the immune response in general [1,2,9,10].
Specifically, research advances on the signalling properties of CD46 have shed light on novel and unexpected links between complement and adaptive immunity and the roles of complement during infection and autoimmune disorders. This review will give an insight into our current understanding of CD46's increasingly diverse roles in innate and adaptive immunity.
CD46 in innate immunity
Complement regulation and related disease states
CD46 is a type I transmembrane glycoprotein identified initially as a complement inhibitor. However, CD46 is involved in additional important biological processes, including sperm–egg fusion during fertilization [11] (for further reading on the function of CD46 in the reproductive system, please see the excellent review by Harris and Morgan [12]), T cell regulation [13] and infection processes due to its pathogen receptor properties [14]. CD46 is expressed on all nucleated cells and alternative splicing of the CD46 gene can generate four commonly expressed isoforms (Fig. 2). CD46 comprehends four short consensus repeats (CCPs), an alternatively spliced, highly glycosylated region (B and/or C region), followed by 12 amino acids of unknown function, a transmembrane region and then one of two possible alternatively spliced cytoplasmic tails (CYT-1 or CYT-2). Both these cytoplasmic domains contain signalling motifs and have been shown to mediate distinct cellular functions [2,15,16].
Fig. 2.

CD46: structure and function. The N-terminus of CD46 consists of four complement control proteins (CCPs) followed by a highly O-glycosylated region generated by alternative splicing and comprehends the B and/or C isoforms. The B and C domains are rich in the amino acids serine, threonine and proline (STP) and are linked to the transmembrane domain through a 12 amino acids region of as yet unknown function. The intracellular region of CD46 is represented by a cytosolic tail. Alternative splicing can generate a CYT-1-bearing isoform or a CYT-2-bearing isoform, both of which contain signalling motifs and which are expressed differentially in inflammatory settings.
CD46 is the only ubiquitously expressed co-factor for the factor-I-mediated cleavage of C3b and C4b (CD35 is expressed much more sporadically) and therefore plays a key role in host cell protection from unwanted complement activation [17]. Attesting to the vital role of CD46 in tonic complement control is the observation that mutations in CD46 affecting its C3b/C4b binding or cleavage ability predispose to atypical haemolytic uraemic syndrome (aHUS) (Table 1). HUS is a pathology induced by uncontrolled complement activation and C3b deposition on host endothelium and characterized by microangiopathic haemolytic anaemia, thrombocytopenia and acute renal failure occurring as consequence of microthrombi in the glomeruli [18–20]. Whether CD46 gene mutations may also be involved in another disease connected with deregulation of complement activation over time, age-related macular degeneration (AMD) is currently being analysed. In addition, given the strong signalling capacity of CD46 in several cell types (see below), the idea that CD46-connected HUS pathology may also potentially be attributed in part to a defect in the signalling component (and not just complement regulation function) is worthy of further investigation.
Table 1.
Human diseases involving defects in CD46-mediated signals or required co-signals
| Disease | Pathology | Aetiology | CD46-related functional defect | References |
|---|---|---|---|---|
| aHUS (atypical haemolytic uraemic syndrome) | Triad of haemolytic anaemia, thrombocytopenia, and acute renal failure | CD46 gene mutations | Defective complement regulation specifically on endothelium | Richards, 2003; Maga, 2010 |
| Multiple sclerosis | Chronic autoimmune disorder leading to cognitive disabilities due to demyelination of axons in the brain and spinal cord | Unknown | Defective regulation of autoreactive effector T cells: | Astier, 2006; Martinez-Forero et al., 2008; Yao, 2010; Vaknin-Dembinsky, 2008 |
| • reduced IL-10 production (lack of Th1 regulation) | ||||
| • excessive Th17 expansion | ||||
| • increased IL-23p19 production by DCs (further amplifying Th17 activation) | ||||
| Rheumatoid arthritis | Chronic and systemic disorder principally leading to the destruction of articular cartilage and ankylosis of the joints | Unknown | Defective regulation of autoreactive effector T cells: | Cardone, Le Friec, 2010 |
| • defective IFN-γ‘shut-down’ | ||||
| • (lack of Th1 regulation) | ||||
| Asthma | Chronic inflammatory disease of the airways characterized by airway obstruction and bronchospasm | Unknown | Defective effector T cell regulation: | Xu et al., 2010 |
| reduced IL-10 production | ||||
| IPEX-like syndrome | Severe immune dysregulation, polyendocrinopathy and enteropathy | †CD25 deficiency – absence of IL-2-mediated signals required for CD46-induced cytokine production | Defective effector T cell induction and regulation: | Caudy, 2007 |
| • ‡absence of regulatory Th1/Tr1 cells | ||||
| Primary C3 deficiency | Recurrent and severe infections due to compromised functions of vital immune cells (including DCs, B cells and T cells) | C3 gene mutation (lack of CD46 ligand expression?) | Defective effector T cell induction and regulation: | Ghannam, 2008 |
| • no IL-10 production and reduced IFN-γ production upon CD46 activation | ||||
| • defective B cell memory compartment |
The cause of CD25 deficiency in this patient is unknown.
The production of IFN-γ after CD3/CD46/IL-2 activation of CD4+ T cells from this patient was not analysed in this study. IL: interleukin; IPEX: immunodysregulation polyendocrinopathy enteropathy X-linked syndrome; Th: T helper; DCs: dendritic cells; IFN: interferon.
Table 1 lists the diseases for which a direct association of CD46 dysfunction has been shown. However, as defective complement regulation is now emerging as a contributory factor for many additional conditions, among those pre-eclampsia [21,22], possibly cardiovascular and related disorders [23] and Alzheimer disease [24], a possible involvement of CD46 in such conditions may be an interesting question to answer.
Exploitation by pathogens
Interestingly, CD46 also functions as an entry receptor for several important human viral and bacterial pathogens [25]. Both measles virus (MV) and adenovirus bind the extracellular domains CCP1 and 2 of CD46 via the viral proteins haemagglutinin and fibre knob, respectively [14,26–28]. The interaction of human herpesvirus-6 (HHV-6) and CD46 requires regions within CCP2 and 3 [29,30]. The M protein of Streptococcus pyogenes is also a ligand for CD46 [31,32], and Neisseria gonorrhoeae and N. meningitidis bind to CD46 via CCP3 and the STP region [33–35]. Further, an important role for CD46 in acute and chronic urinal tract infections has also been suggested: C3b- opsonized uropathogenic Escherichia coli exploit the binding of C3b to CD46 and thereby infect human kidney tubular epithelial cells [36].
The exact reason(s) for CD46's attractiveness to pathogens is not understood fully. One logical explanation, however, may be that CD46 has immunoregulatory properties which could be ‘high-jacked’ by microbes. For instance, MV and HHV-6 binding to CD46 inhibits macrophage production of interleukin (IL)-12 (Fig. 3a), a cytokine required for T helper type 1 (Th1) differentiation and activation of both natural killer (NK) and T cells [37,38] and also down-modulates nitric oxid production, an important messenger molecule regulating cell homeostasis [39,40]. Similarly, the CD46/Streptococci interaction on human CD4+ T cell induces high amounts of immunosuppressive IL-10 and the generation of regulatory T cells (see below) [41].
Fig. 3.
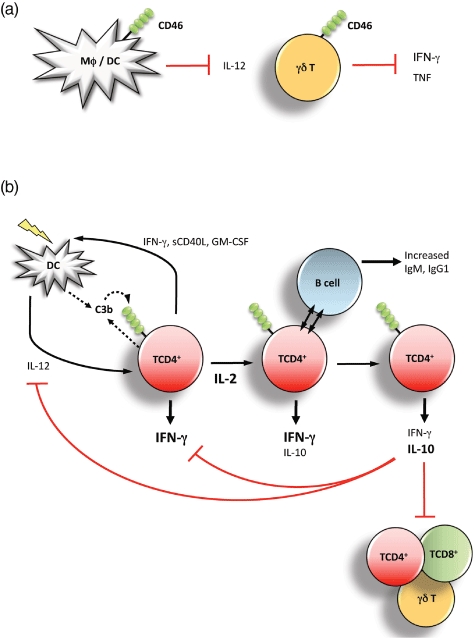
CD46-mediated signals regulate innate and adaptive immune cell function. (a) CD46 modulates innate cell functions. CD46 cross-linking by natural and pathogenic ligands suppresses interleukin (IL)-12 production by macrophages and dendritic cells (DCs) in mice and humans. Further, CD46-mediated signals directly inhibit the production of the effector cytokine interferon (IFN)-γ from γδ T cells (γδ T cells are often considered part of innate immunity because of their more broad activation modus and sentinel function at the host/environment border). (b) Regulation of T helper type 1 (Th1) IFN-γ to IL-10 switching and regulation of adaptive T cell responses. In the induction phase of Th1 responses, IL-2 and C3b production are low and support proliferating prototypic IFN-γ-secreting Th1 cells controlling infections. The expansion of Th1 effector cells raises environmental IL-2 sufficiently to induce IL-10 co-expression and the regulatory phases, coinciding with IFN-γ and IL-2 shut-down and suppression of neighbouring CD4+, CD8+ and γδ T cells as well as IL-12 production by DCs in the vicinity. This, in sum, leads to the contraction of a successful Th1 immune response and the prevention of tissue pathology.
Also, as CD46 seems to participate in the maintenance of epithelial barrier integrity [42–44], binding of pathogens to CD46 may facilitate direct cell entry of transgression through the protective epithelium, thereby fostering pathogen protection and dissemination [29,45,46]. This observation is in line with a recent finding that CD46 participates in the regulation of epithelial cell growth and wound healing by modulating tight junction formation [42]. Further, pathogenic E. coli take advantage of this housekeeping function of CD46 on gut epithelium and induce a dramatic decrease in transepithelial resistance in conjunction with an increase of bacterial paramigration after their binding to CD46 [42]. Interestingly, CD46 engagement by MV and group A streptococci also triggers CD46-mediated cell autophagy and increased death of infected cells: an elegant example of the host's measures to counteract pathogen abuse [47,48].
Unexplored areas of innate immunity
Although we are beginning to understand the role of CD46-mediated signals on macrophages, dendritic and epithelial cells, virtually nothing is known about the role of CD46 on other innate immune cells including NK cells, NK T cells, mast cells, neutrophils and monocytes. Only a single in vitro study has explored CD46 engagement on NK cells and demonstrated reduced killing activity upon CD46 activation [49]. Also, cross-talk between CD46 and other innate danger-sensing systems such as the Toll-like receptor (TLR), NOD-like receptor (NLR) or retinoic acid-inducible gene 1 protein (RIG-I) proteins is highly likely, and will provide rich ground for exciting new discoveries with regard to our expanding understanding of the diverse roles of CD46 in immunity.
CD46 in adaptive immunity
Th1 and γδ T cell regulation
About a decade ago, Astier et al. observed that CD46 functions as a co-stimulatory molecule on human CD4+ T cells inducing high cell proliferation upon activation during antigen presentation [50,51]. This seminal finding was then followed by a series of studies that demonstrated ultimately that CD46 engagement during T cell activation induces the activation or generation of IL-10-secreting regulatory T cells (Tregs) [9,13,52]. Recently, immunologists and the biochemical industry have invested significant resources in the understanding of Tregs due to their therapeutic potential in autoimmune diseases and graft rejection. Tregs can suppress and control the activation of effector Th1, Th2 and Th17 T cells and are currently subdivided into two main subsets: natural CD25 and forkhead box P3 (FoxP3)-positive Tregs that are thymus-derived and pivotal in self-tolerance [53], and adaptive Tregs, induced in the periphery and active in immunosuppression towards self- and non-self antigens [54]. Adaptive Treg functionality is induced by T cell receptor engagement (antigen presence) and appropriate co-stimulation and depends heavily upon the cytokine milieu, as most adaptive Tregs require high IL-2 for activation and function [13,55–57]. While natural Tregs mediate their suppressive properties via cell/cell contact, adaptive Tregs function through the secretion of high amounts of either IL-10 (Tr1 cells and IL-10-secreting Th1 cells) and/or transforming growth factor (TGF)-β (Th3 cells).
The identification of CD46 as a unique co-receptor for the TCR in the induction of IL-10 production by CD4+ T cells (CD46 activation and signalling on CD8+ T cells is so far unexplored) has been the first evidence bridging complement to T cell-mediated immunosuppression and created much interest in CD46 as novel link between innate and adaptive immunity [51,58]. However, the finding that CD3/CD46-activated Tregs suppressed bystander CD4+, CD8+[13] and γδ T cell activation [59] also raised questions with regard to the paradigmatic role of complement activation. In fact, with the complement system usually viewed as the prime danger-sensing and fighting system, down-regulation of the important anti-pathogenic T cell responses by CD46 seemed counterintuitive.
An exciting new set of data extending on the initial observations, however, now delivers a physiologically relevant explanation for CD46-mediated high IL-10 production. We observed that CD46 activation on CD4+ T cells [via C3b, pathogen interaction or monoclonal antibody (mAb) cross-linking] during antigen presentation in the presence of low concentrations of IL-2 induces a Th1 response characterized by high IFN-γ production but no IL-10 secretion. In the presence of high micro-environmental IL-2, signalling through CD46 then initiates the switch of Th1 cells via an IFN-γ+/IL-10+ intermediate population to IFN-γ-/IL-10+ Tr1-like cells. Both the intermediate and the Tr1-like cells are suppressive, while the IFN-γ+/IL-10- Th1 cells are non-suppressive [52]. Most importantly, T cell receptor (TCR) activation results in the secretion of C3 by the T cell itself and the subsequent local generation of the CD46 ligand C3b. Further, CD46 and not CD28-mediated co-stimulation is required for IL-10 production [52] and T cells from a C3-deficient patient do not produce IL-10 upon TCR activation [60]. This suggests a model in which TCR engagement induces locally the generation of CD46 ligands, providing a means for CD46 activation early during T cell activation (Fig. 3b). Such activated cells are now poised to integrate a third signal, high environmental IL-2 reflecting a successfully expanded Th1 response, and switch appropriately into the Treg or contraction phase (Fig. 3b). This would be in excellent agreement with the current view that control of IFN-γ-secreting CD4+ Th1 effector T cells by intrinsic IL-10 production is vitally required for protection against immunopathologies caused by exuberant inflammation during infection and the prevention of unwanted responses to self/innocuous antigens [61]. For example, IL10−/− mice succumb to colitis because of their inability to regulate immune responses against gut flora [62]; similarly, humans with IL-10 receptor gene mutations are highly susceptible to colitis [63]. Moreover, while murine IL-10 deficiency accelerates clearance of Toxoplasma gondii or Trypanosoma cruzi infections, such mice die of severe tissue damage due to IFN-γ overproduction [64,65]. Thus, the timely induction of an IFN-γ to IL-10 switch during the Th1 life cycle by CD46 further supports the emerging role of local complement activation as an important regulator of adaptive immunity [1,66,67]. In accordance with this notion, we have also shown that CD46 negatively regulates the production of IFN-γ by Vγ9Vδ2 cells (Fig. 3a), the main γδ T cell subset [68], which plays a specific role in the host defence at mucosal sites.
The latter observation is in line with the idea that CD46 activation of T cells could play a specific role at the host/environmental interfaces. Rubtsov et al. [69] have shown convincingly in a mouse model that adaptive IL-10-secreting Tregs reside and function mainly in the lung, skin and gut. Similarly, IL-10-secreting Th1 cells have been isolated from the skin and gut in humans [70,71]. We have indeed observed that CD3/CD46-activated CD4+ T cells up-regulate gut-homing markers such as integrin α4β7, the chemokine receptor CCR9 and the ‘intestinal’ cytokine LIGHT (TNFSF14) [72]. These data suggest that CD46-induced lymphocytes might play a role in the balance of gastrointestinal immune homeostasis.
Several groups have observed over the years that CD46-activated CD4+ T cell produce both IFN-γ and IL-10 [13,58,73,74]. However, the finding that secretion of these cytokines with opposing functional properties is not simultaneous, but rather temporally and spatially regulated, allows us to reconcile now previously controversial studies. For example, IL-10 interferes with the transition of immature dendritic cells (DCs) to their matured functional phenotype [75] and ‘conventional’ Tr1 cells usually inhibit DC maturation and thereby further T effector cell activation [61,76]. In contrast, we found that supernatants from CD46-activated Tregs allow DC maturation, antigen presentation and subsequent T effector cell induction because of the accumulation of soluble CD40 ligand (sCD40L) and granulocyte–macrophage colony-stimulating factor (GM-CSF) in 3-day cultures (at that time it was not discovered that CD46 activation induced IFN-γ+/IL-10-, IFN-γ+/IL-10+ and IFN-γ-/IL-10+ subpopulations) [77]; these two factors neutralize the usually inhibitory effect of IL-10 on DCs. Given the new observation, we would now predict that the CD46-induced IFN-γ+/IL-10- cells [which are also the major source of sCD40L and GM-CSF; data not shown] are strengthening the initiation of Th1 responses by supporting DC maturation while the IFN-γ+/IL-10+ T cells and IFN-γ-/IL-10+ T cells will indeed be suppressive towards DCs (as has been shown for IL-10-secreting Th1 cells [76]). Thus, the CD46-mediated switch from IFN-γ to IL-10 production in Th1 cells not only initiates the collapse of T cell responses directly but also via the inhibition of DCs in the vicinity (Fig. 3b).
Knowledge about the regulation of Th2, Th9 and Th17 T cell responses by CD46 is limited. CD46 clearly does not induce or support Th2 responses [13,52], but rather down-modulates IL-4 and IL-5 (unpublished data). CD46 signalling in Th9 cell is currently unexplored, and there is one report suggesting that CD46 may induce IL-17 in human CD4+ T cells [78] (see below).
B cell responses
While there is currently no indication that CD46-mediated signals impact directly on either B cell proliferation, survival or antibody production [79], the effect of CD46-activated T cells on B cells has been investigated. The co-culture of CD46-activated T cells with either autologous or allogeneic B cells showed that CD46-activated T cells are prime B cell activators because they induced significantly increased immunoglobulin (Ig)M and IgG1 production by B cells compared to co-cultures with ‘classically’ CD3/CD28-activated T cells. The mechanisms underlying such a boost in antibody production is not understood fully but requires cell–cell contact, secretion of functional IL-10 and also additional yet currently unidentified factors [79]. This finding is in good agreement with the observation that (a), T cells from a CD46-deficient patient are impaired in promoting Ig production and (b), that ca. 30% of CD46-deficient patients develop common variable immunodeficiency (CVID), a syndrome characterized by hypogammaglobulinaemia [80].
CD46 and human autoimmune disease
Analysing the exact role(s) of CD46 during an immune response in an in vivo model has been hampered by the fact that neither mice, rats nor guinea pigs express CD46 on somatic cells [15]. Mice express crry/p65, which shares some of the complement regulatory properties of CD46 but does not mimic the role of CD46 in T cell activation. Although mice strains transgenic for human CD46 exist, none of the CD46-mediated signalling events inducing Th1 to Treg switching can be recapitulated fully in these animals [9]. However, the correlation of CD46's dysfunction with a number of human pathologies has shed light on the in vivo relevance of CD46 to human immune homeostasis [17] (Table 1).
Multiple sclerosis (MS), rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE)
MS is a neurological disease affecting the central nervous system (CNS). The aetiology of MS is unknown but is thought to result from genetic and environmental risk factors [81]. However, because Tregs have been found to be impaired functionally in MS patients, it is generally accepted that T effector cells drive an uncontrolled immune response towards autoantigens expressed in the CNS in this disease [82].
A study by David Hafler's group was the first to provide evidence that deregulation of CD46-mediated signals in human CD4+ T cells correlates with human disease [74]. These studies showed that CD46-induced Tr1 cells from lymphocytes of MS patients have a defect in the production of IL-10 when compared with healthy donors [83]. A functional comparison between CD3/CD46-activated T cells from MS and healthy donors then highlighted that in the setting of MS, Tr1 cells lacked the ability to suppress autoreactive T cells appropriately. Importantly, here the production of the proinflammatory cytokine IFN-γ was not affected. These initial data have now been confirmed in experimental autoimmune encephalomyelitis (EAE) induced in Cynomolgus monkeys, the animal model of MS, where T cell proliferation and IL-10 secretion, but not IFN-γ production, was impaired upon CD46 activation [84].
Another interesting study suggests that changes in CD46-mediated signals on DCs may also contribute to MS. DCs from MS patients produced much higher amounts of the proinflammatory cytokine IL-23p19 upon CD46 activation compared to those from healthy donors. This increased IL-23 production then induced increased production of IL-17 by T cells in turn [85]. IL-17 has been identified recently as a component responsible for the damage to the CNS and Th17 cells seem to have a pivotal role in driving MS [78]. Thus, defective CD46 signalling seems to be involved in the expansion of Th17 cells in MS.
Inappropriate regulation of effector T cells including Th1 and Th17 are also pivotal in RA, an autoimmune disorder that affects the joints [86]. We found that the CD3/CD46/IL-2-mediated Th1 to Treg switching is defective in RA patients. T cells from these patients do not ‘shut down’ IFN-γ upon CD46-activation, therefore lack the IL-10+ cell subpopulation and produce up to 20 times more IFN-γ compared to T cells from healthy individuals. Further, the IFN-γ+/IL-10+ T cell population from RA patients is non-suppressive, while those from healthy donors suppress bystander T cell activation [52].
Although an association between complement deregulation and aberrant CD4+ T cell responses in the pathogenesis of SLE has been described [87], the exact role of complement-mediated modulation of T cell activity in the pathogenesis of SLE is still unclear. However, three interesting observations suggest that CD46-mediated signals on T cells may indeed impact upon SLE pathogenesis. First, CD46-activated Th1 cells are prime supporters of B cell activation [79], thus deregulation of T cell responses will probably impact upon antibody production (specifically in a setting of antibody production to self, as in SLE). Secondly, IL-2 levels, that finely regulate the CD46-induced switch of CD4+ T cells into a regulatory phenotype and then into the contraction phase (see above), are known to be decreased in SLE patients through abnormally high expression of the cAMP response element modulator (CREM), which inhibits IL-2 transcription by direct binding of the promoter [88]. Such consequential low environmental IL-2 concentrations would promote the Th1 phenotype induced by CD46 and impair transition to the IL-10 Treg phase and could induce/add to disease pathology. Thirdly, C4 deficiency has been associated strongly with SLE [89]. This is particularly interesting because C4b is not only the second physiological ligand for CD46 (beside C3b) but, in fact, C4b binds much more efficiently to CD46 compared to C3b [90], warranting a future ‘closer look’ into the CD46/C4b interaction in disease settings.
Allergies (asthma), immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX) and infections
Appropriate CD46-mediated signals are also important in down-modulating unwanted responses against innocuous non-self antigens and the prevention of allergies as indicated by the observation of reduced IL-10 production, but not IFN-γ secretion after CD46 activation of T cells from asthmatic patients [91]. Dysfunction of immunosuppressive T cells has been reported in allergic diseases including asthma [92] and the beneficial effects of vitamin D3, and glucocorticoids in the promotion of IL-10 have also been proved [93]. Interestingly, combining dexamethasone and CD46 co-stimulation of T cells from asthma patients restores IL-10 production to normal levels, suggesting a functional cross-talk between CD46 and glucocorticoid receptor pathways [91].
Pivotal in driving the switch of Th1 into Tr1-like cells through CD46 is the IL-2 signalling via the high-affinity receptor chain CD25 [13,74]. Human CD25 deficiency causes an autoimmune pathology resembling (but not identical with) the IPEX [94] induced by genetic mutations in the foxp3 loci and the complete loss of natural Tregs[95]. CD4+ T cells from FoxP3−/− IPEX patients produce normal IL-10 upon CD3/CD46/IL-2 activation and thus show that CD46-mediated signals are FoxP3-independent [94]. Conversely, CD25-deficient patients present with normal frequencies of FoxP3+ natural Tregs but, as expected, none of their T cells produce IL-10 upon CD3/CD46/IL-2 activation [94]. Further, the authors reported a higher susceptibility to infections of the CD25-deficient patient as the main difference compared to the IPEX-syndrome patient. These observations are fully in line with a defining role of CD25 and CD46 in the initiation and subsequent regulation of immune responses to self- and non-self antigens.
Concluding remarks
The complement system has come a long way from its early beginnings as a ‘sole pathogen detection and killing system’ to an integral part of the immune network instrumental in the induction and instruction of adaptive immunity. Moreover, recent advances in our understanding of complement's expanding functions imply that it also provides essential dampeners to inflammatory immune responses, thereby being vital in immune homeostasis. This realization is reflected by the growing list of disease states not connected previously with erroneous or deregulated complement activation. There is clearly more to be expected in this area (for example, the novel role of CD46 in growth control warrants analysis for its potential involvement in malignant transformation) and the increasing awareness of the clinical significance of appropriate complement function supports a current resurgence and refocus in complement research.
In this regard, the now tight connection of CD46 with autoimmune settings via faulty Th1 regulation creates a vital platform for the development of novel or improved therapeutics tackling such disease states. Thus, we are currently working on the identification of the molecular signatures and pathways that characterize both the effector and regulatory phases of CD46-induced Th1 differentiation. The identification of key proteins required for IFN-γ to IL-10 switching should then provide us with a framework for actively ‘inducing and locking’ these cells into the desired functional state in the clinical setting. In addition, recent data indicate that this T cell defect is reversible upon treatment with anti-tumour necrosis factor (TNF) therapy (unpublished data), suggesting that remission in RA patients is associated with the capacity to regenerate the Il-10+ T cell subpopulation RA patients – and further, indicates strongly that this pathway is indeed amendable to therapeutic intervention.
Additional important questions to be addressed include the still enigmatic role of complement-induced granzyme B production and its potential role in T cell regulation. CD46-activation on T cells results in very high granzyme B production detectable within the cell [96,97], as well as on the cell surface and in the cell culture media [59]. Granzyme B has been shown to have both detrimental as well as protective properties in disease and transplant settings [98] and it is clear that, similar to the complement system, its functions are vital in immunity but much more complex than acknowledged previously. Progress here will probably be beneficial for a better understanding of a variety of immune-mediated processes.
Finally, although systemic complement C3 activity is undoubtedly a key mediator in the defence against pathogenic microbes, local production of complement now takes centre stage in immune cell regulation [67], and its control may provide an additional realistic target to impact wilfully adaptive immune responses in therapeutic settings.
Acknowledgments
Research in the Kemper laboratory is supported by the MRC Centre for Transplantation, Guy's Hospital, King's College, and the Department of Health, National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.
Disclosure
The authors declare that they have no financial conflict of interest.
References
- 1.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 2.Le Friec G, Kemper C. Complement: coming full circle. Arch Immunol Ther Exp (Warsz) 2009;57:393–407. doi: 10.1007/s00005-009-0047-4. [DOI] [PubMed] [Google Scholar]
- 3.Volanakis JE, Frank MM. The human complement system in health and disease. New York: Marcel Dekker, Inc.; 1998. [Google Scholar]
- 4.Pickering MC, Cook HT. Translational mini-review series on complement factor H: renal diseases associated with complement factor H: novel insights from humans and animals. Clin Exp Immunol. 2008;151:210–30. doi: 10.1111/j.1365-2249.2007.03574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan BP, Harris CL. Complement regulatory proteins. London: Academic Press; 1999. [Google Scholar]
- 6.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–40. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 7.He JQ, Wiesmann C, van Lookeren Campagne M. A role of macrophage complement receptor CRIg in immune clearance and inflammation. Mol Immunol. 2008;45:4041–7. doi: 10.1016/j.molimm.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 8.Harris CL, Spiller OB, Morgan BP. Human and rodent decay-accelerating factors (CD55) are not species restricted in their complement-inhibiting activities. Immunology. 2000;100:462–70. doi: 10.1046/j.1365-2567.2000.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemper C, Atkinson JP. T-cell regulation: with complements from innate immunity. Nat Rev Immunol. 2007;7:9–18. doi: 10.1038/nri1994. [DOI] [PubMed] [Google Scholar]
- 10.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riley RC, Kemper C, Leung M, Atkinson JP. Characterization of human membrane cofactor protein (MCP; CD46) on spermatozoa. Mol Reprod Dev. 2002;62:534–46. doi: 10.1002/mrd.10144. [DOI] [PubMed] [Google Scholar]
- 12.Harris CL, Mizuno M, Morgan BP. Complement and complement regulators in the male reproductive system. Mol Immunol. 2006;43:57–67. doi: 10.1016/j.molimm.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 13.Kemper C, Chan AC, Green JM, Brett KA, Murphy KM, Atkinson JP. Activation of human CD4+ cells with CD3 and CD46 induces a T-regulatory cell 1 phenotype. Nature. 2003;421:388–92. doi: 10.1038/nature01315. [DOI] [PubMed] [Google Scholar]
- 14.Gaggar A, Shayakhmetov DM, Lieber A. CD46 is a cellular receptor for group B adenoviruses. Nat Med. 2003;9:1408–12. doi: 10.1038/nm952. [DOI] [PubMed] [Google Scholar]
- 15.Riley-Vargas RC, Gill DB, Kemper C, Liszewski MK, Atkinson JP. CD46: expanding beyond complement regulation. Trends Immunol. 2004;25:496–503. doi: 10.1016/j.it.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Astier AL. T-cell regulation by CD46 and its relevance in multiple sclerosis. Immunology. 2008;124:149–54. doi: 10.1111/j.1365-2567.2008.02821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kemper C, Verbsky JW, Price JD, Atkinson JP. T-cell stimulation and regulation: with complements from CD46. Immunol Res. 2005;32:31–43. doi: 10.1385/IR:32:1-3:031. [DOI] [PubMed] [Google Scholar]
- 18.Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966–71. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maga TK, Meyer NC, Belsha C, Nishimura CJ, Zhang Y, Smith RJ. A novel deletion in the RCA gene cluster causes atypical hemolytic uremic syndrome. Nephrol Dial Transplant. 2010;26:739–41. doi: 10.1093/ndt/gfq658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liszewski MK, Fang CJ, Atkinson JP. Inhibiting complement activation on cells at the step of C3 cleavage. Vaccine. 2008;26(Suppl. 8):I22–7. doi: 10.1016/j.vaccine.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derzsy Z, Prohaszka Z, Rigo J, Jr, Fust G, Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. 2010;47:1500–6. doi: 10.1016/j.molimm.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 22.Lynch AM, Salmon JE. Dysregulated complement activation as a common pathway of injury in preeclampsia and other pregnancy complications. Placenta. 2010;31:561–7. doi: 10.1016/j.placenta.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oksjoki R, Kovanen PT, Meri S, Pentikainen MO. Function and regulation of the complement system in cardiovascular diseases. Front Biosci. 2007;12:4696–708. doi: 10.2741/2419. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 25.Cattaneo R. Four viruses, two bacteria, and one receptor: membrane cofactor protein (CD46) as pathogens' magnet. J Virol. 2004;78:4385–8. doi: 10.1128/JVI.78.9.4385-4388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naniche D, Wild TF, Rabourdin-Combe C, Gerlier D. Measles virus haemagglutinin induces down-regulation of gp57/67, a molecule involved in virus binding. J Gen Virol. 1993;74(Pt 6):1073–9. doi: 10.1099/0022-1317-74-6-1073. [DOI] [PubMed] [Google Scholar]
- 27.Manchester M, Valsamakis A, Kaufman R, et al. Measles virus and C3 binding sites are distinct on membrane cofactor protein (CD46) Proc Natl Acad Sci USA. 1995;92:2303–7. doi: 10.1073/pnas.92.6.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Segerman A, Atkinson JP, Marttila M, Dennerquist V, Wadell G, Arnberg N. Adenovirus type 11 uses CD46 as a cellular receptor. J Virol. 2003;77:9183–91. doi: 10.1128/JVI.77.17.9183-9191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P. CD46 is a cellular receptor for human herpesvirus 6. Cell. 1999;99:817–27. doi: 10.1016/s0092-8674(00)81678-5. [DOI] [PubMed] [Google Scholar]
- 30.Greenstone HL, Santoro F, Lusso P, Berger EA. Human herpesvirus 6 and measles virus employ distinct CD46 domains for receptor function. J Biol Chem. 2002;277:39112–18. doi: 10.1074/jbc.M206488200. [DOI] [PubMed] [Google Scholar]
- 31.Okada N, Liszewski MK, Atkinson JP, Caparon M. Membrane cofactor protein (CD46) is a keratinocyte receptor for the M protein of the group A streptococcus. Proc Natl Acad Sci USA. 1995;92:2489–93. doi: 10.1073/pnas.92.7.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giannakis E, Jokiranta TS, Ormsby RJ, et al. Identification of the streptococcal M protein binding site on membrane cofactor protein (CD46) J Immunol. 2002;168:4585–92. doi: 10.4049/jimmunol.168.9.4585. [DOI] [PubMed] [Google Scholar]
- 33.Kallstrom H, Liszewski MK, Atkinson JP, Jonsson AB. Membrane cofactor protein (MCP or CD46) is a cellular pilus receptor for pathogenic Neisseria. Mol Microbiol. 1997;25:639–47. doi: 10.1046/j.1365-2958.1997.4841857.x. [DOI] [PubMed] [Google Scholar]
- 34.Kallstrom H, Blackmer Gill D, Albiger B, Liszewski MK, Atkinson JP, Jonsson AB. Attachment of Neisseria gonorrhoeae to the cellular pilus receptor CD46: identification of domains important for bacterial adherence. Cell Microbiol. 2001;3:133–43. doi: 10.1046/j.1462-5822.2001.00095.x. [DOI] [PubMed] [Google Scholar]
- 35.Persson BD, Schmitz NB, Santiago C, et al. Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLoS Pathog. 2010;6:e1001122. doi: 10.1371/journal.ppat.1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li K, Feito MJ, Sacks SH, Sheerin NS. CD46 (membrane cofactor protein) acts as a human epithelial cell receptor for internalization of opsonized uropathogenic Escherichia coli. J Immunol. 2006;177:2543–51. doi: 10.4049/jimmunol.177.4.2543. [DOI] [PubMed] [Google Scholar]
- 37.Karp CL, Wysocka M, Wahl LM, et al. Mechanism of suppression of cell-mediated immunity by measles virus. Science. 1996;273:228–31. doi: 10.1126/science.273.5272.228. [DOI] [PubMed] [Google Scholar]
- 38.Smith A, Santoro F, Di Lullo G, Dagna L, Verani A, Lusso P. Selective suppression of IL-12 production by human herpesvirus 6. Blood. 2003;102:2877–84. doi: 10.1182/blood-2002-10-3152. [DOI] [PubMed] [Google Scholar]
- 39.Kurita-Taniguchi M, Fukui A, Hazeki K, et al. Functional modulation of human macrophages through CD46 (measles virus receptor): production of IL-12 p40 and nitric oxide in association with recruitment of protein-tyrosine phosphatase SHP-1 to CD46. J Immunol. 2000;165:5143–52. doi: 10.4049/jimmunol.165.9.5143. [DOI] [PubMed] [Google Scholar]
- 40.Kurita-Taniguchi M, Hazeki K, Murabayashi N, et al. Molecular assembly of CD46 with CD9, alpha3-beta1 integrin and protein tyrosine phosphatase SHP-1 in human macrophages through differentiation by GM-CSF. Mol Immunol. 2002;38:689–700. doi: 10.1016/s0161-5890(01)00100-6. [DOI] [PubMed] [Google Scholar]
- 41.Price JD, Schaumburg J, Sandin C, Atkinson JP, Lindahl G, Kemper C. Induction of a regulatory phenotype in human CD4+ T cells by streptococcal M protein. J Immunol. 2005;175:677–84. doi: 10.4049/jimmunol.175.2.677. [DOI] [PubMed] [Google Scholar]
- 42.Al-Shouli S, Cardone J, Sacks S, Kemper C. Lets connect: a novel role for CD46 in tight junction regulation. Mol Immunol. 2010;47:2252–3. [Google Scholar]
- 43.Ludford-Menting MJ, Thomas SJ, Crimeen B, et al. A functional interaction between CD46 and DLG4: a role for DLG4 in epithelial polarization. J Biol Chem. 2002;277:4477–84. doi: 10.1074/jbc.M108479200. [DOI] [PubMed] [Google Scholar]
- 44.Johansson L, Rytkonen A, Bergman P, et al. CD46 in meningococcal disease. Science. 2003;301:373–5. doi: 10.1126/science.1086476. [DOI] [PubMed] [Google Scholar]
- 45.Galbraith SE, Tiwari A, Baron MD, Lund BT, Barrett T, Cosby SL. Morbillivirus downregulation of CD46. J Virol. 1998;72:10292–7. doi: 10.1128/jvi.72.12.10292-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakurai F, Akitomo K, Kawabata K, Hayakawa T, Mizuguchi H. Downregulation of human CD46 by adenovirus serotype 35 vectors. Gene Ther. 2007;14:912–19. doi: 10.1038/sj.gt.3302946. [DOI] [PubMed] [Google Scholar]
- 47.Joubert PE, Meiffren G, Gregoire IP, et al. Autophagy induction by the pathogen receptor CD46. Cell Host Microbe. 2009;6:354–66. doi: 10.1016/j.chom.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Meiffren G, Joubert PE, Gregoire IP, Codogno P, Rabourdin-Combe C, Faure M. Pathogen recognition by the cell surface receptor CD46 induces autophagy. Autophagy. 2010;6:299–300. doi: 10.4161/auto.6.2.11132. [DOI] [PubMed] [Google Scholar]
- 49.Oliaro J, Pasam A, Waterhouse NJ, et al. Ligation of the cell surface receptor, CD46, alters T cell polarity and response to antigen presentation. Proc Natl Acad Sci USA. 2006;103:18685–90. doi: 10.1073/pnas.0602458103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Astier A, Trescol-Biemont MC, Azocar O, Lamouille B, Rabourdin-Combe C. Cutting edge: CD46, a new costimulatory molecule for T cells, that induces p120CBL and LAT phosphorylation. J Immunol. 2000;164:6091–5. doi: 10.4049/jimmunol.164.12.6091. [DOI] [PubMed] [Google Scholar]
- 51.Zaffran Y, Destaing O, Roux A, et al. CD46/CD3 costimulation induces morphological changes of human T cells and activation of Vav, Rac, and extracellular signal-regulated kinase mitogen-activated protein kinase. J Immunol. 2001;167:6780–5. doi: 10.4049/jimmunol.167.12.6780. [DOI] [PubMed] [Google Scholar]
- 52.Cardone J, Le Friec G, Vantourout P, et al. Complement regulator CD46 temporally regulates cytokine production by conventional and unconventional T cells. Nat Immunol. 2010;11:862–71. doi: 10.1038/ni.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 54.Allan SE, Broady R, Gregori S, et al. CD4+ T-regulatory cells: toward therapy for human diseases. Immunol Rev. 2008;223:391–421. doi: 10.1111/j.1600-065X.2008.00634.x. [DOI] [PubMed] [Google Scholar]
- 55.Groux H, O'Garra A, Bigler M, et al. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–42. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 56.Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–16. doi: 10.1084/jem.20011629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–22. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marie JC, Astier AL, Rivailler P, Rabourdin-Combe C, Wild TF, Horvat B. Linking innate and acquired immunity: divergent role of CD46 cytoplasmic domains in T cell induced inflammation. Nat Immunol. 2002;3:659–66. doi: 10.1038/ni810. [DOI] [PubMed] [Google Scholar]
- 59.Truscott SM, Abate G, Price JD, Kemper C, Atkinson JP, Hoft DF. CD46 engagement on human CD4+ T cells produces T regulatory type 1-like regulation of antimycobacterial T cell responses. Infect Immun. 2010;78:5295–306. doi: 10.1128/IAI.00513-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghannam A, Pernollet M, Fauquert JL, et al. Human C3 deficiency associated with impairments in dendritic cell differentiation, memory B cells, and regulatory T cells. J Immunol. 2008;181:5158–66. doi: 10.4049/jimmunol.181.7.5158. [DOI] [PubMed] [Google Scholar]
- 61.O'Garra A, Vieira P. T(H)1 cells control themselves by producing interleukin-10. Nat Rev Immunol. 2007;7:425–8. doi: 10.1038/nri2097. [DOI] [PubMed] [Google Scholar]
- 62.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 63.Franke A, Balschun T, Karlsen TH, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 64.Gazzinelli RT, Wysocka M, Hieny S, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 65.Hunter CA, Ellis-Neyes LA, Slifer T, et al. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol. 1997;158:3311–16. [PubMed] [Google Scholar]
- 66.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–35. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raedler H, Heeger PS. Complement regulation of T-cell alloimmunity. Curr Opin Organ Transplant. 2011;16:54–60. doi: 10.1097/MOT.0b013e3283425419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hayday AC. [Gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. 2000;18:975–1026. doi: 10.1146/annurev.immunol.18.1.975. [DOI] [PubMed] [Google Scholar]
- 69.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–58. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 70.O'Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C. Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev. 2008;223:114–31. doi: 10.1111/j.1600-065X.2008.00635.x. [DOI] [PubMed] [Google Scholar]
- 71.Hawrylowicz CM, O'Garra A. Potential role of interleukin-10-secreting regulatory T cells in allergy and asthma. Nat Rev Immunol. 2005;5:271–83. doi: 10.1038/nri1589. [DOI] [PubMed] [Google Scholar]
- 72.Alford SK, Longmore GD, Stenson WF, Kemper C. CD46-induced immunomodulatory CD4+ T cells express the adhesion molecule and chemokine receptor pattern of intestinal T cells. J Immunol. 2008;181:2544–55. doi: 10.4049/jimmunol.181.4.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sanchez A, Feito MJ, Rojo JM. CD46-mediated costimulation induces a Th1-biased response and enhances early TCR/CD3 signaling in human CD4+ T lymphocytes. Eur J Immunol. 2004;34:2439–48. doi: 10.1002/eji.200324259. [DOI] [PubMed] [Google Scholar]
- 74.Astier AL, Meiffren G, Freeman S, Hafler DA. Alterations in CD46-mediated Tr1 regulatory T cells in patients with multiple sclerosis. J Clin Invest. 2006;116:3252–7. doi: 10.1172/JCI29251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 2002;99:2468–76. doi: 10.1182/blood.v99.7.2468. [DOI] [PubMed] [Google Scholar]
- 76.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–81. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 77.Barchet W, Price JD, Cella M, et al. Complement-induced regulatory T cells suppress T-cell responses but allow for dendritic-cell maturation. Blood. 2006;107:1497–504. doi: 10.1182/blood-2005-07-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao K, Graham J, Akahata Y, Oh U, Jacobson S. Mechanism of neuroinflammation: enhanced cytotoxicity and IL-17 production via CD46 binding. J Neuroimmune Pharmacol. 2010;5:469–78. doi: 10.1007/s11481-010-9232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fuchs A, Atkinson JP, Fremeaux-Bacchi V, Kemper C. CD46-induced human Treg enhance B-cell responses. Eur J Immunol. 2009;39:3097–109. doi: 10.1002/eji.200939392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fremeaux-Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017–25. doi: 10.1681/ASN.2005101051. [DOI] [PubMed] [Google Scholar]
- 81.Ramagopalan SV, Dobson R, Meier UC, Giovannoni G. Multiple sclerosis: risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010;9:727–39. doi: 10.1016/S1474-4422(10)70094-6. [DOI] [PubMed] [Google Scholar]
- 82.Haas J, Hug A, Viehover A, et al. Reduced suppressive effect of CD4+CD25high regulatory T cells on the T cell immune response against myelin oligodendrocyte glycoprotein in patients with multiple sclerosis. Eur J Immunol. 2005;35:3343–52. doi: 10.1002/eji.200526065. [DOI] [PubMed] [Google Scholar]
- 83.Martinez-Forero I, Garcia-Munoz R, Martinez-Pasamar S, et al. IL-10 suppressor activity and ex vivo Tr1 cell function are impaired in multiple sclerosis. Eur J Immunol. 2008;38:576–86. doi: 10.1002/eji.200737271. [DOI] [PubMed] [Google Scholar]
- 84.Ma A, Xiong Z, Hu Y, et al. Dysfunction of IL-10-producing type 1 regulatory T cells and CD4(+)CD25(+) regulatory T cells in a mimic model of human multiple sclerosis in Cynomolgus monkeys. Int Immunopharmacol. 2009;9:599–608. doi: 10.1016/j.intimp.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 85.Vaknin-Dembinsky A, Murugaiyan G, Hafler DA, Astier AL, Weiner HL. Increased IL-23 secretion and altered chemokine production by dendritic cells upon CD46 activation in patients with multiple sclerosis. J Neuroimmunol. 2008;195:140–5. doi: 10.1016/j.jneuroim.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5:549–53. doi: 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 87.Manderson AP, Botto M, Walport MJ. The role of complement in the development of systemic lupus erythematosus. Annu Rev Immunol. 2004;22:431–56. doi: 10.1146/annurev.immunol.22.012703.104549. [DOI] [PubMed] [Google Scholar]
- 88.Juang YT, Wang Y, Solomou EE, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest. 2005;115:996–1005. doi: 10.1172/JCI200522854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fielder AH, Walport MJ, Batchelor JR, et al. Family study of the major histocompatibility complex in patients with systemic lupus erythematosus: importance of null alleles of C4A and C4B in determining disease susceptibility. BMJ. 1983;286:425–8. doi: 10.1136/bmj.286.6363.425. Clin Res Ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liszewski MK, Atkinson JP. Membrane cofactor protein (MCP; CD46). Isoforms differ in protection against the classical pathway of complement. J Immunol. 1996;156:4415–21. [PubMed] [Google Scholar]
- 91.Xu YQ, Gao YD, Yang J, Guo W. A defect of CD4+CD25+ regulatory T cells in inducing interleukin-10 production from CD4+ T cells under CD46 costimulation in asthma patients. J Asthma. 2010;47:367–73. doi: 10.3109/02770903.2010.481340. [DOI] [PubMed] [Google Scholar]
- 92.Ray A, Khare A, Krishnamoorthy N, Qi Z, Ray P. Regulatory T cells in many flavors control asthma. Mucosal Immunol. 2010;3:216–29. doi: 10.1038/mi.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xystrakis E, Kusumakar S, Boswell S, et al. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest. 2006;116:146–55. doi: 10.1172/JCI21759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol. 2007;119:482–7. doi: 10.1016/j.jaci.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 95.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 96.Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 97.Grossman WJ, Verbsky JW, Tollefsen BL, Kemper C, Atkinson JP, Ley TJ. Differential expression of granzymes A and B in human cytotoxic lymphocyte subsets and T regulatory cells. Blood. 2004;104:2840–8. doi: 10.1182/blood-2004-03-0859. [DOI] [PubMed] [Google Scholar]
- 98.Gondek DC, Devries V, Nowak EC, et al. Transplantation survival is maintained by granzyme B+ regulatory cells and adaptive regulatory T cells. J Immunol. 2008;181:4752–60. doi: 10.4049/jimmunol.181.7.4752. [DOI] [PMC free article] [PubMed] [Google Scholar]