

Abstract
Pseudomonas fluorescens is an opportunistic indoor pathogen that can cause severe airway proinflammatory responses. Pulmonary epithelium, like other mucosal epithelial linings of the body, constitutes the first line of defense against airway microbial pathogens. Mucosal epithelial cells can be a sentinel of pathogenic bacteria via stimulation of specific cell surface receptors, including the epidermal growth factor receptor (EGFR) and Toll-like receptor (TLR). This study addressed the involvement of EGFR in airway epithelial pathogenesis by P. fluorescens. Human A549 pneumocytes showed prolonged production of proinflammatory interleukin-8 (IL-8) in response to infection with P. fluorescens, which was via the nuclear factor-kappa B (NF-κB) signaling pathway. Production of proinflammatory cytokine IL-8 was not mediated by P. fluorescens lipopolysaccharide, a representative TLR4 agonist, but was mediated through EGFR-linked signals activated by the opportunistic bacteria. Moreover, EGFR signals were involved in NF-κB signal-mediated production of proinflammatory cytokines. Along with persistent NF-κB activation, P. fluorescens enhanced the EGFR phosphorylation and subsequent activation of downstream mediators, including protein kinase B or extracellular-signal-regulated kinases 1/2. Blocking of EGFR-linked signals increased epithelial susceptibility to pathogen-induced epithelial cell death, suggesting protective roles of EGFR signals. Thus, airway epithelial exposure to P. fluorescens can trigger antiapoptotic responses via EGFR and proinflammatory responses via TLR4-independent NF-κB signaling pathway in human pneumocytes.
INTRODUCTION
Pseudomonas fluorescens is generally a nonpathogenic saprophyte that inhabits soil, water, and plant surface environments and is also a competitive commensal against plant and fish pathogens (10, 33). As these environmentally versatile bacteria possess diverse metabolic activities, they or their genetically engineered strains have been explored for their bioremediation capacity (15, 20). P. fluorescens was recently implicated as a potently opportunistic indoor pathogen (11, 13), indicating that it can exploit a breach in the host defense to initiate an inflammatory response after infection. As an opportunistic pathogen, P. fluorescens also may be involved in the pathogenesis of Crohn's disease (32). Pseudomonas species are the most important cause of lung infections in patients with cystic fibrosis, and over 90% of the mortality in cystic fibrosis patients is due to chronic infections leading to bronchiectasis and respiratory failure (29).
Airway epithelia initiate the immune response to inhaled bacteria by recruiting white blood cells from the bloodstream to fight potential infection. Pseudomonas infection effectively induces innate immune responses after contact with lung epithelial cells. One of the major roles of the pulmonary epithelium is as a sentinel barrier between the lumen and the underlying submucosa by modulating proinflammatory cytokine profiles, including those of interleukin-4 (IL-4), IL-13, and IL-8. Lung epithelial IL-8 affects expression of two major airway mucin genes, MUC5AC and MUC5B (3). Whereas IL-8 produced by airway epithelial cells is a potent neutrophil chemoattractant that can be important as a defensive tool, it can also exaggerate the inflammatory responses leading to chronic progression of disease (28). Indoor inhalation of P. fluorescens may be harmful due to cytokine production (11, 13). In particular, P. fluorescens coexisting with fungal strains in moisture-damaged buildings can trigger production of proinflammatory cytokines, such as IL-6 and tumor necrosis factor alpha (TNF-α), by lung epithelial cells and macrophages, producing cytotoxic effects on immune-related cells. Mechanistically, production of proinflammatory cytokines in response to epithelial translocation of Pseudomonas spp. is mediated by the Toll-like receptor 4 (TLR4)-linked signaling pathway (8).
Epidermal growth factor receptor (EGFR; also known as ErbB-1 or HER) is widely expressed in mammalian epithelial tissues. It is a type I transmembrane glycoprotein with an extracellular ligand-binding ectodomain and an intracellular cytoplasmic domain (12, 16, 23, 34). Ligand binding induces the dimerization, autophosphorylation, and transactivation of the tyrosine kinase activity of EGFR, providing a variety of binding sites for a series of proteins, thereby initiating the activation of downstream signaling pathways. EGFR phosphorylation activates EGFR downstream effectors, such as protein kinase B (PKB) and extracellular-signal-regulated kinases 1/2 (ERK1/2) (24). EGFR-mediated signaling pathways are crucial to the human epithelial cell survival response by preventing injury-induced apoptosis. EGFR activation also induces the cell cycle promoter cyclin D1, which confers a growth advantage to lung epithelial cells (27). Inhibition of EGFR diminishes growth and survival of cells by several mechanisms that include arresting cell cycle progression in G1 due to increased expression of the cyclin-dependent kinase inhibitor p27kip (26), increasing cell susceptibility to apoptosis by altering expression of multiple proapoptotic and antiapoptotic genes (17), and stimulating epithelial differentiation (4).
Along with its important roles in regulating the diverse cellular processes of proliferation, differentiation, and apoptosis, EGFR has also been implicated in the bacterial infection-associated signaling pathway (18, 21, 35). In particular, repair of bacterium-triggered wounds is mediated via EGFR-linked signaling pathways. On the basis of the assumption that EGFR and its associated recognition are crucial in airway infection of P. fluorescens, the present study was undertaken to address the involvement of EGFR-associated signals in lung epithelial pathogenesis, including cytokine production and epithelial injuries. The study was performed using the A549 human alveolar epithelial cell culture model to provide molecular insight into Pseudomonas infection and its disease progression.
MATERIALS AND METHODS
Cell cultures and P. fluorescens infection.
Human type II pneumocyte cell line A549 was obtained from the South Korean Cell Line Bank (KCLB; Seoul, South Korea). Cells were maintained in RPMI 1640 medium containing 10% fetal bovine serum (FBS; Welgene, Taegu, South Korea) and 1% penicillin-streptomycin (Welgene). Cells were maintained at 37°C in humidified 5% CO2 incubators. P. fluorescens was maintained on LB agar (Duchefa Biochemie, Haarlem, Netherlands). Infection experiments utilized an established model for infecting epithelial cells (13). Briefly, bacteria were shaken in LB broth (Duchefa Biochemie) at 27°C overnight and then subcultured in antibiotic- and serum-free RPMI 1640 medium until the absorbance at 600 nm reached an optical density (OD) of 0.6 to 0.8. Bacteria were loaded onto the apical surface of A549 cells at a bacterium/cell ratio of 50:1.
Extraction of LPS.
A lipopolysaccharide (LPS) extraction kit (Intron Biotechnology, Seoul, South Korea) was used to isolate endotoxin from P. fluorescens according to the manufacturer's protocol. In brief, cells were collected by centrifuging 2 to 5 ml of culture at 13,000 rpm at room temperature. Lysis buffer was added, and the components were vigorously mixed. After addition of chloroform and vigorous vortexing of the mixture for 10 to 20 s, the mixture was incubated at room temperature for 5 min. Finally, the mixture was centrifuged at 13,000 rpm for 10 min at 4°C. The supernatant was mixed with the purification buffer, and the mixture was incubated for 10 min at −20°C. After the solution was centrifuged at 13,000 rpm for 15 min at 4°C, the layer overlying the pelleted LPS was removed. The LPS pellet was washed, dried, and dissolved in 10 mM Tris-HCl buffer by boiling for 2 min. LPS extracted from bacteria cultured with human epithelial cells was quantified using the Limulus amoebocyte lysate (LAL) assay. The indicated doses of test specimens were mixed separately with LAL reagent, and then the mixture was incubated at 37°C for 30 min, after which the activities were determined using quantitative chromogenic assays.
Western blotting.
Protein expression was compared by Western immunoblot analysis using rabbit polyclonal anti-human β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA), anti-IκBα, anti-phospho-ERK, and anti-poly(ADP ribose)polymerase (PARP) antibodies (Santa Cruz Biotechnology), and anti-phospho-PKB antibody (Cell Signaling Technology, Beverly, MA). Cells were washed with ice-cold phosphate buffer, lysed in boiling lysis buffer (1% [wt/vol] sodium dodecyl sulfate [SDS], 1.0 mM sodium ortho-vanadate, and 10 mM Tris, pH 7.4), and sonicated for 5 s. Protein in each cell lysate was quantified using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Fifty micrograms of protein was separated by 10% SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were transferred onto a polyvinylidene fluoride membrane (Amersham Pharmacia Biotech, Piscataway, NJ), and the blots were blocked for 1 h with 5% skim milk in Tris-buffered saline plus 0.05% Tween 20 (TBST) and probed with each antibody for a further 2 h at room temperature or overnight at 4°C. After the blots were washed three times with TBST, they were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h and washed with TBST a further three times. Protein was detected using an enhanced chemiluminescence substrate (ELPIS Biotech, Taejon, South Korea).
Coimmunoprecipitation assay.
Cell lysates were prepared in IP lysis buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1 mM EDTA, 400 mM Na3VO4, 2.5 mM phenylmethylsulfonyl fluoride) containing 0.1% Triton X-100 and incubated in ice for 40 min. Rabbit monoclonal anti-phospho-EGFR was added to the cell lysate supernatant, which was incubated by rotation overnight at 4°C. Each sample was mixed with protein G-Sepharose and rotated at 4°C for 3 h. The mixture was washed three times using protein G-Sepharose and added to 2× SDS sample buffer. Immunoprecipitates were collected by centrifugation and subjected to 10% SDS-PAGE for Western blot analysis.
Reverse transcription-PCR (RT-PCR).
RNA was extracted with Qiazol lysis reagent (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA from each sample was transcribed to cDNA by Prime Moloney murine leukemia virus reverse transcriptase (Genet Bio, Nonsan, South Korea). The amplification was performed with EX Prime Taq DNA polymerase (Genet Bio) in a Mycycler thermal cycler (Bio-Rad Laboratories, Hercules, CA) using the following parameters: denaturation at 95°C for 5 min and 25 cycles of denaturation reaction at 95°C for 10 s, annealing at 59°C for 30 s, and elongation at 72°C for 45 s. An aliquot of each PCR product was subjected to 1% (wt/vol) agarose gel electrophoresis and visualized by staining with ethidium bromide. The 5′ forward and 3′ reverse-complement PCR primers for amplification of each gene were human IL-8 forward primer 5′-ATGACTTCCAAGCTGGCCGTGGCT-3′, IL-8 reverse primer 5′-TCTCAGCCCTCTTCAAAAACTTCTT-3′, human glyceraldehyde3-phosphate dehydrogenase (GAPDH) forward primer 5′-TCAACGGATTTGGTCGTATT-3′, and GAPDH reverse primer 5′-CTGTGGTCATGAGTCCTTCC-3′. In real-time PCR, 6-carboxyfluorescein (FAM) was used as the fluorescent reporter dye and was conjugated to the 5′ ends of the probes to detect amplified cDNA in an iCycler thermal cycler (Bio-Rad) using the following parameters: denaturation at 94°C for 2 min and 40 cycles of reactions of denaturation at 98°C for 10 s, annealing at 59°C for 30 s, and elongation at 72°C for 45 s. Each sample was tested in triplicate to ensure statistical significance. The relative quantification of gene expression was performed using the comparative threshold cycle (CT) method. The CT value is defined as the point where a statistically significant increase in the fluorescence has occurred. The number of PCR cycles (CT) required for the FAM intensities to exceed a threshold just above the background was calculated for the test and reference reactions. In all experiments, GAPDH was used as the endogenous control. Results were analyzed in a relative quantitation study with the vehicle control.
IL-8 ELISA.
IL-8 was quantified from each cell supernatant using enzyme-linked immunosorbent assay (ELISA). A549 cells (3 × 104) were dispensed in each well of a 24-well plate. After infection with P. fluorescens or signal pathway inhibitors, the medium was collected and centrifuged to remove cell debris. Levels of IL-8 were determined by ELISA using an OptEIA human IL-8 ELISA kit (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's instructions. Briefly, capture antibody was coated onto ELISA plates overnight at 4°C. After the plate was washed with phosphate-buffered saline (PBS) containing Tween 20 (PBST) and blocked with PBS supplemented with 10% (vol/vol) FBS overnight at 4°C, plates were incubated with serial dilutions of IL-8 and standards. After treatment with detection antibody and tetramethylbenzidine (TMB) substrate, absorbance was measured at 405 nm using an ELISA reader. The assay detection limit was 3.1 pg/ml of IL-8.
shRNA and transfection.
Cytomegalovirus (CMV)-driven small hairpin interference RNA (shRNA) was constructed by inserting shRNA into the pSilencer 4.1-CMV-neo vector (Ambion, Austin, TX). The empty vector and EGFR shRNA-containing vectors were constructed. EGFR shRNA targeted the sequence CTCTGGAGGAAAAGAAAGT. Cells were transfected with a mixture of plasmids using Lipofectamine 2000 (Invitrogen) or Carrigene (Kinovate Life Sciences, Oceanside, CA) reagent according to the manufacturer's protocol. Transfection efficiency was maintained at about 50 to 60%, which was confirmed with a pMX-enhanced green fluorescent protein (GFP) vector. To create stable cell lines, cells were transfected using Lipofectamine 2000 reagent. After 48 h, cells were subjected to selection for stable integrants by exposure to 1,000 μg/ml G418 (Invitrogen) in complete medium containing 10% FBS. Selection was continued until monolayer colonies were formed. The transfectants were then maintained in medium supplemented with 10% FBS and 500 μg/ml G418.
Confocal microscopy.
Cells were incubated in a glass-bottom culture dish. After treatment with P. fluorescens, cells were fixed with 4% paraformaldehyde diluted in PBS. Fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min. After 2 h of blocking with 3% bovine serum albumin (BSA) in PBS, cells were incubated with the same BSA-PBS solution containing a 1:200 dilution of rabbit polyclonal anti-phospho-EGFR (pY1068) antibody (Epitomics, Burlingame, CA) at room temperature for 1.5 h and repeatedly washed using PBS. Incubation of Alexa Fluor 488 anti-rabbit IgG (H+L) was done for 1.5 h at room temperature, followed by repeated washes using PBS. After subsequent staining with 100 ng/ml 4′,6-diamidino-2-phenylindole (DAPI) in PBS for 30 min, which has an absorbance at 405 nm, confocal images were obtained using a FV1000 confocal microscope (Olympus, Tokyo, Japan) using single-line excitation (488 nm) or multitrack sequential excitation (488 nm and 633 nm). Images were acquired and processed with FV10-ASW software. Measured EGFR represented a relative value of the ratio of total phosphorylated EGFR staining area under the infection condition minus the ratio of phosphorylated EGFR staining area under the uninfected condition per three to four cells. It was statistically analyzed from four values of phosphorylated EGFR.
Cell cycle analysis and apoptosis quantification by flow cytometry.
Trypsinized cells (5 × 105) were prepared and resuspended in 0.2 ml PBS. Following addition of 0.2 ml heat-inactivated FBS, the cells were immediately fixed by slow dropwise addition of 1.2 ml ice-cold 70% (vol/vol) ethanol with gentle mixing and were then held at 4°C overnight. The cells were washed and incubated in 1 ml propidium iodide (PI) DNA staining reagent (PBS containing 50 μg/ml PI, 50 μg/ml RNase A, 0.1 mM EDTA, and 0.1% [vol/vol] Triton X-100) on ice until they were analyzed. The cell cycle distribution for single cells was measured with a FACSCalibur apparatus (Becton Dickinson, San Jose, CA). Data from 10,000 cells were collected in the list mode. The 488-nm line of an argon laser was used to excite PI, and fluorescence was detected at 615 to 645 nm. The cell cycle of individual cells was studied using a doublet discrimination gating to eliminate doublets and cell aggregates based on the DNA fluorescence. The gate was drawn to include hypofluorescent cells. Cells in the DNA histogram with hypofluorescent DNA were designated apoptotic. All other cells distributed themselves in a normal cell cycle profile.
DNA fragmentation analysis.
DNA was extracted from the alveolar epithelial cell line. In brief, cells (2 × 106) in PBS were centrifuged (2,000 rpm) for 5 min at 4°C, and the pellet was suspended in 0.1 ml hypotonic lysis buffer (10 mM Tris, pH 7.4, 10 mM EDTA, and pH 8.0, 0.5% [vol/vol] Triton X-100). Cells were incubated for 15 min at 4°C. The resulting lysate was centrifuged (13,000 rpm) for 30 min at 4°C. The supernatant containing fragmented DNA was digested for 1 h at 37°C with 0.04 mg/ml of RNase A (Bio Basic, Markham, ON, Canada) and then incubated for an additional 1 h at the same temperature with 0.04 mg/ml of proteinase K (Sigma-Aldrich, St. Louis, MO). DNA was precipitated in 50% (vol/vol) isopropanol in 0.4 M NaCl at −20°C overnight. The precipitate was centrifuged at 13,000 rpm for 30 min at 4°C. The resultant pellet was air dried and resuspended in TE (Tris-EDTA) buffer. An aliquot equivalent to 2 × 106 cells was electrophoresed at 40 V for 3 h in a 2% (wt/vol) agarose gel in 90 mM Tris-glacial acetic acid buffer containing 2 mM EDTA (pH 8.0). After electrophoresis, the gel was stained with ethidium bromide (1 μg/ml) and nucleic acids were visualized with a UV transilluminator. A 1Kb+ DNA ladder (SolGent, Daejeon, South Korea) was used for molecular sizing.
Statistical analysis.
Data were analyzed using Sigma Stat for Windows (Jandel Scientific, San Rafael, CA). For comparative analysis of two groups of data, Student's t test was performed. For comparative analysis of multiple groups of data, data were subjected to analysis of variance (ANOVA) and pairwise comparisons were made by the Student-Newman-Keuls (SNK) method. Data not meeting the normality assumption were subjected to a Kruskal-Wallace ANOVA on ranks, and then pairwise comparisons were made by the SNK method.
RESULTS
P. fluorescens induces prolonged production of proinflammatory IL-8 via NF-κB pathway in A549 cells.
Several indoor-dwelling bacteria have been demonstrated to induce proinflammatory cytokines. In particular, upregulation of IL-8 expression by mucosal epithelial cells triggers inflammatory cascades by recruiting leukocytes in response to infection (2). Among the indoor-dwelling bacteria tested, the cytokine-stimulatory action of P. fluorescens is the most potent (13). Although P. fluorescens is not a well-defined pathogen, in the present study P. fluorescens infection enhanced IL-8 gene expression in human airway pneumocytes, such as A549 and H1299 cells (Fig. 1 A). In contrast, nonpathogenic bacteria like Escherichia coli K-12 did not induce IL-8 gene expression (see Fig. S1A in the supplemental material). IL-8 can be also induced in monocyte-derived cells by bacterial pathogen-associated molecular pattern (PAMP) molecules, including LPS. The influence of P. fluorescens and Escherichia coli LPS on IL-8 induction in A549 epithelial cells was assessed in the present study. As shown in Fig. 1B, IL-8 mRNA expression was not induced by LPS itself in A549 cells, whereas exposure to whole bacteria did enhance transcription of IL-8. P. fluorescens endotoxin-triggering TLR4 signals were not involved in IL-8 induction, but epithelial cells directly infected with bacteria showed prolonged activation of nuclear factor-kappa B (NF-κB) signals via degradation of the inhibitory molecule IκBα (Fig. 1C). In contrast, LPS showed transient activation of the NF-κB signal (Fig. 1D). It was also demonstrated that LPS-neutralizing polymyxin B only marginally decreased the IL-8 induction by P. fluorescens, indicating little contribution of bacterial LPS to IL-8 induction in human airway cells (see Fig. S1B in the supplemental material). Moreover, when the persistent NF-κB signals were suppressed by the chemical inhibitor BAY 11-7082, P. fluorescens-induced IL-8 expression was almost completely reduced to the basal level (Fig. 1E), suggesting the crucial involvement of the NF-κB signaling pathway in bacterium-induced IL-8 expression.
Fig. 1.

Prolonged induction of epithelial IL-8 by P. fluorescens. (A) A549 cells grown to 90% confluence were infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for each time period. Each mRNA was measured using RT-PCR. (B) P. fluorescens and 2 μg/ml LPS from P. fluorescens and E. coli were individually incubated with A549 cells. The amount of LPS was administered with the equivalent dose of live bacteria (CFU). Each mRNA was measured using RT-PCR. (C) A549 cells were infected with P. fluorescens for each time period. Cell lysate of each sample was subjected to Western blot analysis. (D) A549 cells were treated with LPS (dose equivalent to 50× ratio of P. fluorescens CFU per human cell number) from P. fluorescens for each time period. Cell lysate of each sample was subjected to Western blot analysis. (E) A549 cells were pretreated with 20 μM BAY 11-7082 for 2 h and infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for 12 h. IL-8 release was measured by ELISA. A different letter over each bar represents a significant difference between two group (P < 0.05).
P. fluorescens stimulates EGFR-linked signaling pathway independently of NF-κB.
Before it was verified whether EGFR-linked signals upstream of NF-κB signals are affected by P. fluorescens, A549 cells were stimulated with P. fluorescens for up to 6 h and the level of EGFR phosphorylation was analyzed by immunofluorescence confocal microscopy. Temporally increased levels of phosphorylated EGFR in lung epithelial cells were observed, with a maximal level being reached about 2 h after infection with P. fluorescens and a subsequent progressive decrease observed thereafter (Fig. 2 A and B). It was also confirmed by detecting total cellular phospho-EGFR using immunoprecipitation and Western blot analyses (Fig. 2C). However, as indicated in Fig. 1, LPS from P. fluorescens did not influence EGFR phosphorylation in A549 human pneumocyte cells.
Fig. 2.

Activation of epithelial EGFR by P. fluorescens. (A) A549 cells grown to 90% confluence were infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for each time period. EGFR was detected using confocal immunofluorescence microscopy. Cells were stained with DAPI and an α-pEGFR monoclonal antibody. (B) Densitometric analysis of immunofluorescence microscopy data. Measured EGFR represents a value described in Materials and Methods. *, significant difference from group of 0 h of incubation (P < 0.05). (C) Extracts from epithelial cells incubated with P. fluorescens or its LPS (dose equivalent to 50× ratio of P. fluorescens CFU per human cell number) were immunoprecipitated for Western blot analysis. Results are representative of those from three independent experiments.
Since EGFR-linked signals can be associated with proinflammatory responses, it was appropriate to assess the effects of perturbed EGFR signaling on P. fluorescens-induced IL-8 expression in A549 cells. When EGFR tyrosine kinase was selectively blocked by AG1478, P. fluorescens-induced IL-8 production was significantly suppressed in the lung epithelial cells (Fig. 3 A). Additionally, bacterium-induced IL-8 mRNA was reduced by the EGFR blocker (Fig. 3B), implicating signaling involvement of EGFR in pathogen-linked IL-8 expression. Moreover, interfering with EGFR expression using shRNA technology also attenuated IL-8 induction by P. fluorescens (Fig. 3C). Since NF-κB is activated by P. fluorescens infection, the signal was tested for its involvement in the EGFR-linked signaling cascade. EGFR blockage did influence NF-κB signals (Fig. 4 A), implicating the EGFR-dependent pathway of the NF-κB cascade in P. fluorescens infection. In addition to the NF-κB signaling pathway, P. fluorescens enhanced phosphorylation of EGFR protein and its downstream responsive signaling mediators, including PKB and ERK1/2, which were activated in response to P. fluorescens infection (Fig. 4B). When EGFR tyrosine kinase was inhibited, the subsequent PKB and ERK1/2 signaling activations were attenuated in bacterium-infected pneumocytes (Fig. 4C).
Fig. 3.

EGFR-independent IL-8 production and NF-κB activation. (A and B) A549 cells were pretreated with vehicle or each dose of AG1478 for 2 h and were then infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for 12 h. IL-8 production was measured by ELISA (A) and RT-PCT (B). (C) A549 cells transfected with control or EGFR shRNA plasmid were infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for 12 h. IL-8 expression was measured by RT-PCR (upper panels). Additionally, protein lysate from A549 cells infected with P. fluorescens was subjected to Western blot analysis (lower panels).
Fig. 4.

Downstream signals affected by EGFR activation by P. fluorescens. (A and C) A549 cells were pretreated with vehicle (dimethyl sulfoxide [DMSO]) or 10 μM AG1478 for 2 h and were then infected with P. fluorescens for the indicated times. Protein lysate from A549 cells infected with P. fluorescens was subjected to Western blot analysis. (B) A549 cells grown to 90% confluence were infected with P. fluorescens at a cell-to-bacterium ratio of 1:50 for each time period. Cell lysate of each sample was subjected to Western blot analysis.
EGFR-mediated signaling pathways are involved in epithelial survival response against pathogen-induced injury.
To address the roles of bacterium-activated EGFR in addition to the functions in the proinflammatory response, blocking of EGFR signals was tested for the effect on epithelial growth in the presence of P. fluorescens. Cell cycle and apoptosis analyses were carried out by flow cytometry of A549 cells infected with P. fluorescens. A 24-hour infection with P. fluorescens caused lung epithelial cell death (Fig. 5 A), which was consistent with the reported cytotoxicity of indoor-associated P. fluorescens (13). The percentage of bacterium-induced apoptotic cells was also determined in the presence of EGFR-linked signal pathway inhibitors, including AG1478 (EGFR tyrosine kinase inhibitor), LY294002 (phosphoinositide 3-kinase inhibitor), U0126 (mitogen-activated protein kinase Erk kinase 1/2 [MEK1/2] inhibitor), and BAY 11-7085 (NF-κB inhibitor). Infected cells tested with all inhibitors except for the NF-κB inhibitor showed more apoptosis than infected cells tested without inhibitor (Fig. 5A). Moreover, blocking of EGFR expression using shRNA also caused more cytotoxicity than infection with the pathogen only (Fig. 5B). Other apoptotic indexes, such as cleavage of PARP and DNA fragmentation, were analyzed. Cleavage of PARP by caspases and subsequent DNA fragmentation are characteristic features of cellular apoptosis, and the timing of apoptosis-associated events is often determined with the onset of PARP cleavage. As expected, there was an increase in PARP cleavage in P. fluorescens-infected A549 cells (Fig. 5C). The bacterium-mediated PARP cleavage was increased by inhibition of EGFR-linked signaling pathways in infected cells, consistent with a protective role of EGFR signals in P. fluorescens-insulted human cell injury. This pattern was also consistent with the DNA fragmentation results (Fig. 5D). Taken together, the data indicate that EGFR-associated signals, including those of ERK1/2 and PKB, protect human lung epithelial cells from infection-induced apoptotic injuries by P. fluorescens.
Fig. 5.

EGFR signals support epithelial survival response to P. fluorescens-induced injuries. (A, C, and D) A549 cells were pretreated with vehicle, 25 μM LY294002 (LY), 2 μM U0126, 10 μM AG1478 (AG), or 20 μM BAY 11-7082 (Bay) for 2 h and were infected with P. fluorescens for 24 h (C and D) or 48 h (A). Cellular apoptosis was measured using flow cytometric analysis (A), protein lysate analysis using Western blotting (B), and analysis of internucleosomal DNA fragmentation (C). (B) A549 cells transfected with control or EGFR shRNA plasmid were infected with P. fluorescens at a cell-to-bacterium ratio of 1:50, and cellular apoptosis was measured using flow cytometric analysis. Each panel on the right indicates data from one representative experiment. *, significant difference from infected control group (P < 0.05).
DISCUSSION
The present study supports a mechanistic link between indoor microbial exposure to P. fluorescens and airway inflammation. P. fluorescens is one of the most frequently detected bacteria in water-damaged buildings (11) and triggers an epithelial inflammatory response and cytotoxicity (11, 13). In particular, P. fluorescens infection of human alveolar epithelial cells in the present study led to survival-associated EGFR activation that elicited both the ERK1/2 and PKB pathways. Moreover, the airway bacteria also triggered proinflammatory cytokine production via the NF-κB signaling pathway in a manner related to EGFR responses (Fig. 6). Interestingly, NF-κB activation was relatively persistent, showing IκBα degradation even until 24 h. This long-term activation of the NF-κB proinflammatory signal can lead to extended inflammatory responses to P. fluorescens infection in the pulmonary system (1). According to Fig. 1, whereas NF-κB activation by isolated P. fluorescens LPS was transient (0.5 to 2 h), direct pathogen-augmented IL-8 mRNA and NF-κB activation continues to increase for up to 24 h. Released C-X-C chemokine IL-8 is a potent chemoattractant for neutrophils and has been implicated in a number of respiratory ailments, including respiratory distress syndrome, chronic obstructive pulmonary disease, and asthma (9, 30, 31). There could be several explanations for the extended activation of NF-κB in response to P. fluorescens infection. First, extended activation of NF-κB could be associated with slow growth due to a nonoptimal temperature for this microbe. Since the most optimal temperature condition for the growth of P. fluorescens is about 25 to 27°C, bacterial cells could grow slowly under a 37°C culture condition, reaching maximum bacterial numbers later than bacteria in a room temperature environment (see Fig. S2A in the supplemental material). Second, the slow increase of early triggering of the proinflammatory response by P. fluorescens may lessen feedback regulation against NF-κB by microbial infection. Third, additional proinflammatory signals may support persistent NF-κB signals in response to P. fluorescens. In order to maintain the extended NF-κB activation, human cells need several additional boosting kinases, including small GTP-dependent p21-activated kinase (25). Moreover, chronic microbial infection with long-standing inflammatory responses can also enhance these kinase signals, in addition to NF-κB stimulation (5, 19).
Fig. 6.
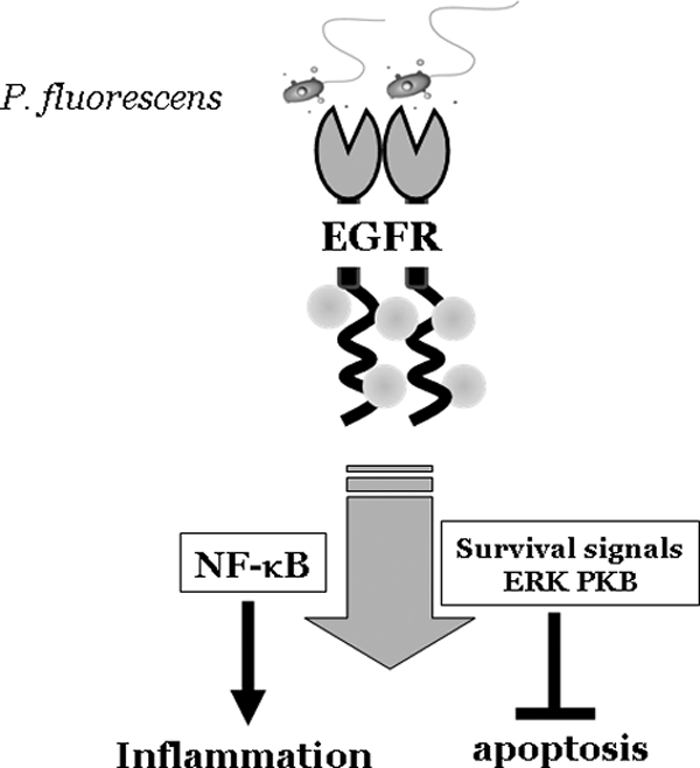
Schematic pattern of airway epithelial injury and proinflammatory responses by P. fluorescens.
It was demonstrated in the present study that inhibition of ERK and PKB activation augmented P. fluorescens-induced apoptosis. P. fluorescens infection activated EGFR and downstream signals, such as the ERK and PKB signaling pathways, which are associated with epithelial survival. In contrast, heat-killed bacteria had little or a transient effect on these signaling pathways (see Fig. S2B in the supplemental material), and additionally, the signaling inhibitors did not influence bacterial growth in the culture medium for human alveolar epithelial cells (see Fig. S1C in the supplemental material). Airway epithelia facing viable microbes can decide their fate between death and survival. Activated EGFR can support cellular survival processes by blocking apoptotic cell death from microbe-induced injuries. Moreover, bacterium-activated NF-κB stimulation can also contribute to epithelial survival as well as proinflammatory responses. However, in our data (Fig. 5A), NF-κB was not associated with an epithelial survival response to P. fluorescens infection, but instead, NF-κB-linked proinflammatory responses were dependent on EGFR-linked signals. Epithelial cell-derived NF-κB can be involved in wound-healing responses (7, 14). In particular, NF-κB promotes the reconstitution of injured epithelial cell monolayer via NF-κB target genes, including inducible nitric oxide synthase and cyclooxygenase-2, which have been extensively investigated as stimulators of epithelial cell migration (6, 22).
In summary, the present study suggests that P. fluorescens, one of the most frequently detected indoor bacteria in moisture-damaged buildings, can trigger EGFR signals and the NF-κB signaling pathway in lung alveolar epithelial cells. Activated EGFR-linked ERK1/2 and PKB support an epithelial cell survival response to P. fluorescens-induced cytotoxic injuries. EGFR activation is also associated with NF-κB signals, which are persistently activated and mediate prolonged proinflammatory IL-8 induction in the alveolar pneumocytes.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from Pusan National University (PNU; Bio-Scientific Research Grant PNU-2008-101-102).
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 22 February 2011.
REFERENCES
- 1. Ait-Ali D., et al. 2008. Tumor necrosis factor (TNF)-alpha persistently activates nuclear factor-kappaB signaling through the type 2 TNF receptor in chromaffin cells: implications for long-term regulation of neuropeptide gene expression in inflammation. Endocrinology 149:2840–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akpinar-Elci M., et al. 2008. Respiratory inflammatory responses among occupants of a water-damaged office building. Indoor Air 18:125–130 [DOI] [PubMed] [Google Scholar]
- 3. Bautista M. V., et al. 2009. IL-8 regulates mucin gene expression at the posttranscriptional level in lung epithelial cells. J. Immunol. 183:2159–2166 [DOI] [PubMed] [Google Scholar]
- 4. Ben-Bassat H., et al. 1997. Inhibitors of epidermal growth factor receptor kinase and of cyclin-dependent kinase 2 activation induce growth arrest, differentiation, and apoptosis of human papilloma virus 16-immortalized human keratinocytes. Cancer Res. 57:3741–3750 [PubMed] [Google Scholar]
- 5. Chen L. M., Bagrodia S., Cerione R. A., Galan J. E. 1999. Requirement of p21-activated kinase (PAK) for Salmonella typhimurium-induced nuclear responses. J. Exp. Med. 189:1479–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cowan M. J., Coll T., Shelhamer J. H. 2006. Polyamine-mediated reduction in human airway epithelial migration in response to wounding is PGE2 dependent through decreases in COX-2 and cPLA2 protein levels. J. Appl. Physiol. 101:1127–1135 [DOI] [PubMed] [Google Scholar]
- 7. Egan L. J., et al. 2003. Nuclear factor-kappa B activation promotes restitution of wounded intestinal epithelial monolayers. Am. J. Physiol. Cell Physiol. 285:C1028–C1035 [DOI] [PubMed] [Google Scholar]
- 8. Fukata M., et al. 2005. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G1055–G1065 [DOI] [PubMed] [Google Scholar]
- 9. Govindaraju V., et al. 2008. The effects of interleukin-8 on airway smooth muscle contraction in cystic fibrosis. Respir. Res. 9:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gram L., Melchiorsen J., Spanggaard B., Huber I., Nielsen T. F. 1999. Inhibition of Vibrio anguillarum by Pseudomonas fluorescens AH2, a possible probiotic treatment of fish. Appl. Environ. Microbiol. 65:969–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirvonen M. R., Huttunen K., Roponen M. 2005. Bacterial strains from moldy buildings are highly potent inducers of inflammatory and cytotoxic effects. Indoor Air 15(Suppl. 9):65–70 [DOI] [PubMed] [Google Scholar]
- 12. Holbro T., Hynes N. E. 2004. ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 44:195–217 [DOI] [PubMed] [Google Scholar]
- 13. Huttunen K., Hyvarinen A., Nevalainen A., Komulainen H., Hirvonen M. R. 2003. Production of proinflammatory mediators by indoor air bacteria and fungal spores in mouse and human cell lines. Environ. Health Perspect. 111:85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ishida Y., Kondo T., Kimura A., Matsushima K., Mukaida N. 2006. Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-kappaB activation and a reciprocal suppression of TGF-beta signal pathway. J. Immunol. 176:5598–5606 [DOI] [PubMed] [Google Scholar]
- 15. Jansen S., Vereecken H., Klumpp E. On the role of metabolic activity on the transport und deposition of Pseudomonas fluorescens in saturated porous media. Water Res. 44:1288–1296 [DOI] [PubMed] [Google Scholar]
- 16. Jorissen R. N., et al. 2003. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 284:31–53 [DOI] [PubMed] [Google Scholar]
- 17. Kari C., Chan T. O., Rocha de Quadros M., Rodeck U. 2003. Targeting the epidermal growth factor receptor in cancer: apoptosis takes center stage. Cancer Res. 63:1–5 [PubMed] [Google Scholar]
- 18. Koff J. L., Shao M. X., Kim S., Ueki I. F., Nadel J. A. 2006. Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J. Immunol. 177:8693–8700 [DOI] [PubMed] [Google Scholar]
- 19. Krall R., Sun J., Pederson K. J., Barbieri J. T. 2002. In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect. Immun. 70:360–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu W. A genetically engineered Pseudomonas fluorescens strain possesses the dual activity against phytopathogenic fungi and insects. J. Microbiol. Biotechnol. 20:281–286 [PubMed] [Google Scholar]
- 21. Nakanaga T., Nadel J. A., Ueki I. F., Koff J. L., Shao M. X. 2007. Regulation of interleukin-8 via an airway epithelial signaling cascade. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L1289–L1296 [DOI] [PubMed] [Google Scholar]
- 22. Noiri E., et al. 1996. Nitric oxide is necessary for a switch from stationary to locomoting phenotype in epithelial cells. Am. J. Physiol. 270:C794–C802 [DOI] [PubMed] [Google Scholar]
- 23. Olayioye M. A., Neve R. M., Lane H. A., Hynes N. E. 2000. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19:3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ono M., et al. 2004. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol. Cancer Ther. 3:465–472 [PubMed] [Google Scholar]
- 25. Orr A. W., Hahn C., Blackman B. R., Schwartz M. A. 2008. p21-activated kinase signaling regulates oxidant-dependent NF-kappa B activation by flow. Circ. Res. 103:671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng D., et al. 1996. Anti-epidermal growth factor receptor monoclonal antibody 225 up-regulates p27KIP1 and induces G1 arrest in prostatic cancer cell line DU145. Cancer Res. 56:3666–3669 [PubMed] [Google Scholar]
- 27. Petty W. J., Dragnev K. H., Dmitrovsky E. 2003. Cyclin D1 as a target for chemoprevention. Lung Cancer 41(Suppl. 1):S155–S161 [DOI] [PubMed] [Google Scholar]
- 28. Richman-Eisenstat J. B., Jorens P. G., Hebert C. A., Ueki I., Nadel J. A. 1993. Interleukin-8: an important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am. J. Physiol. 264:L413–L418 [DOI] [PubMed] [Google Scholar]
- 29. Saiman L., et al. 2003. Evaluation of MicroScan Autoscan for identification of Pseudomonas aeruginosa isolates from cystic fibrosis patients. J. Clin. Microbiol. 41:492–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Su B. H., Chiu H. Y., Lin T. W., Lin H. C. 2005. Interleukin-8 in bronchoalveolar lavage fluid of premature infants at risk of chronic lung disease. J. Formos. Med. Assoc. 104:244–248 [PubMed] [Google Scholar]
- 31. Takasaki J., Ogawa Y. 2001. Anti-interleukin-8 autoantibody in the tracheobronchial aspirate of infants with chronic lung disease. Pediatr. Int. 43:48–52 [DOI] [PubMed] [Google Scholar]
- 32. Wei B., et al. 2002. Pseudomonas fluorescens encodes the Crohn's disease-associated I2 sequence and T-cell superantigen. Infect. Immun. 70:6567–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Weller D. M., Raaijmakers J. M., Gardener B. B., Thomashow L. S. 2002. Microbial populations responsible for specific soil suppressiveness to plant pathogens. Annu. Rev. Phytopathol. 40:309–348 [DOI] [PubMed] [Google Scholar]
- 34. Yarden Y., Sliwkowski M. X. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137 [DOI] [PubMed] [Google Scholar]
- 35. Zhang J., et al. 2004. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 45:2569–2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.