

Abstract
Nerve agents are chiral organophosphate compounds (OPs) that exert their acute toxicity by phosphorylating the catalytic serine of acetylcholinesterase (AChE). The inhibited cholinesterases can be reactivated using oximes, but a spontaneous time-dependent process called aging alters the adduct, leading to resistance toward oxime reactivation. Human butyrylcholinesterase (BChE) functions as a bioscavenger, protecting the cholinergic system against OPs. The stereoselectivity of BChE is an important parameter for its efficiency at scavenging the most toxic OPs enantiomer for AChE. Crystals of BChE inhibited in solution or in cristallo with racemic V-agents (VX, Russian VX, and Chinese VX) systematically show the formation of the PS adduct. In this configuration, no catalysis of aging seems possible as confirmed by the three-dimensional structures of the three conjugates incubated over a period exceeding a week. Crystals of BChE soaked in optically pure VXR-(+) and VXS-(−) solutions lead to the formation of the PS and PR adduct, respectively. These structural data support an in-line phosphonylation mechanism. Additionally, they show that BChE reacts with VXR-(+) in the presence of racemic mixture of V-agents, at odds with earlier kinetic results showing a moderate higher inhibition rate for VXS-(−). These combined results suggest that the simultaneous presence of both enantiomers alters the enzyme stereoselectivity. In summary, the three-dimensional data show that BChE reacts preferentially with PR enantiomer of V-agents and does not age, in complete contrast to AChE, which is selectively inhibited by the PS enantiomer and ages.
Keywords: Acetylcholinesterase, Enzyme Inactivation, Enzyme Mechanisms, Enzyme Structure, Protein Structure, X-ray Crystallography, Butyrylcholinesterase, Nerve Agents, Stereoselectivity
Introduction
The acute toxicity of organophosphorus nerve agents (OPs)2 is due to rapid phosphorylation of acetylcholinesterase (AChE; EC 3.1.1.7) at the neuronal synapses and neuromuscular junctions (1, 2). What follows is an accumulation of acetylcholine that leads, among other cholinergic symptoms, to respiratory failure and even death. One strategy to prevent AChE inhibition is to scavenge the nerve agent before it can reach its synaptic target. Butyrylcholinesterase (BChE; EC 3.1.1.8), abundant in human (3), functions as a natural bioscavenger of nerve agents (4, 5). A large amount of BChE injected intravenously or intramuscularly scavenges the nerve agents and protect animals against 3–5 LD50 of soman and VX (6). BChE purified from human plasma (Baxter Healthcare Corporation) is one enzyme source. This product is under consideration for its development as a stoichiometric bioscavenger for pretreatment of OP intoxication.
The catalytic serine of cholinesterases is located at the bottom of a gorge, surrounded by pockets, named from the part of the native substrate they bind. In AChE, the acyl-binding pocket is much smaller than the choline-binding pocket. Therefore, AChE exerts a strong enantioselectivity on chiral OPs, bearing substituents of different sizes. For example, hAChE reacts about 5 × 104 times more rapidly with PS diastereoisomers of soman because the large pinacolyl and small methyl substituents fit, respectively, in the choline-binding pocket and acyl-binding pocket (7). The acyl-binding pocket of BChE is much wider than that of AChE and is therefore expected to be less selective for some OPs. This is important in regard to the amount of enzyme required to scavenge one equivalent of racemic nerve agent. Indeed, half an equivalent of BChE is sufficient if the enzyme binds preferably the same enantiomer as AChE. At least one equivalent is necessary if BChE binds equally both enantiomers or preferably the less toxic enantiomer.
Several lines of evidence suggest that BChE and hAChE have a different stereoselectivity for V-agents: VX, Russian VX (VR), and Chinese VX (CVX) (Scheme 1). One piece of evidence comes from a spontaneous time-dependent dealkylation of the V-agent adduct of cholinesterases, called aging, which leads to a resistance toward oxime reactivation (8). The dealkylation mechanism for alkoxy-OP adducts is stereoselective because it involves residues located in the choline-binding pocket of BChE. Accordingly, no aging will occur if the alkoxy substituent of V-agents is not pointing toward this pocket. A marginal aging rate is reported for VX-inhibited BChE (t½ = 77 h), and no aging could be detected for VR-inhibited BChE (9). Furthermore, mass spectrometry analysis shows that no aged adduct was detectable for BChE treated with an excess solution of racemic VX or VR (10). This suggests that for these adducts, the alkoxy substituent is usually not located in the choline-binding pocket. This contrasts with the view offered by the x-ray structure of Torpedo californica AChE inhibited by VX with the ethoxy substituent located in the choline-binding pocket and able to age (11). This leads to the conclusion that BChE and AChE have opposite stereoselectivity.
SCHEME 1.

Chemical structures of V-agents, VX, VR, and CVX.
However, measurements of the inhibition rate of separated VX isomers by independent laboratory showed that VXS-(−) inhibits BChE a few-fold faster than VXR-(+), whereas the difference in rate is more than 2 orders of magnitude for hAChE (12, 13). This in turn leads to the conclusion that both cholinesterases share the same stereoselectivity for VXS-(−) but that AChE is much more stereoselective than BChE. This is consistent with recent in vivo experiment showing that a molar equivalent of BChE is required to protect against racemic VX (14).
Interestingly, these early results puzzlingly suggest that the stereoselectivity of BChE differs when exposed to racemic or optically pure solutions of VX. In this structural study, we investigate, at the molecular level, which enantiomer of VX, VR, and CVX reacts preferentially with BChE. We determine the inhibition mechanism for both enantiomers of VX and give a rational explanation for the absence of aging in crystallography and mass spectrometry experiments.
EXPERIMENTAL PROCEDURES
Caution
V-agents (VX, CVX, and VR) are highly toxic and are classified as a schedule 1 chemical as defined in the Chemical Weapons Convention. The handling of V-agents is dangerous and requires suitable personal protection, training, and facilities.
Chemicals
Racemic VX, O-ethyl-S-[2[bis(1-methyl-ethyl)amino] ethyl] methylphosphonothioate, VR, and CVX, were obtained from the Centre d'Étude du Bouchet-Maîtrise NRBC (Vert-le-Petit, France). Optically pure enantiomers of VXR-(+) and VXS-(−) were obtained from TNO (Rijswijk, The Netherlands). For the assignment of absolute configuration R or S of VX-(+/−), see Ref. 15.
Recombinant Human Butyrylcholinesterase
BChE was expressed in Chinese hamster ovary (CHO) cells and secreted into serum-free culture medium, and purified by affinity and ion-exchange chromatography as described earlier (16). The BChE enzyme was a truncated monomer containing residues 1–529 whose tetramerization domain was deleted.
Measurement of Inhibition Rate Constants for Pure Enantiomers of VX
BChE activities were assayed according to Ellman's method (17). BChE solution (17 nm final) was mixed with VXS-(−) or VXR-(+) (respectively, 21 and 88 nm final) in 100 mm phosphate buffer, pH 8.0. Each minute, 5 μl of the mixture was added to a well (96-well plate) filled with 100 μl of 0.8 mm 5,5′-dithiobis-(2-nitrobenzoic acid) in 100 mm phosphate buffer, pH 8.0. After the 7th aliquot (7 min), the wells were filled with 100 μl of 0.8 mm butyrylthiocholine in water. Absorbance was read at 412 nm immediately, then 5 min later. The net raise of absorbance is a direct measure of BChE activity. The initial concentration of VXS-(−) is less than 2-fold the initial concentration of BChE which means that the inhibition follows second-order kinetics. The inhibition rate kiS of BChE by VXS-(−) was determined with the following equation,
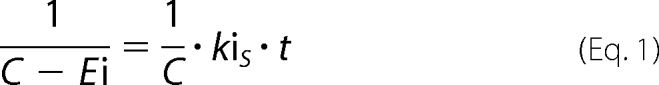 |
where C is the average concentration of enzyme and inhibitor (enzyme+inhibitor/2), Ei is the concentration of the inhibited enzyme, kiS is the inhibition rate, and t is time.
The initial concentration of VXR-(+) is more than 5 times the initial concentration of BChE which means that the inhibition follows pseudo-first-order kinetics. The inhibition rate kiR of BChE by VXR-(+) was determined with the following equation,
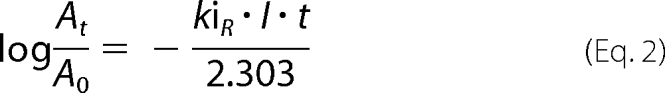 |
where At is the activity of human BChE at time t and A0 at time 0, I is the initial concentration of VXR-(+), kiR is the inhibition rate, and t is time.
Crystals of V-agent-inhibited BChE Conjugates
BChE crystallized at a concentration of 8 mg/ml from 0.1 m MES, pH 6.5, supplemented with 2.1 m ammonium sulfate, using the hanging-drop system. The VX, CVX, and VR stock solutions were at 10 mm in 2-propyl alcohol. Crystals of conjugates were obtained using three different procedures.
Flash Soaking
Crystals of native BChE were flash-soaked into a solution containing 1 mm racemic V-agents or pure enantiomers VX for 5 min and flash-cooled in liquid nitrogen to prevent postinhibition reactions such as aging and/or spontaneous reactivation. Three soaking solutions were tested for racemic V-agents, from either 0.1 m MES, pH 6.5, or 0.1 m phosphate buffer, pH 7.4 or pH 8.0.
Long Soaking
Crystals were long-soaked into a solution containing 1 mm racemic V-agents for 10 days to allow sufficient time for in cristallo aging.
Crystallization after Inhibition
Crystals were obtained from a solution of BChE first inhibited by 0.4 mm racemic V-agents in 5 mm MES, pH 6.5. In all three cases, the crystals were washed with a cryoprotectant solution (0.1 m MES, pH 6.5, with 2.3 m ammonium sulfate, containing 20% glycerol) and then flash-cooled in liquid nitrogen.
X-ray Data Collection and Structure of V-agent-BChE Conjugates
Diffraction data were collected at the European Synchrotron Radiation Facility (ESRF, Grenoble, France), at the ID29, ID23-1, ID14-1, ID14-2, and ID14-4 beam lines. All datasets were processed with XDS (18). The structures were solved by use of the CCP4 suite (19). An initial solution model was determined by molecular replacement, starting from the recombinant BChE structure (Protein Data Bank (PDB) entry 1P0I) from which all ligands (butyrate, glycerol, ions) and glycan chains were removed. For all diffraction data sets, the model was refined with REFMAC5 (20). An initial rigid body refinement was followed by iterative cycles of model building with Coot (21), and then restrained and TLS refinement was carried out with REFMAC5. The bound ligands and their descriptions were built using the Dundee PRODRG 2.5 server including energy minimization using GROMOS 96.1 force field.
Significant drops in R-factor and Rfree occurred with TLS refinement. TLS groups were defined with the help of the TLS Motion Determination server (22). Refined TLS parameters are included in the deposited PDB file for each entry. Simulated annealing composite omit maps were calculated using Phenix (23) to check any bias in the model. Protein structures were illustrated using the program PyMOL.
RESULTS
Inhibition Rate Constants for Optically Pure VX Enantiomers
The bimolecular rate constant for the inhibition of recombinant BChE is 1.09 ± 0.07 × 107 min−1 · m−1 (n = 12) for VXS-(−) and 2.01 ± 0.10 × 106 min−1 · m−1 (n = 12) for VXR-(+). Thus, VXS-(−) is only 5.4-fold more potent than VXR-(+), in good agreement with literature data (12, 13).
X-ray Structure of Racemic V-agent-BChE Conjugates
We followed three different procedures to obtain the crystal structures of the conjugates. The flash-soaking procedure was aimed at obtaining the conjugate structure before any aging or spontaneous reactivation can take place. The long-soaking procedure intended to obtain the structure of the aged conjugate, and the procedure using inhibition in solution before crystallization was aimed at avoiding a possible enantioselectivity or aging bias introduced by the crystal packing. Still, we cannot exclude that a bias could persist during crystallization, if for example one enantiomeric conjugate crystallizes more favorably.
Whatever the procedure used, we systematically obtained identical conjugate structures with all three racemic V-agents, without any evidence for aging. The crystals belonged to the usual space group I422. The structures were refined at resolutions ranging from 2.1 to 2.6 Å. Data and refinement statistics for one dataset per V-agent are presented in Table 1. For each structure, a strong peak of electron density (>11σ) is observed in the Fo − Fc map at covalent bonding distance of the catalytic serine Oγ, in agreement with the presence of the bound inhibitor. The oxygen of the phosphonate moiety is nested in the oxyanion hole, well stabilized by three hydrogen bonds with the main chain amide nitrogen of Gly-116, Gly-117, and Ala-199 (Fig. 1).
TABLE 1.
X-ray data collection and refinement statistics
| Enzyme | Racemate |
Pure enantiomer |
|||
|---|---|---|---|---|---|
| VX in cristallo | VR in solution | CVX in solution | VXS-(−) | VXR-(+) | |
| PDB entry code | 2XQF | 2XQG | 2XQI | 2XQK | 2XQJ |
| Data collection | |||||
| Space group | I422 | I422 | I422 | I422 | I422 |
| Unit cell axes, a = b, c (Å) | 155.1 128.1 | 154.6 127.6 | 155.2 127.0 | 154.9 127.4 | 155.8 128.3 |
| X-ray source | ID14-eh4 (λ = 0.981) | ID14-eh1 (λ = 0.933) | ID14-eh2 (λ = 0.933) | ID23-eh1 (λ = 0.954) | |
| No. of reflections | 409,037 | 249,879 | 223,406 | 214,798 | 284,598 |
| Unique reflections | 45,286 | 34,420 | 23,438 | 30,395 | 31,042 |
| Resolution (Å) | 48.0-2.1 (2.5-2.1) | 41.5-2.3 (2.5-2.3) | 49.1-2.6 (2.9-2.6) | 41.5-2.4 (2.5-2.4) | 49.3-2.4 (2.5-2.4) |
| Completeness (%) | 99.2 (98.5) | 99.7 (99.6) | 96.0 (97.7) | 99.5 (99.6) | 99.9 (99.9) |
| Rmeasa (%) | 6.8 (30.8) | 7.2 (48.8) | 15.5 (50.4) | 7.9 (48.9) | 8.1 (41.5) |
| I/ó(I) | 22.3 (7.9) | 27.3 (4.8) | 9.9 (4.1) | 20.5 (5.3) | 19.9 (6.2) |
| Redundancy | 9.0 (8.7) | 7.3 (7.4) | 9.5 (9.0) | 7.1 (6.9) | 9.2 (9.7) |
| Refinement statistics | |||||
| R-factorb (Rfree)c | 15.0 (18.9) | 16.2 (21.4) | 18.4 (24.7) | 15.7 (21.7) | 16.7 (22.3) |
| No. of atoms | |||||
| Protein | 4269 | 4258 | 4258 | 4265 | 4246 |
| Solvent | 432 | 419 | 247 | 323 | 346 |
| Others | 193 | 185 | 186 | 183 | 161 |
| Mean B-factor (Å2) | 37.1 | 38.1 | 54.0 | 41.2 | 39.7 |
| Root mean square deviation from ideality | |||||
| Bond length (Å) | 0.030 | 0.023 | 0.020 | 0.022 | 0.022 |
| Angles (deg) | 2.291 | 2.051 | 1.877 | 1.954 | 1.976 |
| Chiral (Å3) | 0.207 | 0.144 | 0.127 | 0.136 | 0.139 |
a Rmeas as defined in Ref. 31.
b R-factor = Σ |Fo − |Fc‖ / Σ |Fo|, Fo, and Fc are observed and calculated structure factors.
c Rfree set uses about 1000 of randomly chosen reflections.
FIGURE 1.

Active site of VX-BChE (A), VR-BChE (B), and CVX-BChE (C) conjugates. Crystals are obtained either by long soaking of BChE crystal in a 1 mm solution of racemic VX (A) or by crystallization of a solution containing 0.1 mm BChE inhibited by 0.4 mm of racemic VR (B) or CVX (C). Key residues are represented by sticks with carbon atoms in blue, oxygen atoms in red, nitrogen atoms in dark blue, and phosphorus atom in orange. Hydrogen bonds are represented by black dashes, and atomic distances are indicated in red (Å). Omit maps are represented in green mesh, contoured at 3.0 σ.
For all conjugates, the methyl group of the phosphonyl adducts points toward the catalytic histidine whereas the alkoxy substituent is located in the acyl-binding pocket delimited by Trp-231, Leu-286, and Val-288 (Fig. 1). There is no doubt about the configuration assignment. The electron density maps show no evidence for the presence of the alternate configuration, i.e. the alkoxy group pointing toward the choline-binding pocket.
Adjustment of the acyl loop residues is observed, depending on the bulkiness of the alkoxy substituent (Fig. 2). Compared with the ethoxy substituent in the VX adduct, the isobutyloxy group of the VR adduct induces a 0.7 Å shift of Leu-286, and the n-butyloxy group of CVX adduct induces a 0.4-Å shift of Val-288 and 0.8-Å shift of Leu-286. Rearrangement of the acyl loop conformation was already observed for soman (24) and phosphoramidyl adducts (25). In addition, strain in the acyl-binding pocket translates into a slightly different position of the phosphonyl head for VX and CVX. The phosphorus atom shifts by 0.3 Å away from the pocket, and the phosphonyl moiety rotates about 20° around the SerOγ-P bond (Fig. 2). No other significant displacement of residues is observed compared with the native enzyme (PDB entry 1P0I).
FIGURE 2.

Superimposition of the acyl-binding pocket region of VX-BChE (cyan), VR-BChE (green), and CVX-BChE (magenta) conjugates. Key residues are represented by sticks with oxygen atoms in red, nitrogen atoms in dark blue, phosphorus atoms in orange. Cα atoms are represented as spheres.
Thus, the absolute configuration of the phosphorus atom for each adduct is PS. The orientation of VX is identical to that observed in the recently solved structure of the VX-G117H mutant of BChE (PDB entry 2XMG) (26). This is the mirror image of the VX-TcAChE adduct which is of configuration PR: in this latter, the methyl substituent is located in the acyl-binding pocket, and the ethoxy group points toward the catalytic histidine inducing a conformational change (11).
The formation of the PS adduct could result either from an in-line attack of VXR-(+) with inversion of the phosphorus or an adjacent attack of VXS-(−) and subsequent pseudorotation (27). In an effort to understand which VX enantiomer leads to the formation of the PS adduct and what is the underlying mechanism, we solved the x-ray structure of BChE crystals flash-soaked in solutions containing pure enantiomers.
X-ray Structure of BChE Inhibited by Pure VX Enantiomers
The structures of VXR-(+) and VXS-(−) conjugates were solved to 2.4-Å resolution. Data and refinement statistics for one dataset per isomer are presented in Table 1.
The ethoxy substituent of VXR-(+)-BChE conjugate is located in the acyl-binding pocket whereas the methyl points toward the catalytic histidine (Fig. 3A). The phosphorus atom is of absolute configuration PS. This conformation is identical to that obtained by inhibition using a racemic mixture and in agreement with in-line phosphorylation (Scheme 2).
FIGURE 3.

Active site of VXR-(+)-BChE (A) and VXS-(−)-BChE (B) conjugates. BChE crystals were flash-soaked in a 1 mm solution of VXR-(+) (A) or VXS-(−) (B). Key residues are represented by sticks with carbon atoms in blue, oxygen atoms in red, nitrogen atoms in dark blue, phosphorus atoms in orange. Hydrogen bonds are represented by black dashes, and atomic distances are indicated in red (Å). Omit maps are represented by a green mesh, contoured at 3.0 σ.
SCHEME 2.

Mechanism of inhibition of BChE by VXR-(+) (A) and VXS-(−) (B).
By contrast, the ethoxy substituent of VXS-(−)-BChE points toward the catalytic histidine whereas the methyl group is located in the acyl-binding pocket (Fig. 3B). The phosphorus atom is of absolute configuration PR, in agreement with in-line phosphorylation (Scheme 2). The two residues known to promote aging by dealkylation are close to the ethoxy group, His-438-Nϵ and the ethoxy oxygen being notably at H-bond distance (3.2 Å). This configuration is identical to that observed when TcAChE was inhibited by racemic VX, except that no conformational change of the catalytic histidine is observed. The absence of conformational change of His-438 shows one more time that this catalytic residue is not mobile in BChE (25). These combined results suggest that in the presence of both enantiomers at submillimolar concentration, BChE will react preferentially with VXR-(+).
DISCUSSION
We do not observe identical stereoselectivity for experiments performed in the presence of one or both enantiomers of V-agents. On the one hand, three different experiments using racemic VX, i.e. in vivo protection (14), mass spectrometry (10), and x-ray crystallography, show that BChE reacts preferentially with VXR-(+) and does not age. On the other hand, kinetic experiments performed with separated VXR-(+) or VXS-(−) show a slight selectivity for VXS-(−). Slow aging has been observed with VXS-(−), t½ ≈ 50 h, and no aging could be detected with VXR-(+).3 The absence of aging with VXR-(+) is in agreement with the structural and mass spectrometry data. Noteworthy, very slow aging has been recently reported for BChE inhibited by racemic VX, t½ = 77 h (9). This half-time value seems to be intermediate between those of the VXR-(+) and the VXS-(−) conjugates.
There can be many different explanations for this discrepancy. A first hypothesis is that a shift in selectivity originates from the ability to bind multiple molecules at the same time in the active site gorge, especially at high concentrations. This is illustrated in the x-ray structure of aged soman-BChE conjugate in complex with butyrylthiocholine (24). Multiple binding affects the catalytic behavior of the enzyme. For example, at a high substrate concentration, binding of a second molecule of butyrylthiocholine in the active site gorge of BChE accelerates the turnover about three times (28). Thus, it is possible that in the presence of a racemic mixture, both enantiomers bind simultaneously in the active site gorge and interfere so that VXR-(+) is in a position more favorable for the phosphonylation. This may be favored by the high concentrations of VX required for x-ray crystallography and mass spectrometry. Aging kinetic of BChE inhibited by racemic VX could provide some clues regarding this hypothesis, but the exact concentration of inhibitor was not reported (9). Although unexplained electron density is visible in the active site gorge of the conjugate, next to Trp-82, it does not correspond to a second molecule of V-agents in which P and S atoms large electron density is easily recognizable. An alternative hypothesis is that an unknown ligand corresponding to this unexplained electron density, often present in the choline-binding pocket of BChE (24–26), could alter the binding of VXS-(−). Yet another hypothesis could be that even if both conjugates form in solution, the VXR-(+)-BChE conjugate crystallizes more easily. In this case there would be a crystallization artifact for inhibited BChE. But this hypothesis also requires that there should be also a crystal packing artifact, VXR-(+) binding more favorably to crystallized BChE. At this point we have no evidence to favor one of these nonmutually exclusive hypotheses.
Predicting the stereoselectivity of BChE for VR and CVX uniquely from the active site topology is not straightforward. The question is whether the large substituent of VR and CVX can fit in the acyl-binding pocket of BChE. Some clues were provided by structural data showing that a diethylamino group (TA1) or a N-propylamino group (TA6) do fit in that pocket (25). Here, the x-ray structures of VR- and CVX-BChE confirm that the long n-butyloxy of CVX or the bulky isobutyloxy of VR do fit as well. This explains why no aging was observed during mass spectrometry analysis of the VR-BChE conjugate or kinetics experiments (9). By extrapolation from VX, the PS adducts results likely from the in-line attack by VRR and CVXR, whereas we expect a strong selectivity of human AChE for VRS and CVXS, knowing that VXS-(−) is at least already 100-fold more reactive than VXR-(+) (12, 13).
From a stereoselectivity point of view, hAChE appears as a better V-agents bioscavenger than hBChE. However, titration studies show that one equivalent of hAChE or hBChE is necessary to neutralize one equivalent of racemic VX in conditions simulating the estimated initial concentrations of VX during in vivo experiments (13). Actually, the phosphonylation rate of both VX enantiomers is higher than 106 m−1 · min−1 for both enzymes, so that the incubation time over 30 min was too long to discriminate their behavior. However, in vivo experiment shows that administration of hAChE gives a better survival rate of VX-exposed mouse (13).
Regarding the stereoselectivity of cholinesterases for G-agents tabun and soman, it has been established that human BChE and human AChE share the same selectivity (24, 29). Regarding sarin, bovine AChE reacts more than 4 × 103-fold faster with sarinS-(−), the isopropyloxy substituent being too bulky for the AChE acyl-binding pocket, whereas the enantiomers inhibit horse serum BChE with virtually equal rates (30). Indeed, the structural data presented here and elsewhere show that the acyl-binding pocket of BChE can accommodate quite large substituents like isobutyloxy (VR) or diethylamino (TA1) (25). Moreover, mass spectrometry shows that the sarin-BChE conjugate does age by dealkylation, albeit partially (10). Aging by dealkylation suggests that the isopropyloxy substituent points toward His-438, i.e. the absolute configuration of the adduct is R, which in turn means that the enzyme reacted with sarinS-(−). Incomplete aging could results either from insufficient reaction time or a significant portion of the enzyme inhibited by sarinR-(+). In the latter case, both the kinetic experiment with horse serum BChE and the mass spectrometry analysis with human BChE show that these enzymes are not sufficiently enantioselective for sarin by contrast to AChE. This remains to be eventually supplemented by a structural study.
This work was supported by Direction Générale de l'Armement under Programme d'Etude Amont Grants 08co501, ANR-06-BLAN-0163, and ANR-09-BLAN-0192 (to F. N.) and Defense Threat Reduction Agency Grant CBDIF07-THER01-2-0038 (to D. N., F. N., and M. G.).
The atomic coordinates and structure factors (codes 2XQF, 2XQG, 2XQI, 2XQJ, and 2XQK) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
F. Worek, unpublished data.
- OP
- organophosphate
- AChE
- acetylcholinesterase
- BChE
- butyrylcholinesterase
- CVX
- Chinese VX
- h
- human
- PDB
- Protein Data Bank
- VR
- Russian VX
- TLS
- translation libration screw motion.
REFERENCES
- 1. Holmstedt B. (1959) Pharmacol. Rev. 11, 567–688 [PubMed] [Google Scholar]
- 2. Silman I., Sussman J. L. (2005) Curr. Opin. Pharmacol. 5, 293–302 [DOI] [PubMed] [Google Scholar]
- 3. Li B., Stribley J. A., Ticu A., Xie W., Schopfer L. M., Hammond P., Brimijoin S., Hinrichs S. H., Lockridge O. (2000) J. Neurochem. 75, 1320–1331 [DOI] [PubMed] [Google Scholar]
- 4. Saxena A., Sun W., Luo C., Myers T. M., Koplovitz I., Lenz D. E., Doctor B. P. (2006) J. Mol. Neurosci. 30, 145–148 [DOI] [PubMed] [Google Scholar]
- 5. Masson P., Lockridge O. (2010) Arch. Biochem. Biophys. 494, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lenz D. E., Maxwell D. M., Koplovitz I., Clark C. R., Capacio B. R., Cerasoli D. M., Federko J. M., Luo C., Saxena A., Doctor B. P., Olson C. (2005) Chem. Biol. Interact. 157–158, 205–210 [DOI] [PubMed] [Google Scholar]
- 7. Ordentlich A., Barak D., Kronman C., Benschop H. P., De Jong L. P., Ariel N., Barak R., Segall Y., Velan B., Shafferman A. (1999) Biochemistry 38, 3055–3066 [DOI] [PubMed] [Google Scholar]
- 8. Masson P., Nachon F., Lockridge O. (2010) Chem. Biol. Interact. 187, 157–162 [DOI] [PubMed] [Google Scholar]
- 9. Aurbek N., Thiermann H., Eyer F., Eyer P., Worek F. (2009) Toxicology 259, 133–139 [DOI] [PubMed] [Google Scholar]
- 10. Li H., Schopfer L. M., Nachon F., Froment M. T., Masson P., Lockridge O. (2007) Toxicol. Sci. 100, 136–145 [DOI] [PubMed] [Google Scholar]
- 11. Millard C. B., Koellner G., Ordentlich A., Shafferman A., Silman I., Sussman J. L. (1999) J. Am. Chem. Soc. 121, 9883–9884 [Google Scholar]
- 12. Reiter G., Mikler J., Hill I., Weatherby K., Thiermann H., Worek F. (2008) J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 873, 86–94 [DOI] [PubMed] [Google Scholar]
- 13. Cohen O., Kronman C., Raveh L., Mazor O., Ordentlich A., Shafferman A. (2006) Mol. Pharmacol. 70, 1121–1131 [DOI] [PubMed] [Google Scholar]
- 14. Kasten S. A., Kajih T., Smith J. R., Oliver Z., Otto T. C., Reeves T. E., Lenz D. E., Cerasoli D. M. (2010) in Proceedings of the 2010 Medical Defense Bioscience Review, Hunt Valley, MD, May 24–27, 2010, p. 46, U.S. Army Medical Research and Development Command, Washington, DC [Google Scholar]
- 15. Hall C. R., Inch T. D., Inns R. H., Muir A. W., Sellers D. J., Smith A. P. (1977) J. Pharm. Pharmacol. 29, 574–576 [DOI] [PubMed] [Google Scholar]
- 16. Nachon F., Nicolet Y., Viguié N., Masson P., Fontecilla-Camps J. C., Lockridge O. (2002) Eur. J. Biochem. 269, 630–637 [DOI] [PubMed] [Google Scholar]
- 17. Ellman G. L., Courtney K. D., Andres V., Jr., Feather-Stone R. M. (1961) Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- 18. Kabsch W. (2010) Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collaborative Computational Project 4 (1994) Acta Crystallogr. D Biol. Crystallogr. 50, 760–76315299374 [Google Scholar]
- 20. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. Painter J., Merritt E. A. (2006) Acta Crystallogr. D Biol. Crystallogr. 62, 439–450 [DOI] [PubMed] [Google Scholar]
- 23. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 24. Nicolet Y., Lockridge O., Masson P., Fontecilla-Camps J. C., Nachon F. (2003) J. Biol. Chem. 278, 41141–41147 [DOI] [PubMed] [Google Scholar]
- 25. Carletti E., Aurbek N., Gillon E., Loiodice M., Nicolet Y., Fontecilla-Camps J. C., Masson P., Thiermann H., Nachon F., Worek F. (2009) Biochem. J. 421, 97–106 [DOI] [PubMed] [Google Scholar]
- 26. Nachon F., Carletti E., Wandhammer M., Nicolet Y., Schopfer L. M., Masson P., Lockridge O. (2011) Biochem. J. 434, 73–82 [DOI] [PubMed] [Google Scholar]
- 27. Nachon F., Carletti E., Worek F., Masson P. (2010) Chem. Biol. Interact. 187, 44–48 [DOI] [PubMed] [Google Scholar]
- 28. Masson P., Legrand P., Bartels C. F., Froment M. T., Schopfer L. M., Lockridge O. (1997) Biochemistry 36, 2266–2277 [DOI] [PubMed] [Google Scholar]
- 29. Carletti E., Colletier J. P., Dupeux F., Trovaslet M., Masson P., Nachon F. (2010) J. Med. Chem. 53, 4002–4008 [DOI] [PubMed] [Google Scholar]
- 30. Boter H. L., van Dijk C. (1969) Biochem. Pharmacol. 18, 2403–2407 [DOI] [PubMed] [Google Scholar]
- 31. Diederichs K., Karplus P. A. (1997) Nat. Struct. Biol. 4, 269–275 [DOI] [PubMed] [Google Scholar]