

Abstract
Acute pancreatitis is an inflammation of the pancreas that may lead to systemic inflammatory response syndrome and death due to multiple organ failure. Acinar cells, together with leukocytes, trigger the inflammatory cascade in response to local damage of the pancreas. Amplification of the inflammatory cascade requires up-regulation of pro-inflammatory cytokines and this process is mediated not only by nuclear factor κB but also by chromatin modifying complexes and chromatin remodeling. Among the different families of histone acetyltransferases, the p300/CBP family seems to be particularly associated with the inflammatory process. cAMP activates gene expression via the cAMP-responsive element (CRE) and the transcription factor CRE-binding protein (CREB). CREB can be phosphorylated and activated by different kinases, such as protein kinase A and MAPK, and then it recruits the histone acetyltransferase co-activator CREB-binding protein (CBP) and its homologue p300. The recruitment of CBP/p300 and changes in the level of histone acetylation are required for transcription activation. Transcriptional repression is also a dynamic and essential mechanism of down-regulation of genes for resolution of inflammation, which seems to be mediated mainly by protein phosphatases (PP1, PP2A and MKP1) and histone deacetylases (HDACs). Class II HDACs are key transcriptional regulators whose activities are controlled via phosphorylation-dependent nucleo/cytoplasmic shuttling. PP2A is responsible for dephosphorylation of class II HDACs, triggering nuclear localization and repression of target genes, whereas phosphorylation triggers cytoplasmic localization leading to activation of target genes. The potential benefit from treatment with phosphodiesterase inhibitors and histone deacetylase inhibitors is discussed.
Keywords: Dual specificity protein phosphatases, Acute pancreatitis, Phosphodiesterase inhibitors, Cytokines, Histone acetylation, Pentoxifylline, PP2A, Serine/threonine protein phosphatases
PATHOPHYSIOLOGY OF ACUTE PANCREATITIS
Acute pancreatitis (AP) is an inflammation of the pancreas that often leads to systemic inflammatory response and complications. The mortality rate is approximately 5% but it rises to 17% in cases of severe AP[1]. Mortality in AP is due to multiple organ failure induced by the systemic inflammatory response.
The major causes of the disease are alcohol and gallstones. Nevertheless, the precise mechanisms triggered by the etiological factors to induce tissue injury and an attack of AP are still a matter of debate. An early event in AP is the intrapancreatic activation of trypsinogen and other zymogen enzymes[2]. However, pancreatic digestive enzymes are not responsible for the conversion of a local inflammatory process into a systemic inflammatory response. Numerous inflammatory mediators, such as pro-inflammatory cytokines, chemokines, free radicals, Ca2+, platelet activating factor, and adenosine, have been involved in the pathogenesis of AP and its derived systemic inflammatory response[3-5]. Therefore, AP should be considered another pathological condition together with sepsis, trauma, burns and surgery, which may lead to the systemic inflammatory response syndrome and multiple organ failure.
AP is characterized by interstitial edema and inflammatory infiltrate of macrophages and neutrophils. Nevertheless, acinar cells also behave as inflammatory cells producing and releasing cytokines, chemokines and adhesion molecules[3,6,7]. Hence, acinar cells together with leukocytes trigger the inflammatory cascade in response to local damage of the pancreas. Tumor necrosis factor α (TNF-α) and interleukin 1-β initiate and propagate almost all the consequences of the systemic inflammatory response in AP[8]. Interleukin-6 (IL-6) is a mediator of the acute-phase response and also a marker of severity of AP[9]. The role of cytokines in AP has already been extensively reviewed[4,5].
Amplification of the inflammatory cascade requires up-regulation of pro-inflammatory cytokines and this process is mediated mainly by nuclear factor κB (NF-κB). Elucidation of the role of chromatin modifying complexes and chromatin remodeling in the inflammatory cascade is currently underway and is the focus of the present review.
CHROMATIN MODIFYING COMPLEXES
Nucleosomes, the basic units of eukaryotic chromatin, consist of a histone core with two copies of each core histones H2A, H2B, H3 and H4 and approximately 147 bp of DNA wrapped around the histone octamers. The nucleosomes are structured along the DNA polymer as “beads on a string”, further compacted in the presence of the linker histone H1 to 30 nm solenoid, and finally to the metaphasic chromosome. Nevertheless, the chromatin structure creates an obstacle for the transcriptional machinery, as transcription factors must gain access to the compact DNA template. In order to bypass the transcriptional constrictions, cells have organized the binding of transcriptional activators that target chromatin modifier enzymes to the promoters allowing changes in the chromatin structure and accessibility.
The chromatin modifier complexes covalently modify the histones, either in the histone tails (acetylation, phosphorylation and methylation) or outside the histone tails (ubiquitylation, glycosylation, SUMOylation and ADP-ribosylation), changing the overall charge or hydrophobicity of histones, and modifying the chromatin structure[10,11]. This review focuses on the dynamics of histone acetylation in acute inflammation, particularly in AP.
The combined action of histone acetyltransferases (HAT) and histone deacetylases (HDAC) maintains, through a subtle balance, the steady-state levels of histone acetylation. HAT are enzymes that catalyze the transfer of an acetyl group from acetyl-coenzyme A to the ε-amino groups of conserved lysine residues mainly in histone N-terminal tails. This process is generally linked to transcriptional activity[12] as the neutralization of the basic charge of the histone tails favours the transcription process[13]. On the other hand, HDAC are enzymes that remove acetyl groups from lysine residues of histones, and other non-histone proteins, and reverse the action of HAT[14].
Among the different families of HAT[15,16], the p300/CBP family seems to be particularly associated with the inflammatory process. It comprises the highly related p300 and CBP proteins that have a bromo-domain and three cysteine/histidine rich domains that serve as protein-protein interacting domains. Members of this family are co-activators for multiple transcription factors and they can also associate with other acetyltransferases, showing that multiple HAT enzymes can be recruited to the promoter to act synergistically during gene activation.
HDACs have been classified into four different subfamilies (I, II, III and IV). Class I and II HDACs contain a zinc cation in the catalytic site and they are sensitive to the inhibitor trichostatin A[17], whereas class III HDACs are insensitive to TSA treatment and require the coenzyme NAD+ as a cofactor[18]. HDAC11 is so far the only class IV member. HDACs have to be localized in the nucleus to be active towards histones, but some of them (class II and III HDACs) can also be found in the cytosol. Nuclear targeting of HDACs takes place by a nuclear localisation signal or by interacting with other proteins that translocate them to the nucleus[19]. For instance, HDAC1, HDAC2 and HDAC3 are exclusively nuclear, while HDAC 4, -5 and -7 are able to shuttle in and out of the nucleus in response to cellular stimuli[20,21].
The lysine and arginine residues of histones can be further modified by methylation. Histone methylation has been associated with both transcriptional activation and repression depending on the methylated residue, the degree of methylation and on the interplay with other modifications[11].
Histone H3 may be subject to MAP kinase-mediated phosphorylation in serine 10 and 28 by mitogen- and stress-activated protein kinases MSK1 and MSK2, which trigger transcription of immediate-early genes[22]. Other histones may also be phosphorylated leading to transcriptional activation.
TRANSCRIPTION ACTIVATION IN THE INFLAMMATORY CASCADE IS MEDIATED BY HISTONE ACETYLATION AND METHYLATION
cAMP activates gene expression via the cAMP-responsive element (CRE) and the transcription factor CRE-binding protein (CREB). CREB can be phosphorylated and activated by different kinases, such as protein kinase A, MAPK and CaMKIV[23], and then it recruits the HATs co-activator CREB-binding protein (CBP) and its homologue p300. The recruitment of CBP/p300 and histone acetylation is required for transcription activation[24]. CREB activation via phosphorylation and subsequent CREB-mediated gene expression seem to play an important role in the inflammatory cascade (Figure 1). Thus, CREB phosphorylation by protein kinase C-theta and DNA-CREB binding are required for up-regulation of IL-2 in T-cells[25].
Figure 1.
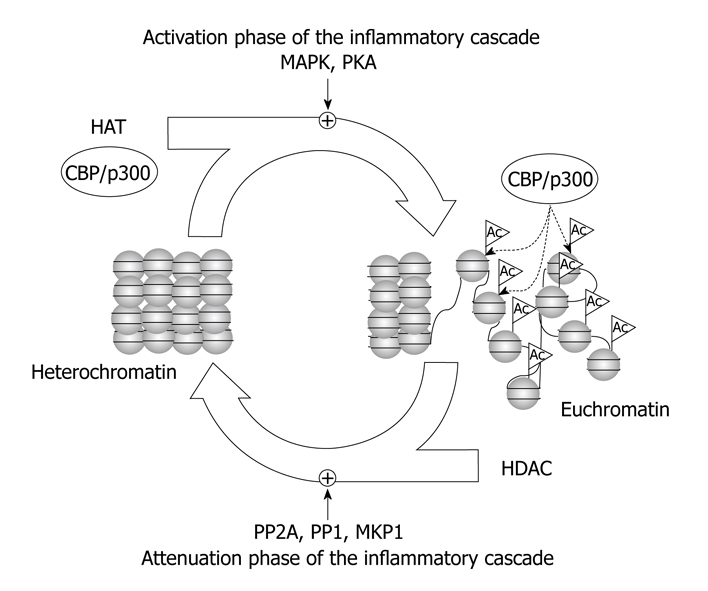
Role of histone acetyltransferases (HATs) and histone deacetylases (HDACs) in the regulation of the inflammatory cascade. HATs and HDACs play central roles, together other chromatin modifying complexes, such as histone methyltransferases, DNA methyltransferases, methyl DNA binding proteins, and heterochromatin proteins, in the activation and attenuation phases of the inflammatory cascade.
CBP and p300 are co-activators of (NF-κB). Accordingly, NF-κB-induced gene transcription is mediated by histone acetylation. NF-κB is the major mediator of TNF-α-induced IL-6 gene expression, which requires CBP/p300 histone acetyltransferase activity[26]. Similar findings were reported for other NF-κB-driven promoters, such as those of IL-8 and endothelial leukocyte adhesion molecule. p300 is also required for transcriptional activation of cyclooxygenase-2 (COX-2) by interleukin-1 β or lipopolysaccharide in macrophages[27].
The E-selectin gene is rapidly and transiently expressed by endothelial cells upon inflammation and it promotes binding and extravasation of leukocytes from the bloodstream. TNF-α induces NF-κB (p65) binding together with histone hyperacetylation via p300/CBP in the E-selectin gene in endothelial cells[28].
Histone H3 acetylation (H3K9 and H3K14), as well as histone H3 methylation (H3R17), are regulated at the promoters of NF-κB-target genes in a CBP/p300 dependent manner[29]. Coactivator-associated arginine methyltransferase-1 (CARM1) is recruited to N-κB-target promoters and participates in NF-κB-mediated transcription through H3 methylation at arginine 17 (H3R17)[29].
STAT 3 is the major signal transducer of IL-6 and hence it mediates acute phase protein induction. IL-6-induced angiotensinogen expression is mediated by association of STAT3 with p300/CBP to trigger histone acetylation and chromatin remodeling[30]. Recently, it has been confirmed that STAT3 transactivates its target genes through recruitment of CBP/p300 co-activators[31].
SHP-1 phosphatase is a key negative regulator of cell signaling. STAT3, DNA methyltransferase 1 and HDAC 1 form complexes that bind to the promoter of SHP-1[32]. Thus, STAT3 may induce methylation of this promoter and epigenetic silencing of SHP-1[32].
ATTENUATION OF THE INFLAMMATORY RESPONSE BY HDAC AND PROTEIN PHOSPHATASES
Transcriptional repression is also a dynamic and essential mechanism of down-regulation of genes for resolution of inflammation. Chromatin modifying complexes act coordinately to regulate cAMP-dependent transcription during the activation phase but also during the attenuation phase. As pointed out by Canettieri et al[33], cAMP-mediated transcription exhibits burst-attenuation kinetics in parallel with PKA-dependent phosphorylation and subsequent PP1-mediated dephosphorylation of CREB. PP1, but not PP2A, blocks CRE-regulated gene expression and transcriptional attenuation of cAMP-induced gene expression requires CREB dephosphorylation by PP1[34]. PP1 is targeted to CREB by binding with class-I HDACs, such as HDAC1 and HDAC8, promoting CREB inactivation by dephosphorylation during pre-stimulus and attenuation phases of the cAMP response[23,33]. Consequently, PP1 and class-I HDACs regulate the duration of CREB-mediated gene transcription (Figure 1). Nevertheless, nuclear PP2A might also be involved in the dephosphorylation of CREB[35].
Attenuation of cAMP-dependent transcription also involves other proteins, such as CRE modulator (CREM)-α, which is a ubiquitously expressed transcription factor responsible for the termination of IL-2 expression in T cells[36]. CREM down-regulates the expression of IL-2 via recruitment of HDAC1 and reduces acetylation of histones[36].
PP2A comprises a family of holoenzyme complexes with diverse biological activities, which mainly depend on individual regulatory subunits. PP2A holoenzymes may form complexes with IKK and negatively regulate NF-κB transcriptional activity[37]. Some PP2A regulatory subunits, such as PR55γ and PR55δ, are inhibitors of JNK and c-SRC[38]. The PP2A heterotrimeric complex formed by the PR65 A subunit, the catalytic subunit (PP2Ac), and G5PR as a regulatory subunit exhibits phosphatase activity on histone H1[39].
Class II HDACs are key transcriptional regulators whose activities are controlled via phosphorylation-dependent nucleo-cytoplasmic shuttling[40]. PP2A is responsible for dephosphorylation of class II HDACs triggering nuclear localization and repression of target genes, whereas phosphorylation triggers cytoplasmatic localization leading to activation of target genes[40,41]. Nitric oxide (NO) induces the formation of a complex between HDAC4, HDAC5, and PP2A that triggers HDAC4 and HDAC5 nuclear shuttling[42]. Thus, NO enhances HDAC activity inhibiting serum-induced histone acetylation in endothelial cells[42].
HDAC3 is a member of class I HDACs involved in the regulation of gene expression that is activated through phosphorylation[43]. HDAC3 seems to act as a scaffold protein for PP2A to dephosphorylate STAT3 and promote inactivation of the STAT3-mediated signaling pathway[44]. In addition, HDAC3 forms a complex with serine/threonine protein phosphatase 4 and its activity is inversely proportional to the cellular levels of this phosphatase[43].
In addition, HDACs down-regulate other inflammatory mediators, such as E-selectin, in the attenuation of the inflammatory cascade. Thus, repression of E-selectin expression is associated with recruitment of HDAC[28].
Dual specificity protein phosphatases, such as MAPK phosphatase-1 (MKP-1) and MKPM-M, also play an important role in the attenuation of the inflammatory cascade, mainly through inactivation of MAP kinases. MKP1 up-regulation is involved in the attenuating response to lipopolysaccharide (LPS) and its induction requires p38 activity[45]. Histone H3 may be phosphorylated by signal transduction pathways and is a novel substrate of MAPK phosphatase-1 (MKP-1)[46].
MKPM-M is rapidly induced by LPS in macrophages and leads to JNK inactivation and decreased TNF-α secretion. LPS causes acetylation of histones H3 and H4 at the promoter of the dual specificity phosphatase MKP-M gene and this effect seems to be mediated by the CREB/CBP pathway[47].
Further studies are needed to elucidate the role of protein phosphatases and HDACs in severe AP in comparison with the mild form of the disease. Indeed, the mechanisms responsible for resolution of inflammation in AP should be identified in order to properly restrain the evolution of the disease.
POTENTIAL BENEFIT FROM TREATMENT WITH PHOSPHODIESTERASE INHIBITORS AND HDAC INHIBITORS IN AP
So far, no specific effective treatment for AP has been reported in clinical trials. Thus the search for novel therapeutic strategies is still focused on experimental models. Phosphodiesterase inhibitors, such as pentoxifylline and rolipram, ameliorate AP in rodents[48-50]. Their beneficial effects seem to be mediated by inhibition of leukocyte activation and migration reducing the up-regulation of TNF-α and IL-6[49,51]. Pentoxifylline exhibits marked anti-inflammatory properties mainly ascribed to the blockade of ERK phosphorylation and inhibition of TNF-α production[48,49]. We have recently demonstrated that it prevents the remarkable loss of serine/threonine phosphatase PP2A activity in pancreas early in the course of AP[52]. This effect was associated with maintenance of cAMP levels, reduced recruitment of histone acetyltransferases and abrogation of up-regulation of pro-inflammatory genes[52]. Consequently, phosphodiesterase inhibitors seem to be beneficial early in the course of AP by down-regulation of the ERK-pathway preventing histone acetylation and the resulting up-regulation of inflammatory mediators.
The action of phosphodiesterase inhibitors may also be mediated by stimulation of dual specificity phosphatases. Dipyridamole is a non-selective phosphodiesterase inhibitor that exerts its anti-inflammatory effect via transient activation of MKP-1, which dephosphorylates and inactivates MAPK and particularly p38[53]. Dypiridamole inhibits the NF-κB signaling pathway blocking up-regulation of IL-6, monocyte chemoattractant protein-1, inducible NO synthase and COX-2 in LPS-activated macrophages[53].
The therapeutic window for treatment with phosphodiesterase inhibitors should be limited to the very early stage of the disease or to prevent the risk of AP induced by endoscopic retrograde cholangiopancreatography. Since an increase in cAMP levels might trigger the PKA/CREB/CBP pathway involved in cytokine up-regulation, the administration of phosphodiesterase inhibitors could not be beneficial when the inflammatory cascade is already ongoing.
Taken into account the involvement of HATs in the up-regulation of pro-inflammatory cytokines and the role of HDACs in the attenuation of inflammation, it seems paradoxical that HDAC inhibitors may exhibit anti-inflammatory properties. Nevertheless, it has been recently reported that histone deacetylase inhibitors reduce inflammation and mortality in mice treated with LPS by blockade of MAPK signaling due to MKP-1 acetylation[54]. Acetylation of MKP-1 is mediated by p300 upon LPS stimulation and it increases its phosphatase activity inhibiting innate immune signaling[54].
However, in the case of AP, the therapeutic window for HDAC inhibitors should be limited to a late stage of the disease in order to promote attenuation of the inflammatory response because they may enhance CREB activity during the burst phase. Nevertheless, further studies are needed to establish the therapeutic windows of phosphodiesterase inhibitors and HDAC inhibitors.
CONCLUSION
In conclusion, amplification of the inflammatory cascade requires up-regulation of pro-inflammatory cytokines and this process is mediated not only by NF-κB but also by chromatin modifying complexes and chromatin remodeling. Transcription activation in the inflammatory cascade is mediated by histone acetylation through recruitment of HAT, particularly CBP and p300. On the other hand, attenuation of the inflammatory response is also a dynamic mechanism that involves protein phosphatases, especially serine threonine phosphatases PP1 and PP2A, as well as recruitment of HDAC. Phosphodiesterase inhibitors have exhibited beneficial effects in the treatment of AP by preventing histone acetylation and the loss of PP2A activity. Inhibitors of HDAC are also proposed as potential therapy for AP. Nevertheless, the therapeutic windows of these therapeutic agents should be established.
Footnotes
Supported by Grants SAF2006-06963, SAF2009-09500 and Consolider CSD-2007-00020 to Sastre J; BFU2007-63120 and CSD2006-49 to López-Rodas G
Peer reviewer: Ilker Tasci, MD, Associate Professor, Department of Internal Medicine, Gulhane School of Medicine, Etlik 06018, Ankara, Turkey
S- Editor Li LF L- Editor Lutze M E- Editor Yang C
References
- 1.Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: bench to the bedside. Gastroenterology. 2007;133:1056.e1–1056.e25. doi: 10.1053/j.gastro.2007.01.055. [DOI] [PubMed] [Google Scholar]
- 2.Grady T, Saluja A, Kaiser A, Steer M. Edema and intrapancreatic trypsinogen activation precede glutathione depletion during caerulein pancreatitis. Am J Physiol. 1996;271:G20–G26. doi: 10.1152/ajpgi.1996.271.1.G20. [DOI] [PubMed] [Google Scholar]
- 3.Grady T, Liang P, Ernst SA, Logsdon CD. Chemokine gene expression in rat pancreatic acinar cells is an early event associated with acute pancreatitis. Gastroenterology. 1997;113:1966–1975. doi: 10.1016/s0016-5085(97)70017-9. [DOI] [PubMed] [Google Scholar]
- 4.Pereda J, Sabater L, Aparisi L, Escobar J, Sandoval J, Viña J, López-Rodas G, Sastre J. Interaction between cytokines and oxidative stress in acute pancreatitis. Curr Med Chem. 2006;13:2775–2787. doi: 10.2174/092986706778522011. [DOI] [PubMed] [Google Scholar]
- 5.Escobar J, Pereda J, Arduini A, Sandoval J, Sabater L, Aparisi L, López-Rodas G, Sastre J. Cross-talk between oxidative stress and pro-inflammatory cytokines in acute pancreatitis: a key role for protein phosphatases. Curr Pharm Des. 2009;15:3027–3042. doi: 10.2174/138161209789058075. [DOI] [PubMed] [Google Scholar]
- 6.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Dios I, Ramudo L, Alonso JR, Recio JS, Garcia-Montero AC, Manso MA. CD45 expression on rat acinar cells: involvement in pro-inflammatory cytokine production. FEBS Lett. 2005;579:6355–6360. doi: 10.1016/j.febslet.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Norman J. The role of cytokines in the pathogenesis of acute pancreatitis. Am J Surg. 1998;175:76–83. doi: 10.1016/s0002-9610(97)00240-7. [DOI] [PubMed] [Google Scholar]
- 9.Leser HG, Gross V, Scheibenbogen C, Heinisch A, Salm R, Lausen M, Rückauer K, Andreesen R, Farthmann EH, Schölmerich J. Elevation of serum interleukin-6 concentration precedes acute-phase response and reflects severity in acute pancreatitis. Gastroenterology. 1991;101:782–785. doi: 10.1016/0016-5085(91)90539-w. [DOI] [PubMed] [Google Scholar]
- 10.Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- 11.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- 12.Cheung WL, Briggs SD, Allis CD. Acetylation and chromosomal functions. Curr Opin Cell Biol. 2000;12:326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- 13.Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 14.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. Eur J Cancer. 2005;41:2381–2402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Marmorstein R, Roth SY. Histone acetyltransferases: function, structure, and catalysis. Curr Opin Genet Dev. 2001;11:155–161. doi: 10.1016/s0959-437x(00)00173-8. [DOI] [PubMed] [Google Scholar]
- 16.Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- 17.Tsai SC, Seto E. Regulation of histone deacetylase 2 by protein kinase CK2. J Biol Chem. 2002;277:31826–31833. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 18.Sengupta N, Seto E. Regulation of histone deacetylase activities. J Cell Biochem. 2004;93:57–67. doi: 10.1002/jcb.20179. [DOI] [PubMed] [Google Scholar]
- 19.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kao HY, Verdel A, Tsai CC, Simon C, Juguilon H, Khochbin S. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J Biol Chem. 2001;276:47496–47507. doi: 10.1074/jbc.M107631200. [DOI] [PubMed] [Google Scholar]
- 21.Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clayton AL, Mahadevan LC. MAP kinase-mediated phosphoacetylation of histone H3 and inducible gene regulation. FEBS Lett. 2003;546:51–58. doi: 10.1016/s0014-5793(03)00451-4. [DOI] [PubMed] [Google Scholar]
- 23.Gao J, Siddoway B, Huang Q, Xia H. Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem Biophys Res Commun. 2009;379:1–5. doi: 10.1016/j.bbrc.2008.11.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yuan LW, Gambee JE. Histone acetylation by p300 is involved in CREB-mediated transcription on chromatin. Biochim Biophys Acta. 2001;1541:161–169. doi: 10.1016/s0167-4889(01)00141-0. [DOI] [PubMed] [Google Scholar]
- 25.Solomou EE, Juang YT, Tsokos GC. Protein kinase C-theta participates in the activation of cyclic AMP-responsive element-binding protein and its subsequent binding to the -180 site of the IL-2 promoter in normal human T lymphocytes. J Immunol. 2001;166:5665–5674. doi: 10.4049/jimmunol.166.9.5665. [DOI] [PubMed] [Google Scholar]
- 26.Vanden Berghe W, De Bosscher K, Boone E, Plaisance S, Haegeman G. The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem. 1999;274:32091–32098. doi: 10.1074/jbc.274.45.32091. [DOI] [PubMed] [Google Scholar]
- 27.Deng WG, Zhu Y, Wu KK. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood. 2004;103:2135–2142. doi: 10.1182/blood-2003-09-3131. [DOI] [PubMed] [Google Scholar]
- 28.Edelstein LC, Pan A, Collins T. Chromatin modification and the endothelial-specific activation of the E-selectin gene. J Biol Chem. 2005;280:11192–11202. doi: 10.1074/jbc.M412997200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miao F, Li S, Chavez V, Lanting L, Natarajan R. Coactivator-associated arginine methyltransferase-1 enhances nuclear factor-kappaB-mediated gene transcription through methylation of histone H3 at arginine 17. Mol Endocrinol. 2006;20:1562–1573. doi: 10.1210/me.2005-0365. [DOI] [PubMed] [Google Scholar]
- 30.Ray S, Sherman CT, Lu M, Brasier AR. Angiotensinogen gene expression is dependent on signal transducer and activator of transcription 3-mediated p300/cAMP response element binding protein-binding protein coactivator recruitment and histone acetyltransferase activity. Mol Endocrinol. 2002;16:824–836. doi: 10.1210/mend.16.4.0811. [DOI] [PubMed] [Google Scholar]
- 31.Cvijic H, Bauer K, Löffler D, Pfeifer G, Blumert C, Kretzschmar AK, Henze C, Brocke-Heidrich K, Horn F. Co-activator SRC-1 is dispensable for transcriptional control by STAT3. Biochem J. 2009;420:123–132. doi: 10.1042/BJ20081989. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Q, Wang HY, Marzec M, Raghunath PN, Nagasawa T, Wasik MA. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc Natl Acad Sci USA. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canettieri G, Morantte I, Guzmán E, Asahara H, Herzig S, Anderson SD, Yates JR 3rd, Montminy M. Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat Struct Biol. 2003;10:175–181. doi: 10.1038/nsb895. [DOI] [PubMed] [Google Scholar]
- 34.Hagiwara M, Alberts A, Brindle P, Meinkoth J, Feramisco J, Deng T, Karin M, Shenolikar S, Montminy M. Transcriptional attenuation following cAMP induction requires PP-1-mediated dephosphorylation of CREB. Cell. 1992;70:105–113. doi: 10.1016/0092-8674(92)90537-m. [DOI] [PubMed] [Google Scholar]
- 35.Wadzinski BE, Wheat WH, Jaspers S, Peruski LF Jr, Lickteig RL, Johnson GL, Klemm DJ. Nuclear protein phosphatase 2A dephosphorylates protein kinase A-phosphorylated CREB and regulates CREB transcriptional stimulation. Mol Cell Biol. 1993;13:2822–2834. doi: 10.1128/mcb.13.5.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tenbrock K, Juang YT, Leukert N, Roth J, Tsokos GC. The transcriptional repressor cAMP response element modulator alpha interacts with histone deacetylase 1 to repress promoter activity. J Immunol. 2006;177:6159–6164. doi: 10.4049/jimmunol.177.9.6159. [DOI] [PubMed] [Google Scholar]
- 37.Li S, Wang L, Berman MA, Zhang Y, Dorf ME. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24:497–509. doi: 10.1016/j.molcel.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eichhorn PJ, Creyghton MP, Wilhelmsen K, van Dam H, Bernards R. A RNA interference screen identifies the protein phosphatase 2A subunit PR55gamma as a stress-sensitive inhibitor of c-SRC. PLoS Genet. 2007;3:e218. doi: 10.1371/journal.pgen.0030218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kono Y, Maeda K, Kuwahara K, Yamamoto H, Miyamoto E, Yonezawa K, Takagi K, Sakaguchi N. MCM3-binding GANP DNA-primase is associated with a novel phosphatase component G5PR. Genes Cells. 2002;7:821–834. doi: 10.1046/j.1365-2443.2002.00562.x. [DOI] [PubMed] [Google Scholar]
- 40.Martin M, Potente M, Janssens V, Vertommen D, Twizere JC, Rider MH, Goris J, Dimmeler S, Kettmann R, Dequiedt F. Protein phosphatase 2A controls the activity of histone deacetylase 7 during T cell apoptosis and angiogenesis. Proc Natl Acad Sci USA. 2008;105:4727–4732. doi: 10.1073/pnas.0708455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paroni G, Cernotta N, Dello Russo C, Gallinari P, Pallaoro M, Foti C, Talamo F, Orsatti L, Steinkühler C, Brancolini C. PP2A regulates HDAC4 nuclear import. Mol Biol Cell. 2008;19:655–667. doi: 10.1091/mbc.E07-06-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Illi B, Dello Russo C, Colussi C, Rosati J, Pallaoro M, Spallotta F, Rotili D, Valente S, Ragone G, Martelli F, et al. Nitric oxide modulates chromatin folding in human endothelial cells via protein phosphatase 2A activation and class II histone deacetylases nuclear shuttling. Circ Res. 2008;102:51–58. doi: 10.1161/CIRCRESAHA.107.157305. [DOI] [PubMed] [Google Scholar]
- 43.Zhang X, Ozawa Y, Lee H, Wen YD, Tan TH, Wadzinski BE, Seto E. Histone deacetylase 3 (HDAC3) activity is regulated by interaction with protein serine/threonine phosphatase 4. Genes Dev. 2005;19:827–839. doi: 10.1101/gad.1286005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Togi S, Kamitani S, Kawakami S, Ikeda O, Muromoto R, Nanbo A, Matsuda T. HDAC3 influences phosphorylation of STAT3 at serine 727 by interacting with PP2A. Biochem Biophys Res Commun. 2009;379:616–620. doi: 10.1016/j.bbrc.2008.12.132. [DOI] [PubMed] [Google Scholar]
- 45.Hu JH, Chen T, Zhuang ZH, Kong L, Yu MC, Liu Y, Zang JW, Ge BX. Feedback control of MKP-1 expression by p38. Cell Signal. 2007;19:393–400. doi: 10.1016/j.cellsig.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 46.Kinney CM, Chandrasekharan UM, Yang L, Shen J, Kinter M, McDermott MS, DiCorleto PE. Histone H3 as a novel substrate for MAP kinase phosphatase-1. Am J Physiol Cell Physiol. 2009;296:C242–C249. doi: 10.1152/ajpcell.00492.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Musikacharoen T, Yoshikai Y, Matsuguchi T. Histone acetylation and activation of cAMP-response element-binding protein regulate transcriptional activation of MKP-M in lipopolysaccharide-stimulated macrophages. J Biol Chem. 2003;278:9167–9175. doi: 10.1074/jbc.M211829200. [DOI] [PubMed] [Google Scholar]
- 48.Gómez-Cambronero L, Camps B, de La Asunción JG, Cerdá M, Pellín A, Pallardó FV, Calvete J, Sweiry JH, Mann GE, Viña J, et al. Pentoxifylline ameliorates cerulein-induced pancreatitis in rats: role of glutathione and nitric oxide. J Pharmacol Exp Ther. 2000;293:670–676. [PubMed] [Google Scholar]
- 49.Pereda J, Sabater L, Cassinello N, Gómez-Cambronero L, Closa D, Folch-Puy E, Aparisi L, Calvete J, Cerdá M, Lledó S, et al. Effect of simultaneous inhibition of TNF-alpha production and xanthine oxidase in experimental acute pancreatitis: the role of mitogen activated protein kinases. Ann Surg. 2004;240:108–116. doi: 10.1097/01.sla.0000129343.47774.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sato T, Otaka M, Odashima M, Kato S, Jin M, Konishi N, Matsuhashi T, Watanabe S. Specific type IV phosphodiesterase inhibitor ameliorates cerulein-induced pancreatitis in rats. Biochem Biophys Res Commun. 2006;346:339–344. doi: 10.1016/j.bbrc.2006.05.133. [DOI] [PubMed] [Google Scholar]
- 51.Haddad JJ, Land SC, Tarnow-Mordi WO, Zembala M, Kowalczyk D, Lauterbach R. Immunopharmacological potential of selective phosphodiesterase inhibition. I. Differential regulation of lipopolysaccharide-mediated proinflammatory cytokine (interleukin-6 and tumor necrosis factor-alpha) biosynthesis in alveolar epithelial cells. J Pharmacol Exp Ther. 2002;300:559–566. doi: 10.1124/jpet.300.2.559. [DOI] [PubMed] [Google Scholar]
- 52.Sandoval J, Escobar J, Pereda J, Sacilotto N, Rodriguez JL, Sabater L, Aparisi L, Franco L, López-Rodas G, Sastre J. Pentoxifylline prevents loss of PP2A phosphatase activity and recruitment of histone acetyltransferases to proinflammatory genes in acute pancreatitis. J Pharmacol Exp Ther. 2009;331:609–617. doi: 10.1124/jpet.109.157537. [DOI] [PubMed] [Google Scholar]
- 53.Chen TH, Kao YC, Chen BC, Chen CH, Chan P, Lee HM. Dipyridamole activation of mitogen-activated protein kinase phosphatase-1 mediates inhibition of lipopolysaccharide-induced cyclooxygenase-2 expression in RAW 264.7 cells. Eur J Pharmacol. 2006;541:138–146. doi: 10.1016/j.ejphar.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–1503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]