

Abstract
Peroxiredoxins (Prx) are thiol peroxidases that exhibit exceptionally high reactivity toward peroxides, but the chemical basis for this is not well understood. We present strong experimental evidence that two highly conserved arginine residues play a vital role in this activity of human Prx2 and Prx3. Point mutation of either ArgI or ArgII (in Prx3 Arg-123 and Arg-146, which are ∼3–4 Å or ∼6–7 Å away from the active site peroxidative cysteine (Cp), respectively) in each case resulted in a 5 orders of magnitude loss in reactivity. A further 2 orders of magnitude decrease in the second-order rate constant was observed for the double arginine mutants of both isoforms, suggesting a cooperative function for these residues. Detailed ab initio theoretical calculations carried out with the high level G4 procedure suggest strong catalytic effects of H-bond-donating functional groups to the Cp sulfur and the reactive and leaving oxygens of the peroxide in a cooperative manner. Using a guanidinium cation in the calculations to mimic the functional group of arginine, we were able to locate two transition structures that indicate rate enhancements consistent with our experimentally observed rate constants. Our results provide strong evidence for a vital role of ArgI in activating the peroxide that also involves H-bonding to ArgII. This mechanism could explain the exceptional reactivity of peroxiredoxins toward H2O2 and may have wider implications for protein thiol reactivity toward peroxides.
Keywords: Computer Modeling, Enzyme Kinetics, Enzyme Mechanisms, Kinetics, Peroxidase, Ab Initio Calculations, Mechanism, Peroxide, Peroxiredoxin
Introduction
Peroxiredoxins (Prx)2 are a family of ubiquitous proteins that are important for antioxidant defense and redox signaling (1, 2). Prx reduce H2O2 extremely rapidly (3–5). They are also highly reactive against peroxynitrite and other peroxides (4, 6, 7), but less so with other typical thiol-reactive reagents such as chloramines or alkylating electrophiles (5, 8). There are six subfamilies (Prx1, Prx6, AhpE (one-cysteine peroxiredoxin from Mycobacterium tuberculosis), Prx5, Tpx, and bacterioferritin comigratory protein), categorized by amino acid sequence, which share a similar catalytic cycle. A number of highly conserved amino acid residues promote similar structures around their active site cysteine (peroxidatic cysteine; Cp) (9). Based on mechanistic considerations they are further subcategorized into 1-Cys and 2-Cys Prx. Prx2 and -3, the focus of this manuscript, are 2-Cys Prx that belong to the Prx1 subfamily.
The reactivity of the Cp thiol toward H2O2 is many orders of magnitude larger than that of free cysteine (e.g. the second-order rate constants for the reaction of human Prx2 and Prx3 with H2O2 are ∼ 107 m−1 s−1 (3, 5), compared with ∼1 m−1 s−1 for free cysteine or GSH (10)). Due to the low pKa < 6 of the Cp sulfhydryl group, it is in its more nucleophilic thiolate form at physiological pH (5, 11, 12). However, this is not sufficient to explain the high reactivity of Prx with peroxides and further lowering of the pKa below 6 would decrease reactivity due to the linear free energy relationship (13). It is likely that conserved amino acid residues at the active site influence the rate of reaction via H-bonding interactions with the Cp sulfur as well as with H2O2. Based on structural considerations, Hall et al. (9, 14) proposed that a highly conserved arginine residue (ArgI) near the Cp (Fig. 1, Prx3 Arg-123) activates the peroxide at the active site of Prx. There is another arginine (ArgII; Fig. 1a, Prx3 Arg-146) close to the active site (6–7 Å from the Cp sulfur, Fig. 1b) present in all members of the Prx1, Prx6, and AhpE subfamilies (see “Experimental Procedures”), which we hypothesize to have a role in the catalytic activity of these proteins.
FIGURE 1.

There are two highly conserved arginine residues at the active site of Prx3, which play major roles in its peroxidase activity. a, active site structure of bovine Prx3 (Protein Data Bank code 1ZYE) around its peroxidative cysteine (Cys-47) residue. b, distances of ArgI (Arg-123) and ArgII (Arg-146) nitrogens from the Cp sulfur in the crystal structure of bovine Prx3 (Protein Data Bank code 1ZYE). c, proposed transition structure for the reaction of H2O2 with Prx3 in which ArgI (Arg-123) donates an H-bond to the Cp (Cys-47) sulfur as well as to Oa and ArgII (Arg-146) assists in the protonation of the leaving ObH− moiety.
Although an activator role for ArgI on H2O2 has been proposed, only one study has tested the role of these arginine residues (along with other conserved amino acids) using site-directed mutagenesis. This showed that barley Prx ArgI and ArgII are important for its peroxidatic activity (15). However, activity was measured using a catalytic assay where the rate-determining step is reduction of the disulfide-linked Prx dimer, so it was not possible to assess the extent to which the reactivity of Cp with H2O2 was decreased. The effects of ArgI and ArgII on the kinetics of the reaction of Cp with H2O2 have not been investigated. In a combined crystallographic and quantum chemical investigation, Nakamura et al. (16) examined the archeal peroxiredoxin (ApTPx) in which a histidine residue (rather than an arginine) is in close proximity to Cp. (This His residue is absent at the active site of mammalian Prx.) Their theoretical study focused on the reaction steps subsequent to the reduction of H2O2, and they concluded that a hypervalent sulfurane intermediate is involved in the formation at that stage of a sulfenic acid (16). In contrast, the theoretical component of the present investigation focuses on the initial catalytic reduction of H2O2 by Cp.
In the current study, we have mutated ArgI and ArgII in human Prx2 and Prx3 and measured their rates of reaction with H2O2. We demonstrate that both arginines play important roles in the reactivities of Prx2 and Prx3, independently from one another as well as in a cooperative fashion. We also used high level ab initio calculations to mechanistically explain our findings and propose a novel model to elucidate the high reactivity of peroxiredoxins with peroxides.
EXPERIMENTAL PROCEDURES
Reagents, preparation of recombinant Prx and site-directed mutagenesis of arginine residues, biophysical characterization of the recombinant Prx, computational details, and additional computational results are described in the supplemental “Methods.”
BLAST Search
To analyze whether Arg-146 of human Prx3 is conserved among other human peroxiredoxin isoforms and whether it is conserved in other organisms, a BLAST search against NCBI protein reference sequences was performed using residues 121–170 of human Prx3 as a query sequence (BLASTP 2.2.24). All of the 250 retrieved reference proteins aligning with the query sequence contained a corresponding arginine at a similar position in the protein.
Analysis of Oxidation State of Prx
Reduced monomer and disulfide-bonded dimer were separated by nonreducing SDS-PAGE on 15% gels as in Ref. 6. Gels were Coomassie Blue-stained and scanned using a Fluor-S® MultiImager (Bio-Rad). Bands were quantified using Quantity One® software from Bio-Rad.
Kinetic Experiments
Prior to each experiment, Prx was reduced with either 25 mm DTT (Prx3) in 50 mm phosphate buffer for 2 h or by tris(2-carboxyethyl)phosphine (Prx2) for 1 h at pH 7.4. Buffer solutions contained 1 mm diethylenetriaminepentaacetic acid to chelate trace metal contaminants. Reduced Prx were separated from excess DTT using Micro Bio-Spin 6 columns (Bio-Rad,) prewashed with 200 μl of 10 mg/ml bovine catalase and equilibrated with 50 mm phosphate buffer containing 100 μm diethylenetriaminepentaacetic acid. Prx concentrations were measured using Bio-Rad DC Protein Assay Reagent and bovine serum albumin as standard. For kinetic analyses, the initial concentration of reduced Prx was calculated based on the relative intensity of the bands representing oxidized and reduced forms determined by gel electrophoresis.
Catalase Competition Assay
As described (3), 8 μm Prx was reacted with equimolar H2O2 in the presence of bovine catalase (0.81–51 μm). Reactions were started by rapid addition of H2O2 at 20 °C, with vigorous stirring. Remaining thiol residues on the Prx were alkylated after ∼15 s by adding 67 mm N-ethylmaleimide in sample buffer (4% SDS, 10% glycerol, and 62.5 mm Tris-HCl, pH 6.8). Samples were analyzed by gel electrophoresis.
HRP Competition Assay
Second-order rate constants of the recombinant WT proteins were measured by competition with HRP (12). Briefly, 10 μm HRP was premixed with each Prx (1.7–10.2 μm). The reactions were started by rapid addition of H2O2 (4.7 μm) at 20 °C, with vigorous stirring. Conversion of HRP to compound I was monitored spectrophotometrically by the loss of absorbance at 403 nm. The second-order rate constant for the reaction of Prx with H2O2 was calculated based on the fractional inhibition and the reported rate constant (k = 1.7 × 107 m−1 s−1) (17) of the reaction of HRP with H2O2.
Measurement of Second-order Rate Constants of Arginine Mutants with H2O2
Reactions were started by the addition of H2O2 to the mutant Prx during vigorous vortex mixing at 20 °C. Reactions were quenched either with 100 mm N-ethylmaleimide or 0.7 mg/ml catalase and analyzed by SDS-PAGE. Kinetic runs were carried out under pseudo first-order conditions with 5–10 μm Prx and 13–50 μm or 2.3–5 mm H2O2 for the single and double mutants, respectively. Pseudo first-order rate constants were obtained by fitting the kinetic traces to a single exponential equation. Second, order rate constants were obtained by correcting the pseudo first-order rate constants with the applied H2O2 concentrations. Kinetic curves were analyzed by SigmaPlot 11.
Ab Initio Calculations
Full computational details are presented in the supplemental data. Here, we note in addition that our discussion is based on enthalpies (ΔH) rather than free energies (ΔG) because our small model approach would not adequately take into account the preorganization provided by the enzyme (18). However, the latter quantities show the same qualitative trends (supplemental Table S2). We refer to the electrophilic oxygen of H2O2 (the one being attacked by HS−) as Oa and to the oxygen of the leaving hydroxide group as Ob (14). The theoretical results reported in this paper correspond to the CPCM-G4//B3-LYP/6–31+G(2df,p) level, which denotes the combination of a conductor-like polarizable continuum model (CPCM) solvation correction on top of a gas-phase G4//B3-LYP/6–31+G(2df,p) enthalpy.
RESULTS
Two Highly Conserved Arginine Residues Play a Key Roles in the Peroxidase Activity of Prx2 and Prx3
Single substitution of Arg-146 (ArgII) with various residues, Arg-123 (ArgI) with glycine, and a double substitution of both arginine residues with glycine, were introduced in Prx3. ArgI (Arg-127) and ArgII (Arg-150) in Prx2 were replaced with lysine, either singly or as a double mutant. WT recombinant proteins were constructed under the same conditions.
Commercially available and recombinant WT and mutant proteins displayed similar CD spectra, suggesting that secondary structures were not affected by mutations (supplemental Fig. S1). Melting points of the recombinant WT and mutant proteins (supplemental Table S1) show that, although R146A and R146H of Prx3 behaved differently, other mutants melted at a similar temperature to the WT.
The reactivities of the WT recombinant proteins with H2O2 were investigated using competition kinetic assays against increasing amounts of bovine catalase. Prx2 and Prx3 are 2-Cys peroxiredoxins that form disulfide bonds between the Cp on one chain and the resolving cysteine on the other (Cys-172 and Cys-168 in Prx2 and Prx3, respectively). Oxidation can therefore be followed as dimer formation on a nonreducing gel. As shown in Fig. 2a, addition of H2O2 converted both Prx from reduced monomers to dimers. Catalase competed poorly against WT Prx2 and Prx3, with only marginal protection against dimerization at the highest concentration. This indicates a fast reaction between Prx and H2O2 and agrees with previous studies using purified human erythrocyte Prx2 (5) and commercially available Prx3 (3).
FIGURE 2.

Kinetic assays of recombinant WT Prx2 and Prx3. Competition of Prx2 (a) and Prx3 (b) with bovine catalase. The first and second lanes show the ratio of reduced and oxidized Prx at time 0 and after 15 s of incubation, respectively. Other lanes represent the ratios after reaction with H2O2 in the presence of stated amount of catalase. Protection is apparent as an increase in the Prx monomer band with increasing amounts of catalase. Gels are representative of three independent experiments. Measurement of the second-order rate constants for reaction of Prx2 (c) and Prx3 (d) with H2O2 by competition with HRP. The second-order rate constants (see Table 1) were calculated from the slopes of linear plots of (F/1 − F)kHRP[HRP] versus [Prx2]; F = fractional inhibition. Error bars represent S.D. of triplicate measurements. The experiment was repeated on a different day with similar results. For experimental details, see “Experimental Procedures.”
Prx also were competed against a constant amount of HRP for H2O2 as described (12). H2O2 reacts rapidly with the ferric form of HRP to give a stable intermediate (compound I) that has a lower Soret band absorbance at 403 nm. The reaction was quantified by measuring the decrease in this absorbance. The decrease was inhibited by increasing concentrations of the recombinant WT Prx (Fig. 2, c and d). Analysis of the competition data gave apparent second-order rate constants for reaction with H2O2 that are similar to those reported for isolated and commercially available Prx (keff ∼ 2 × 107 m−1 s−1; Table 1) (3, 5).
TABLE 1.
Second-order rate constants for the reactions of H2O2 with Prx2 and Prx3 WT and mutant proteins
Values are means ± S.D. from three to seven independent experiments using different H2O2 and Prx concentrations. Data are from experiments described in Figs. 2 and 3. (For details, see “Experimental Procedures.”)
| keff | |
|---|---|
| m−1 s−1 | |
| Prx3 | |
| WT | (1.9 ± 0.1) × 107 |
| R146A | (2 ± 0.5) × 102 |
| R146H | (3 ± 1) × 102 |
| R146K | (4 ± 2) × 102 |
| R146G | (2 ± 0.9) × 102 |
| R123G | (7 ± 2) × 102 |
| R123G/R146G | 2 ± 0.5 |
| Prx2 | |
| WT | (1.6 ± 0.1) × 107 |
| R150K | (6 ± 3) × 102 |
| R127K | (3 ± 2) × 102 |
| R127K/R150K | 3 ± 1 |
With the arginine mutants, catalase was fully protective even at the lowest concentration, and the Prx mutants did not inhibit the formation of compound I by HRP. This indicates that all of the arginine mutants are several orders of magnitude less reactive than the WT proteins. Reactions with these mutants are sufficiently slow to be followed over 15 min under pseudo first-order conditions (excess of H2O2), using gel electrophoresis to quantify the relative amounts of the monomeric (reduced) versus dimeric (oxidized) forms (Fig. 3). Dimer formation is a two-step process. The reaction of H2O2 with the Cp results in the formation of a sulfenic acid intermediate (Reaction 1), which reacts with the resolving cysteine of another protein molecule to give a disulfide-linked dimer (Reaction 2).
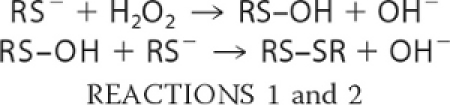 |
FIGURE 3.

Time course for conversion of reduced (monomer) to oxidized (dimer) of the ArgI, ArgII, and ArgI/ArgII mutants for reaction with H2O2. The arginine residues of Prx3 (a–c) and Prx2 (d–f) were converted to Gly in Prx3 or Lys in Prx2. Data points on the kinetic traces represent the loss of reduced Prx as a % of total Prx (dimer + monomer; obtained from the depicted gels). Second-order rate constants obtained from the single exponential fits are shown in Table 1. Reactions were quenched by catalase (a) or N-ethylmaleimide (b–f). For experimental details, see “Experimental Procedures.”
Reactions were quenched with catalase or by alkylating the reactive thiol groups of the proteins with N-ethylmaleimide. Both methods gave similar rate constants, suggesting that Reaction 1 (between H2O2 and Cp) was being measured, and not Reaction 2 (which is expected to be rapid) (19, 20). A potential complication is that the Prx dimer bands on the gels represent both the single and double disulfide-bonded isoforms of the protein. Therefore, a proportionality constant needs to be incorporated into the rate law. This constant should have a value between 1 (if the reactivity of the dimer isoform with one disulfide bond is much greater than the monomer) and 2 (if the dimer is much less reactive). This introduces a 50% error in the apparent second-order rate constants for the reactions of H2O2 with Cp.
The kinetic traces that were obtained by following the loss of the monomeric forms fit well to single exponential equations, indicating a first-order dependence on Prx concentration (Fig. 3). The second-order rate constants (Table 1) were obtained by correcting the pseudo first-order rate constants with the H2O2 concentrations. Second-order rate constants were similar at different H2O2 concentrations (in the range of 13–50 μm), suggesting overall second-order kinetics for the reaction. When ArgI was mutated, the second-order rate constants (for Prx3R123G and Prx2R127K) dropped by 5 orders of magnitude (to ∼ 102 m−1 s−1) compared with to WT. ArgI is therefore vital for the high reactivity of Prx.
Point mutation of ArgII (in Prx3R146A, Prx3R146H, Prx3R146K, Prx3R146G, and Prx2R150K) resulted in a similar decrease in the rate constant, demonstrating that ArgII is as important for the catalytic activity as ArgI. With both ArgI and ArgII mutated (Prx3R123G/R146G and Prx2R127K/R150K), the rate constants were a further 2 orders of magnitude smaller than for single mutants. This comprises a total of 7 orders of magnitude difference between double mutant and WT proteins (see Table 1), comparable with the difference in reactivity between free cysteine and Cp.
Ab Initio Calculations
High level calculations using the composite Gaussian-4 (G4) procedure were performed to assess possible H-bonding interactions at the active site that would lower the activation energy and increase the rate of the reaction of Cp with H2O2. For reasons of computational efficiency, the Cp and arginine residues were modeled by HS− and guanidinium cation, respectively. All of the uncatalyzed and catalyzed reductions of H2O2 by HS− are found to proceed via an SN2 mechanism. Fig. 4 and supplemental Fig. S2 display the reactant complexes, transition structures (TS), and product complexes located along the uncatalyzed and catalyzed reaction pathways. Table 2 gives the calculated reaction barriers (ΔH‡) at 298 K.
FIGURE 4.

Ab initio calculations for the catalytic role of H-bonding interactions by guanidinium, HF, and H2O moieties in the reaction of HS− with H2O2. Calculated reactant complexes (RC), TS, and product complexes (PC) located on the potential energy surface for the uncatalyzed reduction and for the reactions catalyzed by guanidinium, HF, and H2O moieties. Hydrogen bonds are shown as dashed lines, and partial bonds in the SN2 transition structures are shown as parallel dashed and solid lines. The atomic color scheme is as follows: white, H; gray, C; blue, N; red, O; cyan, F; yellow, S).
TABLE 2.
Barrier heights associated with the reactions in Fig. 4 and supplemental Fig. S2
Values are given as kJ mol−1, calculated at the CPCM-G4//B3-LYP/6–31+G(2df,p) level in a simulated protein-like phase.
| Reactiona,b | ΔH‡ |
|---|---|
| Uncatc | 67.2d |
| Uncat | 70.6 |
| [FH···Oa] | 56.8 |
| [Ob···HF] | 55.7 |
| [FH···S] | 65.1 |
| [FH···Oa,Ob···HF] | 52.1 |
| [FH···S,Oa···HF] | 56.6 |
| [FH···S,Ob···HF] | 47.8 |
| [2FH···S,Oa,Ob···HF] | 49.7 |
| [Gua+···S,Oa,Ob] | 44.8 |
| [H2C = NH···S,Oa,Ob···Gua+] | 40.4 |
| [H2NH···S,Oa,Ob···Gua+] | 38.3 |
| [HOH···S,Oa,Ob···Gua+] | 32.2 |
| [FH···S,Oa,Ob···Gua+] | 26.4 |
| [Gua+···S,Oa] | 48.2 |
| [Gua+···S,Oa,Ob···HOH] | 40.1 |
| [Gua+···S,Oa,Ob···HF] | 27.8 |
a Unless otherwise specified, the barriers are taken relative to the reactant complex.
b In enzyme-like medium (ϵ = 4.0), unless otherwise noted.
c In aqueous solution (ϵ = 78.4).
d Relative to the free reactants (see text).
In our analysis, the catalytic efficiency or rate enhancement of the catalyst is taken as the difference in barrier between the uncatalyzed process in aqueous solution and the enzymatic reaction (i.e. it is given by ΔΔH‡ = ΔH(uncat,aq)‡ − ΔH(cat,enz)‡), where the enzyme-like environment in the catalyzed reaction is simulated by a homogeneous polarizable continuum model with the dielectric constant ϵ = 4.0. We note that, according to the Arrhenius equation, a change of 5.7 kJ mol−1 in the barrier corresponds to a change of 1 order of magnitude in the reaction rate at 298 K.
Formation of the reactant complex (Fig. 4, Uncat-RC) is thermodynamically unfavorable in aqueous solution (ΔH(aq) = +5.2 kJ mol−1). Thus, the enthalpic barrier in aqueous solution is taken relative to the free reactants (ΔH(uncat,aq)‡ = 67.2 kJ mol−1). In all other cases, the barrier is taken as the energy of the TS relative to that of the reactant complex.
Probing the Effect of Hydrogen Bonding
The partial protonation or deprotonation that accompanies H-bonding can play an important role in facilitating enzymatic reactions (18). To consider the effect on the reaction profile of H-bonding at the oxygen centers of the H2O2, HF was chosen as a model H-bond donor (reactions [FH···Oa], [Ob···HF], and [FH···Oa,Ob···HF] in supplemental Fig. S2). H-bonding of HF to Oa in the transition structure will render the oxygen center a better electrophile, whereas H-bonding to Ob will facilitate departure of the hydroxide moiety. Indeed, we observe catalytic enhancements of ΔΔH‡ = 10.4, 11.5, and 15.1 kJ mol−1 for the [FH···Oa], [Ob···HF], and [FH···Oa,Ob···HF] reactions, respectively.
An additional H-bonding interaction at the S center in [Ob···HF] (reaction [FH···S,Ob···HF], supplemental Fig. S2) increases the catalytic enhancement from ΔΔH‡ = 11.5 to 19.4 kJ mol−1. However, interaction at the sulfur atom in [FH···Oa], Uncat, and [FH···Oa,Ob···HF] results in little or no reduction of the barrier heights (namely, by 0.2–2.4 kJ mol−1). Finally, we note that in all cases, the hydrogen bonds at the various O or S centers stabilize the transition structures more than they stabilize the reactant complexes (see supplemental data).
Single-arginine Model
To model the effects of ArgI, we incorporated a single guanidinium cation moiety into our calculations instead of HF. We located two reaction pathways, shown as reactions [Gua+···S,Oa,Ob] and [Gua+···S,Oa] in Fig. 4. The transition structure in [Gua+···S,Oa] is qualitatively similar to the insightful structure proposed by Hall et al. (14) for a related system. The two transition structures are fairly close in energy; in the enzyme environment [Gua+···S,Oa,Ob]-TS lies lower in energy by 3.4 kJ mol−1. In [Gua+···S,Oa,Ob]-TS, the guanidinium cation interacts with all three S, Oa, and Ob centers, as evident from hydrogen bond distances of 2.14 (Hα···S), 2.18 (Hα···Oa), 2.03 (Hβ···Oa), and 1.80 Å (Hβ···Ob), where Hα and Hβ are the hydrogens closer to the S and Ob atoms, respectively. On the other hand, in [Gua+···S,Oa]-TS the guanidinium cation interacts mainly with S and Oa (the Hα···S and Hβ···Oa distances are 2.12 and 1.61 Å, respectively) (see also supplemental Table S4).
For the [Gua+···S,Oa] reaction, we obtain ΔH(cat,enz)‡ = 48.2 kJ mol−1, thus ΔΔH‡ = 19.0 kJ mol−1, which corresponds to a rate enhancement of ∼3 orders of magnitude. Reaction [Gua+···S,Oa,Ob] has a slightly lower barrier (ΔH(cat,enz)‡ = 44.8 kJ mol−1), which leads to ΔΔH‡ = 22.4 kJ mol−1 and corresponds to a rate enhancement of ∼4 orders of magnitude.
Single-arginine Model Plus an Extra H-bond Donor Moiety
The computational experiments (found under Probing the Effect of Hydrogen Bonding) suggest that a strong hydrogen-bonding interaction at the S center in [Gua+···S,Oa,Ob]-TS or at the Ob center in [Gua+···S,Oa]-TS will further enhance the catalytic activity of the guanidinium catalyst. Indeed, if we consider the simultaneous interaction of a guanidinium at Oa and Ob and an HF molecule at S (reaction [FH···S,Oa,Ob···Gua+], Fig. 4), we find a barrier of just 26.4 kJ mol−1 (Table 2). The catalytic efficiency is thus increased in this way to ΔΔH‡ = 40.8 kJ mol−1, which corresponds to a rate enhancement of ∼7 orders of magnitude relative to the uncatalyzed reaction in water. Consideration of weaker hydrogen-bond donors at S (namely H2O, NH3 or H2C=NH, Table 2) results in smaller catalytic enhancements.
When the transition structure involves a simultaneous interaction of a guanidinium at S and Oa and an HF molecule at Ob (reaction [Gua+···S,Oa,Ob···HF]), we find a barrier of 27.8 kJ mol−1. The catalytic efficiency is thus increased in this way to ΔΔH‡ = 39.4 kJ mol−1. A weaker hydrogen bond donor at Ob (e.g. [Gua+···S,Oa,Ob···HOH]) reduces ΔΔH‡ to 27.1 kJ mol−1.
DISCUSSION
H2O2 is a strong oxidant. However, its reactions with most thiols have a high activation barrier and therefore, although thermodynamically favored, are relatively slow. The structures of Prx overcome this barrier. This study investigated the roles of two highly conserved arginine residues (Fig. 1) in the exceptional reactivity of Prx with H2O2. Kinetic data obtained with various mutants of human Prx2 and Prx3 provided strong kinetic evidence that both arginine residues are vital for the peroxidase activities of these Prx. ArgI has been proposed to contribute significantly to the catalytic reaction. Indeed, we show mutations of ArgI in Prx2 and Prx3 result in a 5 orders of magnitude drop in reactivity with H2O2. Surprisingly, comparable losses in reactivity were also observed in ArgII mutants of both Prx. Importantly, simultaneous mutation of both arginine residues in Prx2 and Prx3 further decreased the rate constant to a total of 7 orders of magnitude less than the WT value.
High level quantum chemistry calculations provide mechanistic insight into the roles of these arginine residues in the reduction of H2O2 by Cp. The calculations indicate that partial protonation through H-bonding at the S, Oa, and Ob centers significantly lowers the activation barrier in a cooperative manner. A model using a single guanidinium cation moiety identifies two reaction pathways for catalysis ([Gua+···S,Oa,Ob] and [Gua+···S,Oa], Fig. 4), with reductions of 22.4 and 19.0 kJ mol−1 in the barrier compared with the uncatalyzed reaction in water.
The presence of an additional H-bond donor such as water lowers the barrier further. We located two relevant transition structures ([Gua+···S,Oa,Ob···HOH]-TS and [HOH···S,Oa,Ob···Gua+]-TS, Fig. 4) that lead to barrier reductions and are consistent with the position of ArgI at the Prx active site. The reductions in the barriers are 27.1 and 35.0 kJ mol−1, respectively, compared with that for the uncatalyzed reaction. This translates to an increase of ∼5–6 orders of magnitude in the second-order rate constant of the reaction. This is consistent with the experimentally observed difference in reactivity between the ArgI mutants and the WT proteins.
H-bonding of a guanidinium nitrogen to Oa and either the leaving group (Ob) or the active site thiol has a profound effect on the catalytic activity. However, to account for the reactivity of the Prx compared with free cysteine, the barrier needs to be lowered further. This is achieved by introduction of a second, stronger H-bond donor (modeled by an HF), interacting at S or Ob ([FH···S,Oa,Ob···Gua+]-TS and [Gua+···S,Oa,Ob···HF]-TS, Fig. 4). The barriers then become 26.4 and 27.8 kJ mol−1, corresponding to a total of 7 orders of magnitude increase in rate constant, as observed experimentally for the double mutant proteins. Thus, the computational results as well as substantiating the proposal (14) that ArgI is crucial in the catalytic mechanism also indicate a critical role for a second protonating agent.
Computational modeling suggests that reduction of H2O2 by cysteine proceeds via an SN2 mechanism with a single transition structure (Scheme 1).
SCHEME 1.

On the basis of our experimental observations and theoretical computations, we propose that ArgI is involved in lowering the electron density of the reacting oxygen (Oa) in H2O2 (Fig. 1c) via H-bonding, making it a better electrophile. Simultaneously, ArgI could also be engaged in an H-bonding interaction with Cp, which would not only contribute to lowering its pKa but would also anchor Oa to Cp (further increasing reactivity in line with theoretical calculations). This role is consistent with structural data on the position of ArgI in the Prx active site and similar to that proposed by Hall et al. (9, 14).
Computational data indicate that additional interaction of either Ob or Cp with a strong H-donor in the Prx structure is needed to explain the high reactivity with H2O2. The dramatic decrease in reaction rate by ArgII mutation also needs to be taken into account. The crystal structures of Prx2 and Prx3 suggest that ArgII would be ideally located to donate an H-bond to Ob. The dimensions of the active site (Fig. 1c) enable H2O2 to sit within bonding distance of both ArgI and ArgII. This would lower the energy of the transition structure, thus facilitating the reaction. An H-bonding network between the two arginine residues and H2O2 would also pull the two peroxide oxygens apart, thereby activating the oxidant by weakening the Oa–Ob bond. This would favor the OH+ transfer to Cp as well as facilitate the departure of OH−.
This role for ArgII in the reactivity of Prx differs from that proposed by Hall et al. (9, 14). They suggest that ArgII assists ArgI to adopt the correct conformation via an Arg-Glu-Arg H-bonding network to exert its catalytic effect. Our observation that point mutation of either arginine residue results in a similar drop in reactivity, for both Prx2 and Prx3, is consistent with this suggestion. However, the further 2 orders of magnitude drop in reactivity observed for double arginine mutants suggests a cooperative function for both residues.
We acknowledge that the orientation of H2O2 in the transition structure of our proposed mechanism is different from that of the H2O2 crystallized in the active site of the archeal peroxiredoxin ApTPx (21). Hall et al. (14) used the ApTpx-H2O2 structure as the basis of their proposed model and also have shown that it is consistent with the orientation of different substrate mimics that have co-crystallized at the active site of other Prx. This model therefore favors involvement of residues other than ArgII in the H-bonding network. Further mechanistic and structural investigations are therefore required to reconcile these observations with our kinetic data.
In conclusion, our kinetic and computational studies substantiate structural studies and provide strong evidence for a vital role of ArgI in the high reactivity of Prx2 and Prx3 with H2O2. Our data also indicate that ArgII is critical in the activity of these proteins, and we propose that it could act through H-bonding to H2O2.
Supplementary Material
Acknowledgments
We thank the NCI National Facility and Intersect Australia, Ltd. for the provision of generous grants of computer time. We are grateful to Andrew Cox who helped with some of the initial analyses and Tong Zhu and Grant Pierce (BioInteraction Center at the University of Canterbury) for help with CD and melting point experiments.
This work was supported by the Marsden Fund, the Health Research Council of New Zealand, and the Australian Research Council.

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Methods,” Tables S1–S5, Figs. S1–S4, and additional references.
- Prx
- peroxiredoxin(s)
- Tpx
- thiol peroxidase
- ApTPx
- archeal peroxiredoxin
- Cp
- peroxidative cysteine
- Oa
- electrophilic oxygen of H2O2 (the one being attacked by HS−)
- Ob
- leaving hydroxide group of H2O2
- G4
- Gaussian-4
- TS
- transition structures
- ΔH‡
- activation enthalpy
- ΔG‡
- activation free energy; ΔΔH‡ = ΔH(uncat,aq)‡ − ΔH(cat,enz)‡
- ϵ
- dielectric constant
- CPCM-G4//B3-LYP/6–31+G(2df,p)
- conductor-like polarizable continuum model solvation correction on top of a gas phase G4//B3-LYP/6–31+G(2df,p) enthalpy
- uncat
- uncatalised.
REFERENCES
- 1. Cox A. G., Winterbourn C. C., Hampton M. B. (2010) Biochem. J. 425, 313–325 [DOI] [PubMed] [Google Scholar]
- 2. Fourquet S., Huang M. E., D'Autreaux B., Toledano M. B. (2008) Antioxid. Redox. Signal. 10, 1565–1576 [DOI] [PubMed] [Google Scholar]
- 3. Cox A. G., Peskin A. V., Paton L. N., Winterbourn C. C., Hampton M. B. (2009) Biochemistry 48, 6495–6501 [DOI] [PubMed] [Google Scholar]
- 4. Parsonage D., Karplus P. A., Poole L. B. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 8209–8214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Peskin A. V., Low F. M., Paton L. N., Maghzal G. J., Hampton M. B., Winterbourn C. C. (2007) J. Biol. Chem. 282, 11885–11892 [DOI] [PubMed] [Google Scholar]
- 6. Peskin A. V., Cox A. G., Nagy P., Morgan P. E., Hampton M. B., Davies M. J., Winterbourn C. C. (2010) Biochem. J. 432, 313–321 [DOI] [PubMed] [Google Scholar]
- 7. Trujillo M., Ferrer-Sueta G., Thomson L., Flohé L., Radi R. (2007) Subcell. Biochem. 44, 83–113 [DOI] [PubMed] [Google Scholar]
- 8. Stacey M. M., Peskin A. V., Vissers M. C., Winterbourn C. C. (2009) Free Radic. Biol. Med. 47, 1468–1476 [DOI] [PubMed] [Google Scholar]
- 9. Hall A., Nelson K., Poole L., Karplus P. A. (2010) Antioxid. Redox. Signal., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winterbourn C. C., Metodiewa D. (1999) Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 11. Nelson K. J., Parsonage D., Hall A., Karplus P. A., Poole L. B. (2008) Biochemistry 47, 12860–12868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ogusucu R., Rettori D., Munhoz D. C., Netto L. E., Augusto O. (2007) Free Radic. Biol. Med. 42, 326–334 [DOI] [PubMed] [Google Scholar]
- 13. Nagy P., Winterbourn C. C. (2010) in Advances in Molecular Toxicology (Fishbein J. C. ed), pp. 183–222, Elsevier [Google Scholar]
- 14. Hall A., Parsonage D., Poole L. B., Karplus P. A. (2010) J. Mol. Biol. 402, 194–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. König J., Lotte K., Plessow R., Brockhinke A., Baier M., Dietz K. J. (2003) J. Biol. Chem. 278, 24409–24420 [DOI] [PubMed] [Google Scholar]
- 16. Nakamura T., Yamamoto T., Abe M., Matsumura H., Hagihara Y., Goto T., Yamaguchi T., Inoue T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6238–6242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dolman D., Newell G. A., Thurlow M. D. (1975) Can. J. Biochem. 53, 495–501 [DOI] [PubMed] [Google Scholar]
- 18. Sandala G. M., Smith D. M., Radom L. (2010) Acc. Chem. Res. 43, 642–651 [DOI] [PubMed] [Google Scholar]
- 19. Nagy P., Lemma K., Ashby M. T. (2007) J. Org. Chem. 72, 8838–8846 [DOI] [PubMed] [Google Scholar]
- 20. Nagy P., Ashby M. T. (2007) J. Am. Chem. Soc. 129, 14082–14091 [DOI] [PubMed] [Google Scholar]
- 21. Nakamura T., Kado Y., Yamaguchi T., Matsumura H., Ishikawa K., Inoue T. (2010) J. Biochem. 147, 109–115 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.