

Abstract
Astrogliosis following spinal cord injury (SCI) involves an early hypertrophic response that is beneficial and a subsequent formation of a dense scar. We investigated the role of bone morphogenetic protein (BMP) signaling in gliosis after SCI and find that BMPR1a and BMPR1b signaling exerts opposing effects on hypertrophy. Conditional ablation of BMPR1a from glial fibrillary acidic protein (GFAP)-expressing cells leads to defective astrocytic hypertrophy, increased infiltration by inflammatory cells, and reduced axon density. BMPR1b-null mice conversely develop “hyperactive” reactive astrocytes and consequently have smaller lesion volumes. The effects of ablation of either receptor are reversed in the double knock-out animals. These findings indicate that BMPR1a and BMPR1b exert directly opposing effects on the initial reactive astrocytic hypertrophy. Also, BMPR1b knock-out mice have an attenuated glial scar in the chronic stages following injury, suggesting that it has a greater role in glial scar progression. To elucidate the differing roles of the two receptors in astrocytes, we examined the effects of ablation of either receptor in serum-derived astrocytes in vitro. We find that the two receptors exert opposing effects on the posttranscriptional regulation of astrocytic microRNA-21. Further, overexpression of microRNA-21 in wild-type serum-derived astrocytes causes a dramatic reduction in cell size accompanied by reduction in GFAP levels. Hence, regulation of microRNA-21 by BMP signaling provides a novel mechanism for regulation of astrocytic size. Targeting specific BMPR subunits for therapeutic purposes may thus provide an approach for manipulating gliosis and enhancing functional outcomes after SCI.
Introduction
Formation of a glial scar following spinal cord injury (SCI) acts as both a physical and a molecular barrier to axonal regeneration (Fawcett and Asher, 1999; Silver and Miller, 2004). However, astrogliosis also plays a crucial role in restoring homeostasis following SCI by facilitating repair of the blood–brain barrier and limiting infiltration of inflammatory cells (Faulkner et al., 2004; Sofroniew, 2005; Okada et al., 2006; Herrmann et al., 2008). Ablation of reactive astrocytes following SCI in mice increased inflammatory infiltration and worsened long-term recovery (Faulkner et al., 2004). Conditional ablation of STAT3 (signal transducer and activator of transcription 3) correspondingly resulted in similar deficits, indicating that STAT signaling is involved in the beneficial effects of gliosis (Okada et al., 2006; Herrmann et al., 2008). Gliosis following spinal cord injury involves an early hypertrophic response and a subsequent hyperplastic response (Fawcett and Asher, 1999; Barnabé-Heider and Frisén, 2008). The beneficial effects attributed to astrogliosis are largely associated with the earlier, mainly hypertrophic response whereas the later hyperplastic response and increase in the number of reactive astrocytes at the lesion site leads to formation of a dense scar. Finding a means of inhibiting the later detrimental effects without hindering the earlier beneficial effects offers a potential means of facilitating axonal regeneration.
The bone morphogenetic proteins (BMPs) are members of the TGFβ superfamily of growth factors. BMP signaling promotes astrocytic lineage commitment in vitro both by subventricular zone progenitor cells (Gross et al., 1996) and by glial restricted progenitor cells (Mabie et al., 1997; Cheng et al., 2007). Transgenic overexpression of BMP4 in the developing brain leads to a significant increase in the number of astrocytes (Gomes et al., 2003). Conversely, disruption of BMP signaling in vivo negatively affects astrogliogenesis (See et al., 2007). BMP levels increase following spinal cord injury (Setoguchi et al., 2001; Chen et al., 2005). Several groups have studied the effects of globally inhibiting BMP signaling after SCI with mixed results. Transplantation into the injured spinal cord of cells engineered to secrete noggin, an antagonist of BMP signaling, resulted in expanded lesion volumes and poor functional recovery (Enzmann et al., 2005). Others report that inhibition of BMP signaling enhances axonal outgrowth and locomotor recovery after SCI (Setoguchi et al., 2004; Matsuura et al., 2008). These observations suggest that BMP signaling may be involved in both the beneficial and the detrimental effects of gliosis.
BMPs exert their biological effects by binding to type I (BMPRIa and BMPRIb) and type II (BMPRII) receptor subunits that are organized with minor modifications of the prototypical TGFβ subclass of serine-threonine kinase receptors (Koenig et al., 1994; ten Dijke et al., 1994; Liu et al., 1995; Nohno et al., 1995; Rosenzweig et al., 1995). Since BMPRIa and BMPRIb may mediate different biological responses in the developing nervous system (Panchision et al., 2001; Gulacsi and Lillien, 2003; Brederlau et al., 2004; Samanta et al., 2007; Yamauchi et al., 2008), we asked whether different type I receptors might serve different roles during astrogliosis following SCI. We find that BMPR1a and BMPR1b signaling exert opposite effects on the early beneficial phases of astrocytic hypertrophy. Further, the two receptors have differing roles in regulation of astrocytic microRNA-21 (miR-21). We find that BMPR1a signaling decreases levels of mature miR-21 and that miR-21, in turn, negatively regulates glial fibrillary acidic protein (GFAP) levels in astrocytes.
Materials and Methods
Transgenic mouse lines.
The GFAP-cre (line 73.12) mice were maintained in C57BL/6J and BMPR1a-fx mice were maintained in a mixed 129SvJ:C57BL/6 background. The generation of GFAP-cre mice (Garcia et al., 2004), BMPR1a +/− (constitutive null mice) (Mishina et al., 1995), and BMPR1a-flox mice (Mishina et al., 2002) and their genotyping has been described previously. We mated GFAP-Cre mice with heterozygous BMPR1a-null mutant (BMPR1a+/−) mice to obtain GFAP-Cre, BMPR1a+/− mice, which were then mated with homozygous floxed-BMPR1a (BMPR1afx/fx) mice to obtain four separate genotypes: GFAP-Cre, BMPR1afx/−, GFAP-Cre, BMPR1afx/+, BMPR1afx/+, and BMPR1afx/− with the last two genotypes being wild type (WT) for the Cre transgene (see supplemental Fig. 3, available at www.jneurosci.org as supplemental material). GFAP Cre mice were crossed to Rosa26 reporter mice (The Jackson Laboratory) to generate GFAP Cre, R26R mice.
The BMPR1b receptor knock-out (KO) mice have been described previously (Yi et al., 2000). We mated heterozygotes to derive BMPR1b +/+ (WT), +/−, and −/− (BMPR1b KO) mice.
Mouse spinal cord injuries and animal care.
All animal procedures were undertaken in accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals. Female 129/SvJ mice (10 weeks of age; The Jackson Laboratory), and 2-month-old WT and KO mice were anesthetized using isoflurane inhalation anesthetic (2.5% in 100% oxygen) administered using a VetEquip Rodent anesthesia machine. After laminectomy at the T10 vertebral segment, the spinal cord was compressed dorsoventrally by the extradural application of a 24 g modified aneurysm clip for 1 min (FEJOTA mouse clip). After SCI, the skin was sutured using AUTOCLIP (9 mm; BD Biosciences). Postoperatively, animals were kept on a heating pad to maintain body temperature. Bladders were manually emptied twice daily throughout the duration of the study. In the event of discomfort, Buprenex (2 mg/kg, s.c., twice daily) was administered. Gentamycin was administered once daily in the event of hematuria (20 mg/kg) subcutaneously for 5 d. Mice that exhibited any hindlimb movement 24 h after the injury were excluded from the study.
Astrocyte cultures.
Primary astrocyte cultures were derived from brains of postnatal animals as previously described (McCarthy and de Vellis, 1980). Briefly, the brains were demeningized, triturated, and plated on tissue culture flasks with airtight lids (BD Biosciences) in DMEM (Invitrogen) supplemented with 10% fetal calf serum (Hyclone) and 1× penicillin, streptomycin, and l-glutamine (Invitrogen). Medium was replaced on the second day and every 4 d thereafter. After overnight shaking on the 10th day, the cells were passaged onto fresh tissue culture flasks. Purity of the cultures was assessed by immunostaining for GFAP, and we found that >95% of the cells were GFAP positive. For evaluating the effects of BMP4 treatment, the cultures were passaged and then plated in serum containing medium for 1 d, following which the medium was replaced with serum-free medium: DMEM + N2 serum supplement (Invitrogen) + penicillin-streptomycin-l-glutamine (Invitrogen), for 7 d. Following this, the astrocytes were left either in serum-free medium or serum-free medium + 20 ng/ml BMP4 (R&D Biosystems) for the time periods described in the text.
Protein extraction and Western blot analysis.
For protein samples from the spinal cord, mice were killed as previously described and perfused with 1× PBS. Samples from injured cords were taken from the injured segment as well as one half vertebral segment rostral and caudal to the lesion site. Tissue samples were collected in T-PER protein extraction buffer (Pierce) supplemented with 1× HALT Protease inhibitor cocktail (Thermo Scientific), and homogenized on ice using a tissue homogenizer (Polytron PT3000, Brinkmann). Samples were spun at 4°C for 5 min and then flash frozen and stored at −80°C until used.
For samples from cultured neurospheres and astrocyte monolayers, the cells were harvested in 1× PBS and pelleted and then homogenized in M-PER protein extraction reagent (Pierce) as described above.
For Western analysis, samples were boiled for 5 min in strong denaturing conditions (1% SDS with 4% β-mercaptoethanol in 40 mm Tris) and loaded on SDS-polyacrylamide gels, following which they were transblotted onto polyvinylidene difluoride membranes at 4°C for 1 h. Membranes were blocked in TBST (Tris-buffered saline with 0.05% Tween 20) with 5% BSA for 1 h at room temperature, following which they were incubated in blocking solution and appropriate primary antibodies overnight at 4°C. On day 2, membranes were washed three times with TBST and then incubated with appropriate HRP-conjugated secondary antibodies (Santa Cruz Biotechnology, 1:5000 in TBST), washed three times again with TBST, and developed using Supersignal West Pico reagent (Thermo Scientific). The primary antibodies used were GFAP (rabbit polyclonal, DAKO; 1:5000), Phospho STAT3 (Tyr 705) and Total STAT3 (rabbit polyclonal; Cell Signaling Technology, 1:1000), phospho-SMAD 1,5,8 (Cell Signaling Technology, 1:1000), and GAPDH (mouse IgG, Millipore Bioscience Research Reagents; 1:5000).
Tissue processing and immunohistochemistry.
Mice were killed using CO2 inhalation and then perfused transcardially with cold 1× PBS followed by 4% paraformaldehyde in 1× PBS. The tissue was dissected and left in the fixative for another 2 h on ice, following which they were dehydrated overnight in 30% sucrose in 1× PBS. Cords were then embedded in Tissue-Tek O.C.T. embedding compound (Sakura), frozen on dry ice, and stored at −80°C until used.
Frozen sections (20 μm thick) were cut on a Leica (CM3050S) cryostat and collected on Superfrost Bond Rite slides (Richard Allen Scientific). Every fifth section was placed on the same slide such that each adjacent section was 80 μm away from its neighbor. Four sections were placed on each slide and hence the width the frozen cord spanned on each slide was 240 μm. We roughly averaged ∼20 slides per cord. Sections were stained with the primary antibodies diluted in 1× PBS + 1% BSA + 0.25% Triton X-100, overnight at 4°C. On the next day, sections were washed three times for 5 min in 1× PBS at room temperature and then incubated in Alexa Fluor-conjugated secondary antibody (Molecular Probes/Invitrogen Labeling and Detection, 1:500 in 1× PBS) for 1 h at room temperature, followed by three washes in 1× PBS; then they were counterstained with Hoechst nuclear stain (1:500 in 1× PBS) for 15 min at room temperature and then mounted using Prolong Gold antifade reagent (Invitrogen). For detection of thymidine analogs, the sections were first boiled in 10 mm sodium citrate for 10 min and allowed to cool to room temperature for 30 min, following which the DNA was denatured using 2N hydrochloric acid application for 45 min at room temperature. Sections were rinsed with PBS and then processed subsequently as described above.
Primary antibodies used were: GFAP (Sigma; mouse IgG1, 1:500), CD11b (Serotec; rat IgG, 1:150), β-galactosidase (Abcam; chk IgY 1:200), BMPR1a (Orbigen; rabbit polyclonal 1:200), phospho-SMAD 1/5/8 (Cell Signaling Technology; rabbit polyclonal 1:100), NeuN (mouse IgG1, 1:500), BrdU (Millipore; mouse IgG2a, 1:1000), CldU (Accurate Chemical and Scientific; rat monoclonal anti-BrdU, 1:250;), and SMI-312 (Sternberger Monoclonals; mouse IgG1 1:500).
Myelin staining.
This was done according to Wolcke's modification of the original Weigert method. Briefly, the sections were kept overnight in 2.5% ferric ammonium-sulfate, rinsed twice and then stained using Weigert's hematoxylin solution (10 cc of 10% basic hematoxylin in ethanol + 7 cc of saturated lithium carbonate + 90 cc of distilled water) for 4 h. Sections were cleared overnight in 70% ethanol and then counterstained with cresyl violet and mounted in DPX (distyrene, tricresyl phosphate, and xylene).
Fluorescence intensity quantification.
The thickness of the cord was determined by counting the total number of longitudinal sections that contained the lesion for each animal. The midline of the cord was then defined by taking the central most section and then two more sections for each animal were picked 200 μm left and right of the midline section. Sections from WT and KO groups were processed simultaneously. A control uninjured cord was processed in a similar manner and the center of this cord was used to provide the baseline for normalizing the fluorescence intensity values. Levels measured in the injured cords from all genotypes were normalized to intensity values obtained from this control spinal cord. Following immunostaining for GFAP, the sections were imaged on a Zeiss uvLSM510 confocal microscope. The glial scar was imaged in the area adjacent to the infiltration site where the highest staining was observed in all cords. Imaging was done using a 20× objective which covered both gray and white matter, and separate scans were taken both rostral and caudal to the lesion. Each section was scanned with identical laser power, amplifier gain, and offset values. All sections were imaged in one sitting to avoid problems of fading. Each confocal Z-stack was converted to monochrome tiff images and then the intensity value for each plane of the Z-stack was measured using MetaMorph 2.6 software. All the values in the stack were integrated for the total intensity of the entire stack and the process was repeated for each stack at the end of which we had integrated pixel values for each Z-stack from all the sections. These values were individually normalized to the intensity values obtained from scans of uninjured spinal cords and then the intensity values were averaged across the WT and KO groups.
For CD11b intensity measurements, we used 40× objectives to scan the area in the gray matter immediately adjacent to the lesion and then in an area 300 μm away from the lesion. The fluorescence intensity for the Z-stacks was measured are as previously described. Since we could not normalize these values to an uninjured cord, the intensity values for each stack are represented in the intensity units that were produced by the software. These values were then averaged between the groups.
Lesion area measurements.
Tissue sections were picked as described above and then imaged with a 10× objective on an Axiovision epifluorescence microscope. The lesion area was defined as the region of dense nuclear staining that was GFAP negative and was flanked by GFAP-positive reactive astrocytes. The heightened GFAP immunoreactivity in comparison with neighboring uninjured areas is easily distinguishable and well defines the edges of the lesion, at both 1 and 5 weeks post-SCI (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Only areas that were flanked by reactive astrocytes that showed nuclear stain were included for analysis. Hence, cavities alone that arose from processing artifacts (that were devoid of nuclei) were not included. An illustration of this measurement has been included in supplemental Figure 8 (available at www.jneurosci.org as supplemental material). The area was then averaged for each animal and then the areas from all the animals were grouped for comparison.
Axon density measurements.
Sections adjacent to the ones used for CD11b and GFAP measurements were used from the groups mentioned. GFAP- and SMI-312R-stained longitudinal sections were then imaged using TissueGnostics imaging acquisition software at the Northwestern University cell imaging facility. We set the GFAP staining as a reference point, and the first point at which we observed GFAP staining was used to determine the rostral and caudal edges of the lesion. We then drew a rectangle 150 μm in width equidistant from the rostral and caudal edges of the lesion spanning the entire dorsoventral thickness of the cord, not including the edges to avoid edge artifacts. Since the GFAP area was difficult to define in the BMPR1a conditional KO (CKO) animals, we felt that this methodology allowed for more consistent sampling, across animals, that was not affected by the poorly defined lesion in the BMPR1a CKO mice. The axon density measurements were done using the Feature J function on the NIH ImageJ software as described previously (Grider et al., 2006). Briefly, the monochrome image of the rectangular area was inverted in Adobe PhotoShop and then imported in ImageJ. We used the “smallest Hessian distribution function” on Feature J. After setting the threshold (minimum and maximum) for each image, the area of all the positive pixels was summed to give the total area of SMI-312R-positive pixels. These were then grouped for all the animals for each genotype and then used for statistical analysis. For the axon density in the uninjured cord, we used an identical rectangular area rostral to the lesion site >2 mm away in each section. The axon density was similarly measured, and then the axon density in the lesion area was expressed as a percentage of the area in the uninjured part of the cord. The percentages of all the sections were then grouped for each animal. The average densities for all the animals were then grouped by genotype for statistical analysis.
GFAP area measurements.
After perfusion, the lesion epicenter was identified in each mouse macroscopically, and then the spinal cord was cut into a rostral and caudal part. We collected 20-μm-thick cross-sections from the rostral part. Every section was collected and placed onto slides, and the cord was cut across 10 slides. This was done such that the adjoining sections on a single slide were 200 μm apart. Sections were processed for GFAP and CD11b immunostaining as described earlier, and then the first section from the epicenter, where we observed GFAP immunostaining, was used to measure the GFAP area. The entire cross-section was imaged using the TissueGnostics image acquisition software at the Northwestern University cell imaging facility. Following this, the entire area of GFAP+ pixels was done using the Feature J function in NIH ImageJ software as described earlier.
RNA extraction and real-time reverse transcription PCR.
Total RNA was collected from spinal cords or cultured astrocytes using the RNAqueous-4PCR kit (Ambion). A total of 0.5 μg of RNA was used for generating cDNA using the Thermoscript reverse transcriptase and oligo-dT primers (Invitrogen). Real-time PCR was performed with the SybrGreen Master Mix (Applied Biosystems) and Realplex2 Mastercycler (Eppendorf) using the following cycling parameters: 95°C, 15 s; 60°C, 60 s for 40 cycles. A total of 2 μl of cDNA was used per PCR.
For miR-21 studies, the total RNA was extracted using an RNAqueous Micro kit (Ambion). For detecting pri- and pre-miR-21, SYBRGreen PCR Master Mix was used with the same conditions as described above, using the following primers: pri-miR21 forward, GGTTTAAAGCAAAGCAAACATCTCTGG, pri-miR21 reverse, TTATTTCAATGTGATTTTCCTTTGACA T; pre-miR21 forward, GATACCAAAATGTCAGACAGCCC, pre-miR21 reverse, TGTACCACCTTGTCGGATAGCT.
For detecting the mature miR-21 as well as snoR202, 10 ng of total RNA was used for the reverse transcription. We used the gene-specific RT primers as well as Taqman detection probes from Applied Biosystems, using the manufacturer's instructions.
Basso, Beattie, and Bresnahan scoring.
Functional assessments were performed at weekly intervals extending up to 5 weeks postinjury during the active phase of the murine sleep/wake cycle. The mice were assessed for functional recovery by using the modified Basso, Beattie, and Bresnahan (BBB) hindlimb locomotor test for mice (Joshi and Fehlings, 2002a), using two independent observers. The KO animals had no difference in coat color or body weight. The experimenters who genotyped the animals were different from the ones who scored the mice. Thus, the observers who scored the mice were blinded to their identity. Mice that exhibited any hindlimb movement at 24 h postinjury were excluded from this study altogether.
Thymidine analog administration.
The thymidine analogs (Sigma)were administered intraperitoneally at the following dosages: bromodeoxyuridine (BrdU), 50 mg/kg; 5-chloro-2′-deoxyuridine (CldU), 42.5 mg/kg. For the subacute phase, the analog was administered once daily. The analogs were diluted in saline for administration.
Statistical analysis.
For all comparisons between BMPR1a WT and CKO, as well as BMPR1b WT and KO animals, a Student's unpaired t test was used. For comparison among the three groups in the double KO analysis, a three-way ANOVA single factor was performed followed by Tukey's post hoc t test for comparison within the groups. Values in graphs are shown as mean ± SEM.
miR-21 cloning, lentivirus generation, and miR-21overexpression.
The miR-21 sequence was identified from the Sanger database. The pri-miR-21 sequence was identified by comparing with the human sequence, amplified using PCR, and then cloned into the XhoI, NotI sites in control plemiR plasmid (Open Biosystems). This results in the miR-21 sequence being placed 3′ to the RFP coding sequence. For generation of the lentivirus, 293 cells were cotransfected with either control lemiR or plenti-miR-21 along with the packaging plasmids psPAX2 and pMD2.G (Addgene).
Wild-type astrocytes were derived from CD1 pups at postnatal day 2 as described earlier. After shaking on day 10, the astrocytes were infected with virus containing medium for 24 h and then refed with normal fresh serum-containing medium on the following day. Cells were refed daily and after 48 h, the medium was replaced with serum containing medium that contained 2 μg/ml puromycin (selection medium). The RFP fluorescence was imaged at 3 d after selection and on the fourth day, cells were lysed and used for RNA and protein analysis.
Results
Astrogliosis follows an initial hypertrophic and subsequent hyperplastic response
Adult 129/SvJ female mice were injured using a clip compression model, in which the initial assault is followed by a persistent compression, a phenomenon that mimics what is often seen in cases of human SCI (Joshi and Fehlings, 2002a,b). We used a severe (24 g clip compression) injury that results in locomotor deficits that are identical to those observed with a complete transection of the cord (Joshi and Fehlings, 2002a,b). The inflammatory response and gliosis following SCI were evaluated by immunostaining for CD11b and GFAP, respectively. At 3 d after SCI, when reactive hypertrophy is just beginning to occur, there is infiltration by CD11b-positive macrophages/microglia into the parenchyma (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The core of the lesion at this time is largely acellular and mostly contains cellular debris and degenerating and necrotic nuclei. However, 5 weeks after SCI, the CD11b-positive cells have been tightly compacted by reactive astrocytes toward the lesion epicenter and are intermingled with a dense glial scar (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
GFAP immunohistochemistry revealed that in the initial stages (4 and 7 d postinjury), the lesion site was populated by characteristic reactive astrocytes (Baldwin and Scheff, 1996) with distinctly hypertrophic cellular processes (Fig. 1A). At 5 weeks postinjury, there was significant astrocytic hyperplasia at the injury site. Consistent with the increased GFAP staining, quantitative reverse transcription (RT)-PCR demonstrated a significant increase in GFAP transcripts in the injured spinal cord compared with uninjured controls (n = 3 animals for each time point analyzed). At 4 d after SCI, there was a ∼3.2-fold increase in GFAP transcript levels, which increased to ∼3.5-fold, 7 d later. (Fig. 1B).
Figure 1.
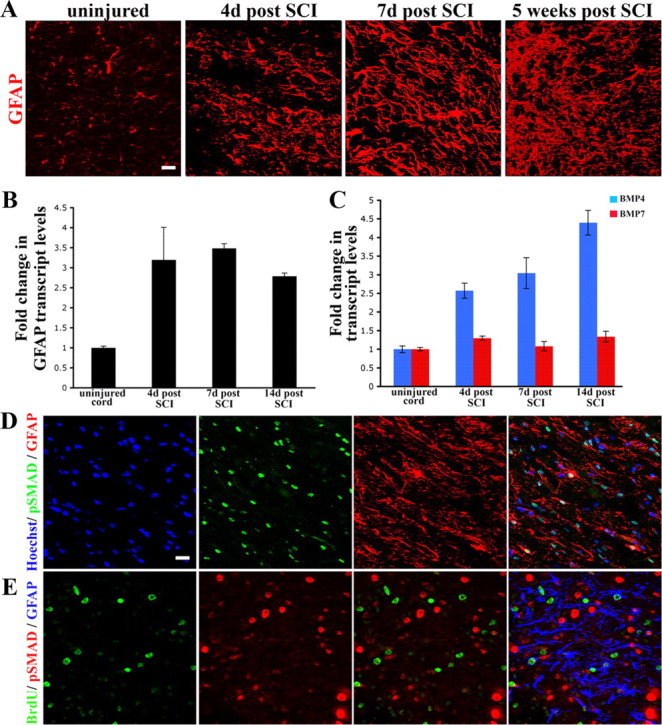
BMP signaling is active during reactive gliosis after spinal cord contusion injury. A, Longitudinal spinal cord sections stained with GFAP (red). Panels represent normal uninjured cord versus the injured cord at 4 d, 1 week, and 5 weeks postinjury. At 3 and 7 d, the reactive astrocytes display long processes (arrowheads) that appear parallel to the long axis of the cord. By 5 weeks, there is a definite increase in the number of astrocytes that are present at the lesion site. For lower-magnification images see supplemental Figure 1 (available at www.jneurosci.org as supplemental material). B, Transcript levels of GFAP (normalized to GAPDH) and expressed as fold changes compared with levels in uninjured controls ± SEM. C, Transcript levels of BMP4 (blue bars) and BMP7 (red bars) were normalized to GAPDH and expressed as fold changes compared with levels in uninjured controls. D, Spinal cords at 4 d postinjury, stained with phospho-SMAD 1/5/8 (green), GFAP (red) and Hoechst nuclear stain (blue). Nuclear phospho-SMAD staining is seen in some, but not all reactive astrocytes. E, Spinal cords at 4 d postinjury, stained with BrdU (green), phospho-SMAD 1/5/8 (red) and GFAP (blue). Merged panel shows that BrdU+ and phospho-SMAD nuclei are mutually exclusive. Astrocytes that are BrdU positive have weak or low levels of nuclear phospho-SMAD. Scale bars (in A, D, E), 20 μm.
BMP signaling is upregulated after spinal cord injury
We hypothesized that BMP signaling may play an important role in this upregulation of GFAP after SCI. Quantitative RT-PCR demonstrated that BMP4 transcript levels were significantly increased at 4 and 7 d (∼2.5-fold) following SCI and even further increased (∼4.3-fold) at 2 weeks postinjury (n = 3 animals for each time point analyzed). BMP7 transcript levels increased modestly at 2 and 4 d (∼1.6-fold), but returned to baseline levels by 7 d following injury (Fig. 1C). BMP2 transcript levels did not change significantly (data not shown). Canonical BMP signaling phosphorylates SMAD 1, SMAD 5, or SMAD 8, which complexes with SMAD 4, translocates to the nucleus, and mediates the transcriptional effects of BMP signaling (Heldin et al., 1997). The intracellular localization of phospho-SMAD 1,5,8 was therefore examined as a measure of activity of this BMP signaling pathway. At 4 d after injury, nuclear phospho-SMAD 1,5,8 (npSMAD) staining was present in reactive astrocytes (Fig. 1D). However, npSMAD was also observed in GFAP-negative cells, indicating that cells other than astrocytes also activate this pathway. To examine the cell cycle status of the cells with npSMAD, mice were injected with the thymidine analog BrdU (50 mg/kg) and killed 1 h later. The cells that displayed npSMAD were BrdU negative (Fig. 1D,E), suggesting that BMP signaling is active in nondividing GFAP-positive reactive astrocytes after SCI.
Reactive astrocytes and not inflammatory cells express the BMPR1a receptor
BMPs exert their biological effects by binding to type I (BMPRIa and BMPRIb) and type II (BMPRII) receptor subunits (Koenig et al., 1994; ten Dijke et al., 1994; Ebendal et al., 1998) with BMPR1a being the predominant receptor in the adult nervous system (Zhang et al., 1998). There were no significant changes in the levels of BMPR1b mRNA following SCI, but BMPR1a transcript levels were increased ∼2-fold at 4 d, rising to nearly 4-fold at 7 d postinjury, compared with levels in uninjured control animals (Fig. 2A), and returned to baseline at 2 weeks postinjury (n = 3 animals for each time point analyzed). Interestingly, the changes in BMPR1a levels followed a course similar to that of GFAP transcript levels (compare Figs. 1B, 2A). This increase occurred at a time when reactive hypertrophy is the predominant astrocytic response, suggesting that BMPR1a signaling may participate in this process.
Figure 2.
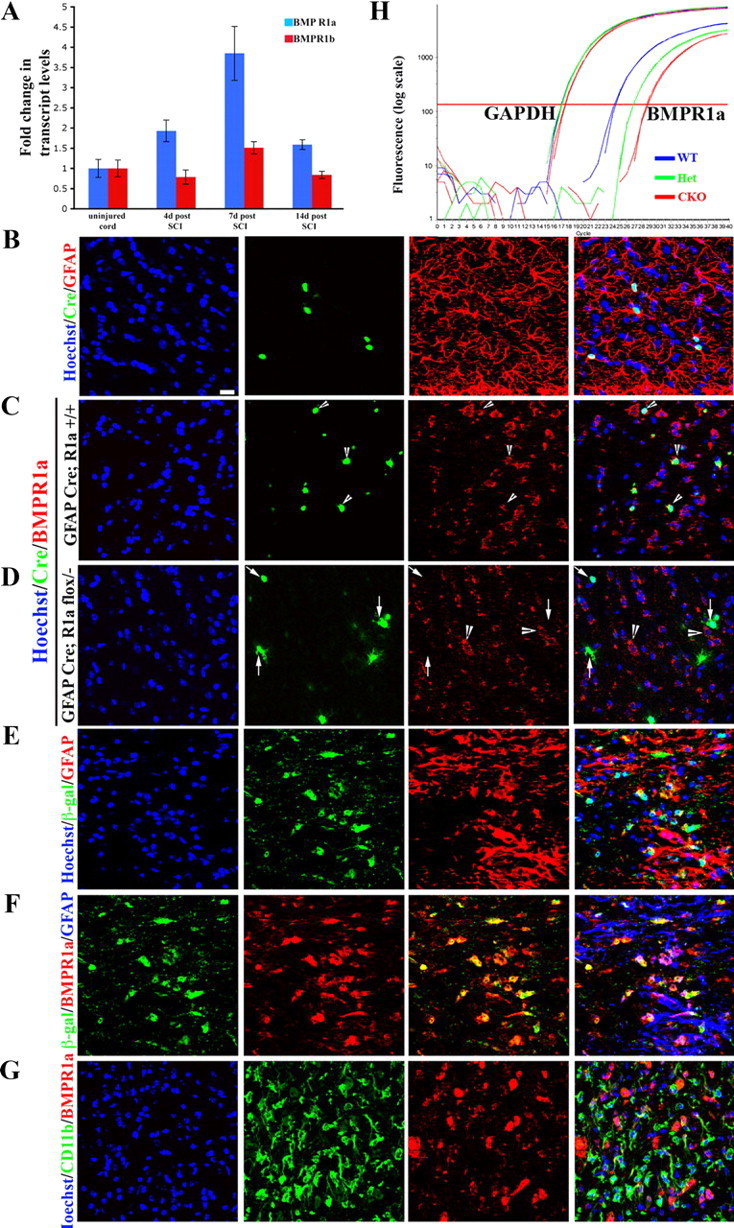
BMPR1a receptor is expressed on reactive astrocytes and not on inflammatory cells. A, Transcript levels of BMPR1a (blue bars) and BMPR1b (red bars) were normalized to GAPDH and expressed as fold changes compared with levels in uninjured controls± SEM. B, WT spinal cords stained with Hoechst nuclear stain (blue), Cre (green) and GFAP (red). Note that the Cre+ cells are GFAP+. C, D, Single confocal Z-sections from normal uninjured spinal cords stained with Hoechst nuclear stain (blue), Cre (green), and BMPR1a (red) from GFAP Cre (C) and GFAP Cre;BMPR1aflox/− (BMPR1a CKO) (D) animals. In GFAP Cre animals there is intact receptor staining seen on Cre+ cells (arrowheads). In the BMPR1a CKO animals, there is no staining for the receptor on Cre+ cells (arrows), while adjacent Cre-negative cells continue to express BMPR1a (D, arrowheads). E, Injured cord at 4 d post-SCI from a GFAP Cre, ROSA lacZ mouse stained with Hoechst nuclear stain (blue), β-galactosidase (green) and GFAP (red) showing that all the β-gal-positive cells are in GFAP-positive astrocytes. F, Sections in E stained with β-galactosidase (blue), BMPR1a (green), and GFAP (red). The β-gal staining clearly outlines the astrocytic soma, which stains for BMPR1a. G, Injured spinal cord sections at 4 d postinjury stained with Hoechst nuclear stain (blue), CD11b (green), and BMPR1a (red). Note that the receptor staining does not overlap with the CD11b. Scale bar (B–G), 20 μm. H, Real-time RT-PCR for exon2 of BMPR1a from total RNA extracted from spinal cords of WT, GFAP cre; BMPR1aflox/+ (het) and GFAP Cre; BMPR1aflox/− (CKO) mice. GAPDH serves as a loading control. There is an approximately 4-cycle reduction in the transcript levels of the floxed exon2.
We examined the expression pattern of BMPR1a in the injured as well as uninjured spinal cord using immunohistochemistry. In the uninjured spinal cord, BMPR1a expression was detected on GFAP+ cells (Fig. 2B,C) as well as on neurons (data not shown). The staining was confirmed to be specific to the receptor since it was absent on Cre-expressing cells in BMPR1a conditional knock-out mice (described in the next section) while the adjacent Cre-negative cells showed intact BMPR1a labeling (Fig. 2D). GFAP Cre;BMPR1a flox/+ heterozygous mice served as littermate controls (Fig. 2C). In the injured spinal cord, double-label immunohistochemistry for GFAP and BMPR1a at 4 d post-SCI revealed that reactive astrocytes express the receptor (Fig. 2E,F). In contrast, no colocalization was observed with CD11b, indicating that BMPR1a is not expressed by macrophages/microglia that populate the injury site (Fig. 2G). These observations suggest that BMPR1a mediates the effects of the increase in BMP4 on reactive astrocytic hypertrophy after SCI.
Conditional ablation of the BMPR1a receptor
BMPR1a-null embryos die at gastrulation (Mishina et al., 1995). We therefore used the GFAP Cre line 73.12 (Garcia et al., 2004) to conditionally ablate BMPR1a in astrocytes to help define the role of the receptor after SCI. This Cre line is expressed only in mature astrocytes in the adult spinal cord and not in neurons, oligodendrocytes, or neurons projecting to the spinal cord (Herrmann et al., 2008). We confirmed this expression pattern by performing immunostaining for Cre and GFAP in the adult uninjured spinal cord and found that Cre expression was restricted to GFAP-expressing cells (Fig. 2B). We obtained conditionally null BMPR1a mice by mating GFAP Cre;BMPR1a +/− transgenic mice with mice homozygous for the BMPR1a floxed allele (flox/flox) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). In the floxed mice, the exon2 of BMPR1a is flanked by LoxP sites. Excision of exon2 results in a receptor lacking the ligand binding domain which is critical for receptor activation and signaling (Mishina et al., 2002). The resulting mice that were GFAP Cre, BMPR1a flox/− are hereafter referred to as BMPR1a CKO mice. Flox/+ mice (Cre negative) were used as wild-type littermate controls.
Adult BMPR1a CKO mice appeared phenotypically normal and had normal body weights (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Further, the distribution and numbers of NeuN (neurons)- and GFAP (astrocytes)-immunoreactive cells in the adult spinal cords of BMPR1a CKO mice were unchanged (supplemental Fig. 2E–H, available at www.jneurosci.org as supplemental material). Astrocytes cultured from the brains of postnatal day 2 BMPR1a CKO animals expressed GFAP (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). Araya et al. (2008) have shown that disruption of astrocytic BMPR1a signaling delays the formation of the blood–brain barrier and that these deficits disappear in adult animals. Consistent with this, we found no incidence of Iba1+ microglia or CD45+ circulating cells in the spinal cords of 2-month-old BMPR1a CKO mice (data not shown).
We confirmed the excision of the floxed allele in the adult spinal cord at 4 d post-SCI using real-time RT-PCR and found a >90% reduction (∼4 cycles; Fig. 2H). Similarly there was a comparable reduction in receptor transcript levels in astrocyte monolayers derived from BMPR1a CKO mice (∼5 cycles; supplemental Fig. 2D, available at www.jneurosci.org as supplemental material). These observations confirm that astrocytes in BMPR1a CKO mice are largely null for the functional receptor and indicate that the majority of BMPR1a is expressed on GFAP-expressing cells in the injured spinal cord.
Reduced astrocytic hypertrophy and increased inflammatory infiltration in BMPR1a CKO mice
GFAP immunohistochemistry of the spinal cords of injured WT mice at 1 week postinjury showed characteristic reactive astrocytes with enlarged soma and elaborate processes (Fig. 3A,C). In contrast, the BMPR1a CKO mice had smaller astrocytes with far fewer processes (Fig. 3A,C; see Fig. 8A,B). Quantification of the GFAP immunofluorescence intensity showed a significant reduction in the KO mice (GFAP ratios: WT, 2.66 ± 0.12; BMPR1a CKO, 1.62 ± 0.14; p < 0.0001; Fig. 3E). Since reactive astrocytosis has been implicated in limiting infiltration of macrophages/microglia, we used CD11b immunohistochemistry to determine whether ablation of BMPR1a altered this process. In WT animals, the majority of the CD11b+ inflammatory cells were compacted immediately adjacent to the lesion, whereas no such compaction of these cells was observed in the BMPR1a CKO animals (Fig. 3C,D). Most CD11b staining in injured WT animals was concentrated within 300 μm of the lesion, with significantly less staining 300–600 μm from the lesion. In contrast, in BMPR1a CKO mice there was no difference in staining intensity between the regions 0–300 μm and 300–600 μm from the lesion (Fig. 3F). The intensity of CD11b staining 300–600 μm from the lesion was significantly higher in the BMPR1a CKO mice, indicating that these cells were present in the parenchyma over greater distances (Fig. 3F, p < 0.004). Further, myelin + cresyl violet staining revealed greater loss of normal tissue in BMPR1a CKO injured cords (Fig. 3B). Since BMPR1a is not expressed by CD11b+ cells (Fig. 2G) and is largely restricted to GFAP+ cells (Fig. 2F) in the injured spinal cord, the increased infiltration of inflammatory cells is an indirect effect reflecting the defect in reactive gliosis.
Figure 3.
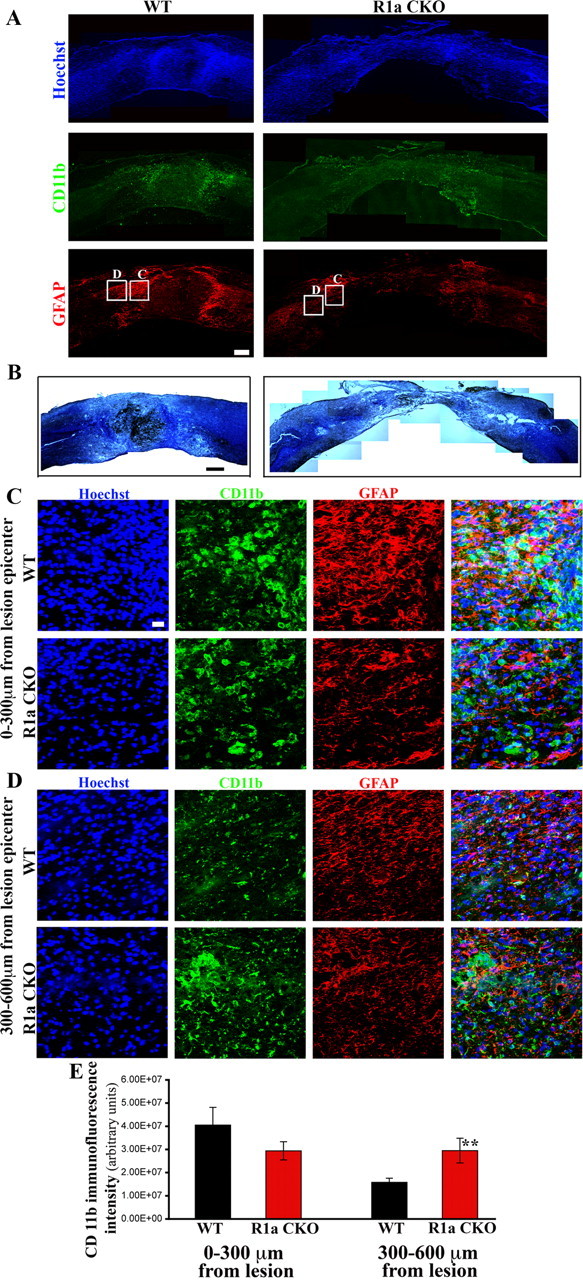
BMPR1a conditional knock-out mice show defective gliosis and increased inflammatory infiltration after spinal cord injury. A, Injured spinal cords at 1 week postinjury from BMPR1a WT and CKO mice. Rostral is to the left and dorsal to the top of the images. WT cords show intense GFAP+ astrocytes (red) staining at the edge of the lesion, a sign of reactive gliosis, that is not observed in the BMPR1a CKO mice. The astrocytes in the WT mice have condensed the CD11b-positive inflammatory cells(green) toward the lesion (intense green seen in and around the GFAP stain while these are still present diffusely in the parenchyma in the BMPR1a CKO mice. (Scale bar: 200 μm). B, WT and BMPR1a CKO spinal cords stained with a modified Weigert protocol (see Materials and Methods) and counterstained with cresyl violet showing greater tissue loss in BMPR1a CKO animals. (Scale bar: 250 μm). C, Higher-magnification (40×) images taken from the boxed regions shown in A. Note that there is intense CD11b staining in this area in the WT mice and the astrocytes have extended long processes. The astrocytes in the BMPR1a CKO mice have smaller, thinner processes. D, Images (40×) of boxed regions in A taken in the region spanning 300–600 μm from the edge of the lesion. Note the increased CD11b staining that persists in this area in the BMPR1a CKO mice that is comparable to the levels seen in C. Scale bars (C, D), 20 μm. Blue, Hoechst; green: CD11b; red, GFAP. E, Quantification of the CD11b staining intensity in the regions described in B and C in WT and BMPR1a CKO mice. There is a significantly higher CD11b staining in the region 300–600 μm from the lesion edge. (n = 3 WT and BMPR1a CKO animals **p < 0.0039 by Student's unpaired t test).
Figure 8.
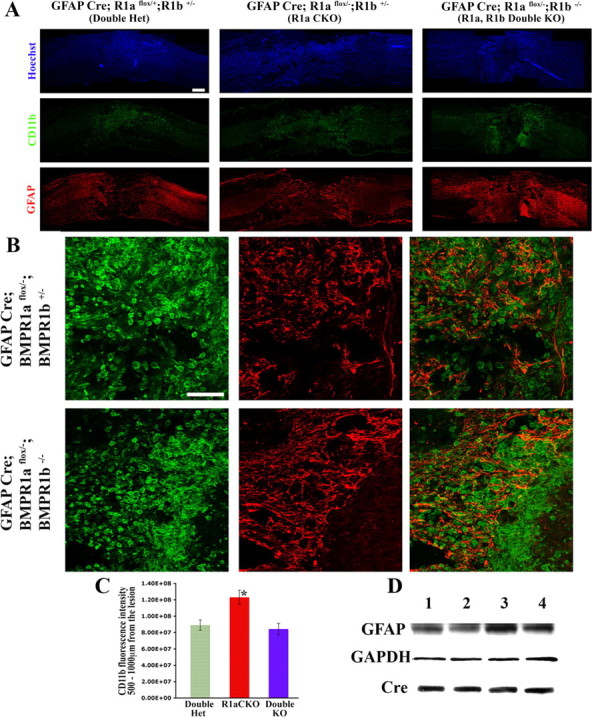
BMPR1a and BMPR1b have antagonistic roles in gliosis and wound closure after spinal cord injury. A, Injured spinal cords at 1 week postinjury from double heterozygotes (Double het), BMPR1a CKO and BMPR1a and BMPR1b double KO animals stained for Hoechst nuclear stain (blue), CD11b (green) and GFAP (red). CD11b+ cells have infiltrated greater distances into the cord in the BMPR1a CKO mice, but are tightly condensed in the double KO animals, similar to WT mice. Scale bar, 200 μm. B, Higher-magnification images, showing poor gliosis in the BMPR1a CKO mice. In contrast, the astrocytes in the double KO animals have normal processes and have formed a sharp boundary. The CD11b+ cells have been compacted toward the lesion and the bulk of these cells are enclosed next to the boundary formed by the reactive astrocytes. Scale bar, 100 μm. C, Quantification of the CD11b staining intensity in the region 500–1000 μm from the lesion edge. There is a significantly higher CD11b staining in this region in the BMPR1a CKO mice, but no difference between the double het and double KO animals (n = 3 animals in each group, p < 0.0001 by Tukey's post hoc t test). D, Western blots for GFAP protein in injured cords at 6 d post-SCI from the following littermates: Lane 1, GFAPCre;BMPR1a flox/+;BMPR1b +/− (double het); lane 2, GFAPCre;BMPR1a flox/−;BMPR1b+/− (BMPR1a KO); lane 3, GFAPCre;BMPR1a flox/+;BMPR1b −/− (BMPR1b KO); lane 4, GFAPCre;BMPR1a flox/−;BMPR1b −/− (double KO). The blot was stripped and reprobed for GAPDH (loading control) and Cre.
In the above experiments, the conditional knock-out mice were being compared with Cre-negative BMPR1a flox/+ littermates. Hence it was possible that the effects on gliosis were due to the presence of the Cre line in the knock-out animals. To rule out this possibility, we compared the gliosis at 1 week postinjury in GFAP Cre;BMPR1a +/+ animals with BMPR1a CKO mice. For this, we used cross-sections of the spinal cord and examined the GFAP+ cells (see Materials and Methods) (Fig. 4A,B). Within the first 200 μm from the lesion epicenter, the Cre+ WT animals showed marked gliosis with the characteristic reactive astrocytes in this region. However, these were strikingly reduced in the BMPR1a CKO mice (Fig. 4A,C). Quantification of the GFAP+ area in these mice showed a significant reduction in the BMPR1a CKO mice (GFAP+ area averages: GFAP Cre;BMPR1a +/+, 1048 ± 68 μm2; BMPR1a CKO, 605 ± 94 μm2, p < 0.01). Hence the effects on gliosis that were observed in BMPR1a CKO animals were not due to the presence of the Cre line. Thus, ablation of BMPR1a caused defective astrocytic hypertrophy with a consequent failure at limiting inflammatory cell infiltration.
Figure 4.
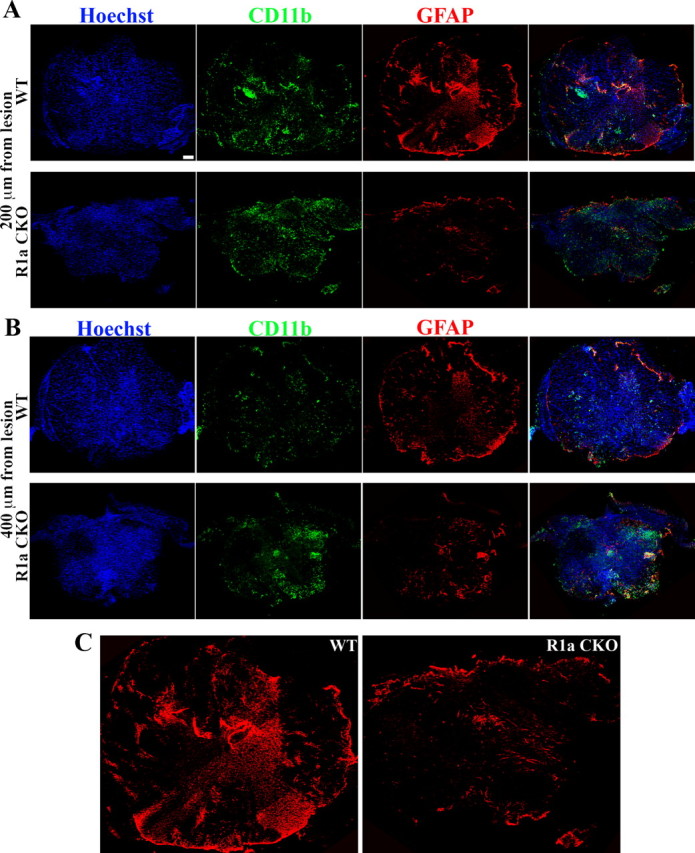
Reduction in GFAP area in BMPR1a CKO mice indicating defective gliosis is independent of the Cre line. A, Cross-sections taken at 200 μm distance from the lesion rostral to it from either GFAP Cre; BMPR1a +/+ (WT) animals or GFAP Cre; BMPR1a flox/− animals (BMPR1a CKO) at 1 week postinjury stained with Hoechst nuclear stain (blue), CD11b (green) and GFAP (red). B, Cross-sections similar to those from the genotypes in A at 400 μm distance from the lesion rostral to it. Scale bars (in A, B), 100 μm. C, Larger images of GFAP-stained sections shown in A showing reduced GFAP staining in the BMPR1a CKO compared with WT animals.
Consistent with the immunohistochemistry, we found a significant reduction in the levels of GFAP protein by Western analysis in BMPR1a CKO spinal cords at 1 week postinjury (Fig. 5A). Transcript levels of BMP4 and BMP7 after SCI did not differ between the groups, suggesting that the defects in gliosis did not reflect differences in the abundance of the ligands (Fig. 5B). In toto, these observations indicate that astrocytic BMPR1a signaling increases GFAP levels in reactive astrocytes.
Figure 5.
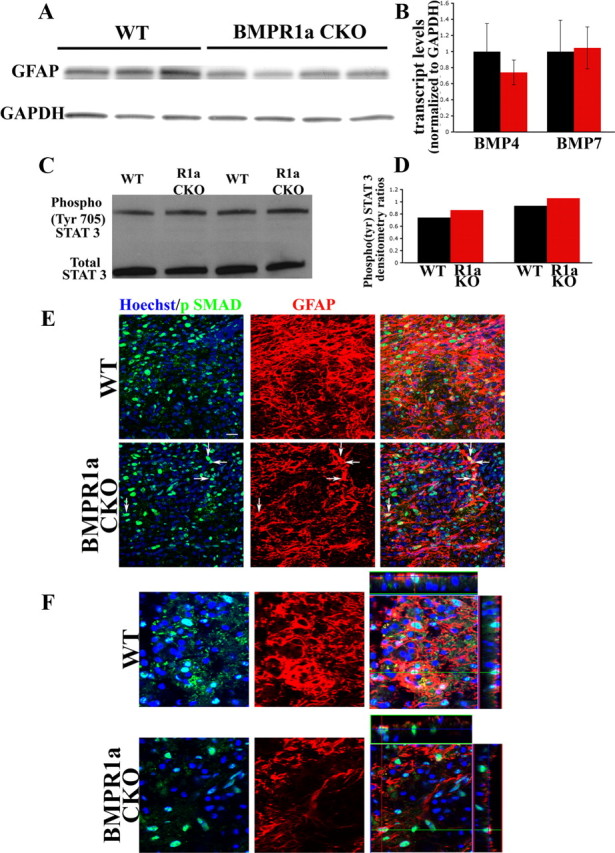
BMPR1a signaling affects GFAP levels independent of the canonical SMAD signaling pathway. A, Western blots for GFAP protein in injured cords from WT and BMPR1a CKO mice at 6 d post-SCI. The blot was stripped and reprobed for GAPDH (loading control). B, Normalized transcript levels of BMP4 and BMP7 in cords from WT and BMPR1a CKO mice at 6 d post-SCI showing no significant difference between the groups. C, Western blot analysis for Phospho (Tyr) STAT3 and total STAT3 in injured cords from 2 sets of littermates of WT and BMPR1a CKO animals at 4 d post-SCI. Activated STAT3 is present in the BMPR1a CKO mice. D, Densitometric analysis of the individual littermate pairs showing a slight increase in the levels of phospho-STAT3 as normalized to total STAT3 in the cords from BMPR1a CKO animals. E, Confocal Z-stacks taken from WT and BMPR1a CKO injured spinal cords (6 d post-SCI) stained for Hoechst nuclear stain (blue), npSMAD (green) and GFAP(red). Defective reactive astrocytes in BMPR1a CKO mice still have robust npSMAD (arrows). Scale bar, 20 μm. F, Single confocal Z-sections (from the stacks shown in E) taken at higher magnification with orthogonal projections showing intact npSMAD staining in the BMPR1a CKO astrocytes.
At 5 weeks post-SCI, quantification of GFAP immunofluorescence revealed a persistent but smaller reduction in the BMPR1a CKO mice (Fig. 6A) that was only borderline significant (GFAP ratios: WT, 2.44 ± 0.23; BMPR1a CKO, 2.0 + 0.16, p < 0.062). However, the lesion area, defined as the core area that is flanked by GFAP-positive astrocytes, was significantly larger in the BMPR1a CKO mice, indicating a larger lesion volume and a persistent failure in wound closure (Fig. 6A; supplemental Fig. 8A, available at www.jneurosci.org as supplemental material). We have refrained from measuring the lesion area at 1 week postinjury since the defect in astrocytic GFAP staining makes it difficult to establish a clear landmark for the lesion area at this stage in BMPR1a CKO mice. However, the increased lesion area at 5 weeks is consistent with the increase in infiltration that was present at 1 week postinjury. In addition, there continued to be a higher density of Iba1+ inflammatory cells in the area of the scar at 5 weeks postinjury (supplemental Fig. 8B,C, available at www.jneurosci.org as supplemental material). These observations suggest that BMPR1a signaling plays a more important role in early reactive astrocytic hypertrophy rather than glial scar progression.
Figure 6.
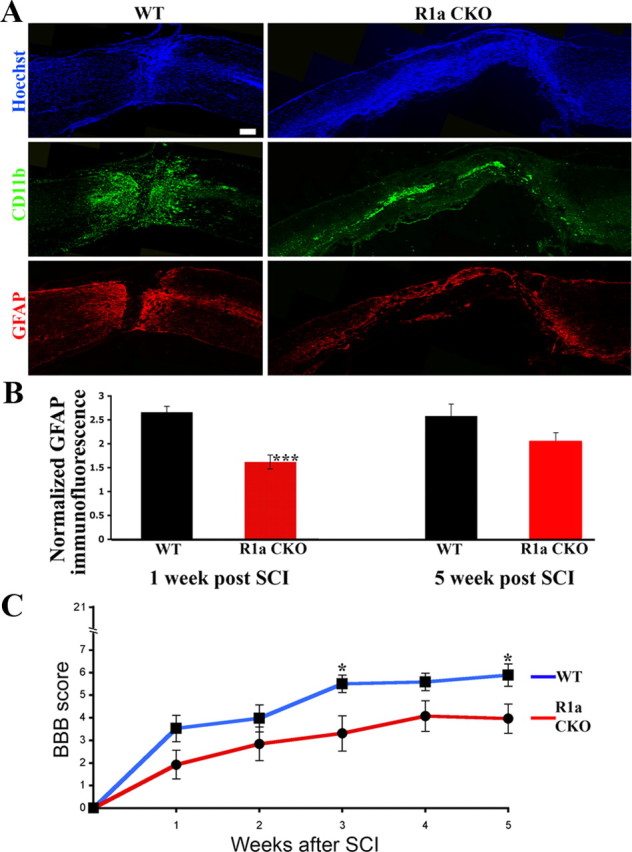
BMPR1a conditional knock-out mice have reduced gliosis and increased lesion volumes in the chronic injured spinal cord and show worsened locomotor recovery. A, WT and BMPR1a CKO mice at 5 weeks postinjury, stained with Hoechst nuclear stain (blue) and GFAP (red). Note the increased lesion area as marked by GFAP-negative area that is populated by the dense nuclear infiltrate and flanked by GFAP-positive astrocytes. Scale bar, 200 μm. B, Quantification of GFAP intensity in the BMPR1a CKO mice, in the area immediately adjacent to the lesion edge. Values are expressed as fold increases over uninjured spinal cord controls (1 week post-SCI: n = 3 WT and BMPR1a CKO animals, ***p < 0.00001 by Student's unpaired t test; 5 weeks post-SCI: n = 4 animals in either group, p < 0.06, by Student's unpaired t test). C, BBB scores of WT and BMPR1a CKO mice at weekly intervals after injury. The KO animals have significantly lower scores at 5 weeks postinjury (WT, 5.89 ± 0.4; KO, 4.0 ± 0.7; *p < 0.05 by Student's unpaired t test, n = 15 WT, n = 12 BMPR1a CKO animals).
BMPR1a regulates astrogliosis via a STAT3- and SMAD-independent pathway
The defect in gliosis with increased inflammatory cell infiltration after SCI in BMPR1a CKO mice is similar to what has been reported for mice with conditional null mutation of STAT3 (Okada et al., 2006; Herrmann et al., 2008). BMP signaling interacts with STAT3 signaling in several systems (Kawamura et al., 2000; Hjertner et al., 2001; Ying et al., 2003), and STAT3 may be involved in the effects of BMP signaling on expression of GFAP (Rajan et al., 2003; Fukuda et al., 2007). We therefore evaluated the phosphorylation status of STAT3 in injured spinal cords from BMPR1a CKO and wild-type mice. STAT3 activation remained intact in the BMPR1a CKO mice; in fact, BMPR1a CKO mice had slightly elevated levels of phosphorylated STAT3 (Fig. 5C,D). The increase in phospho-STAT3 after treatment with BMP4 was also intact in BMPR1a CKO postnatal day 2 neural stem cells (supplemental Fig. 4, available at www.jneurosci.org as supplemental material) despite a significant reduction in levels of BMPR1a. These observations suggest that activation of STAT3 by BMP4 does not require the BMPR1a receptor and that the reduction in gliosis in BMPR1a CKO mice occurs via a STAT3-independent pathway.
We also evaluated the localization of phospho-SMAD 1/5/8 in the injured spinal cord in WT and BMPR1a CKO animals. At 6 d post-SCI, the BMPR1a CKO astrocytes still displayed pronounced nuclear localized phospho-SMAD 1/5/8 (Fig. 5E,F), suggesting that effects of BMPR1a on reactive hypertrophy are not dependent on nuclear effects of SMAD signaling.
Accelerated wound closure and increased gliosis in BMPR1b knock-out mice
BMPR1b is expressed in the adult nervous system, although it is less abundant than BMPR1a (Zhang et al., 1998). BMPR1b-null (BMPR1b KO) mice are viable, survive to adulthood, and have no apparent phenotype in the nervous system (Yi et al., 2000). We also looked at the numbers of Neurons (NeuN) and astrocytes (GFAP) in adult WT and BMPR1b KO spinal cords and found no differences between the groups (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). We therefore examined effects of SCI in BMPR1b KO and used WT littermates as controls. At 1 week postinjury, BMPR1b KO mice developed reactive hypertrophic astrocytes (Fig. 7A). Immunohistochemistry for GFAP revealed hypertrophic astrocytes that did not appear different from their counterparts in WT littermates (Fig. 7A). However, Western blot analysis revealed significantly higher levels of GFAP protein in the BMPR1b KO spinal cords compared with WT controls [Fig. 7C; densitometric measurements of the GFAP protein levels (normalized to GAPDH) showed significantly higher levels in BMPR1b KO animals; p < 0.04 by Student's unpaired t test], suggesting an enhanced state of reactive astrogliosis. Consistent with this observation, the lesion area that was compacted by the reactive astrocytes was significantly smaller (Fig. 7A), and there was a smaller area of demyelination (Fig. 7B). Quantification of the GFAP-negative area in BMPR1b KO mice showed a lesion area that was only one-fourth that of WT controls (Fig. 7F; lesion area averages: WT, 977,140.45 ± 78,550 μm2; BMPR1b KO, 253,384 ± 27,205 μm2; p < 10−6). At this time, reactive astrocytes in WT animals had begun to compact CD11b+ cells toward the lesion, but the lesion core itself was still largely acellular, with the majority of the CD11b-positive cells intermingled with GFAP-positive astrocytes at the lesion's edge (Fig. 7A; also compare with WT BMPR1a animals in Fig. 3A). In contrast, in BMPR1b KO mice, the GFAP-negative area flanked by the reactive astrocytes was not acellular, and the lesion core showed dense staining for CD11b. Hence, there was not only a smaller lesion area in BMPR1b KO mice, but the CD11b+ cells were compacted further than in WT animals. Thus, the acutely injured BMPR1b KO spinal cord had a phenotype that was directly opposite to that in the BMPR1a CKO mice, with higher levels of GFAP protein and reduced infiltration by CD11b+ cells.
Figure 7.

BMPR1b knock-out mice have increased GFAP levels and reduced lesion volumes in acute stages following injury, but a reduced glial scar in chronic stages following injury. A, Low-magnification (10×) montages of WT and BMPR1b KO mouse spinal cords at 1 week postinjury, stained with Hoechst nuclear stain(blue), CD11b (green) and GFAP (red). The reactive gliosis at the edge of the lesion appears similar in WT and KO mice. However, the area compacted between the wall of reactive astrocytes is smaller in the KO. CD11b-positive cells have already been largely compacted into this area while this region continues to be mostly acellular in the WT mice as evidenced by the Hoechst stain. Scale bar, 200 μm. B, Spinal cord sections stained for myelin and cresyl violet showing smaller lesion size in BMPR1b KO mice. Scale bar, 250 μm. C, Western blot for GFAP from total protein extracted from injured spinal cords at 6 d postinjury from WT and BMPR1b KO animals. D, WT and BMPR1b KO spinal cords at 5 weeks postinjury, stained with Hoechst nuclear stain (blue) and GFAP (red). The GFAP-positive astrocytes have walled off the inflammatory cells but there are fewer astrocytes in the area adjacent to them. (Scale bar: 200 μm). E, Higher-magnification images of the area adjacent to the lesion (in D) showing reduced gliosis in the BMPR1b KO mice. Scale bar, 100 μm. F, Quantification of the GFAP-negative lesion area in WT and BMPR1b KO mice at 1 and 5 weeks postinjury.
Attenuated glial scar in BMPR1b KO mice
At 5 weeks there were no significant differences between WT and BMPR1b KO mice in the size of the GFAP-negative area (Fig. 7F). However, in BMPR1b KO mice there were fewer astrocytes in the parenchyma adjacent to the lesion (Fig. 7D,E), and quantitation of GFAP immunofluorescence revealed a significant decrease in the BMPR1b KO mice (average intensity ratios: WT, 2.69 ± 0.15; BMPR1b KO, 2.12 ± 0.18; p < 0.002). This suggests that BMPR1b signaling is involved in progression of the glial scar. The decreased gliosis in BMPR1b KO mice at 5 weeks could reflect either decreased astrocytic proliferation or diminished recruitment of astrocytes to the glial scar. To define possible effects on proliferation, mice were injected with the thymidine analog CldU daily from day 7 to day 14, and labeling was examined at 5 weeks post-SCI. There was no difference in CldU counts between WT and KO mice in the area of gliosis (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material), suggesting that the reduction in the glial scar at 5 weeks does not arise from reduced proliferation of astrocytes. In toto, these observations indicate that BMPR1a and BMPR1b signaling exert directly opposing effects on astrocytic hypertrophy and thereby on infiltration by inflammatory cells as well. Further, they have differing roles in the subsequent glial scar progression.
BMPR1a and BMPR1b exert antagonistic effects on wound closure and astrogliosis after injury
We next examined the effect of ablation of both BMPR1a and BMPR1b (see supplemental Fig. 6, available at www.jneurosci.org as supplemental material, for double knock-out strategy). In contrast to the reduced gliosis and increased inflammatory cell infiltration in BMPR1a CKO animals (Fig. 8A,C), the double KO animals surprisingly had normal-appearing reactive astrocytes (Fig. 8A,B) and the distribution of CD11b-positive cells resembled that in the double heterozygotes (Fig. 8C). We also examined the boundary formed by the reactive astrocytes in relation to the inflammatory cells. In the BMPR1a CKO animals, the reactive astrocytes had smaller and fewer processes and hence there was no sharp boundary formed as the inflammatory cells had invaded into this area (Fig. 8B). In contrast, astrocytes in the double KO animals formed a sharp boundary with the bulk of the CD11b cells compacted toward the lesion epicenter, similar to WT animals (Fig. 8B). Quantification of CD11b immunofluorescence intensity 500–1000 μm from the lesion epicenter was significantly higher in the BMPR1a CKO animals (p < 0.001), but was reduced in the double KO animals to levels indistinguishable from the double heterozygotes (Fig. 8C). These findings suggest that defects in wound closure in BMPR1a CKO mice, were due to increased BMPR1b signaling. Further, the lesion area in the double KO animals was no longer different from the WT animals (lesion area averages: double heterozygotes, 589,246 ± 154,035 μm2; double KO, 764,893 ± 95,399 μm2), suggesting that the accelerated wound closure observed in the BMPR1b KO animals (Fig. 7A,F) reflects increased BMPR1a signaling.
We finally performed Western blot analyses on protein extracts from injured cords (at 1 week post-SCI) from littermates obtained from the mating strategy designed to derive double knock-out animals. All the animals used were GFAP Cre positive to rule out the effects of the Cre line. We examined GFAP levels from double heterozygotes, BMPR1a CKO, BMPR1b KO and double KO animals (Fig. 8D). We found that BMPR1a CKO mice had reduced while BMPR1b KO mice had elevated GFAP protein levels. These effects were negated in the double knock-out mice, which had levels comparable to the double heterozygotes (Fig. 8D). This shows that BMPR1a and BMPR1b signaling exert opposite effects on GFAP levels and therefore on astrogliosis following injury. This further indicates that the regulation of GFAP postinjury by the two receptors occurs regardless of the background strain of mice. In toto, these findings show that BMPR1a and BMPR1b signaling exert opposing effects on astrogliosis and wound closure after SCI. BMPR1a positively and BMPR1b negatively regulates this process; disruption in the signaling from one receptor shifts the balance in favor of the effects mediated by the unaffected receptor (Fig. 9).
Figure 9.

Model showing the opposing effects of BMPR1a and BMPR1b signaling after spinal cord injury. Schematic representation of the effects of BMPR1a and BMPR1b signaling on reactive gliosis in the acutely injured spinal cord. SCI causes an upregulation of BMPR1a in reactive astrocytes (blue arrow in top row) in the WT cord, which promotes reactive astrocytic hypertrophy and limits infiltration by inflammatory cells (green ovals). BMPR1b signaling negatively regulates this process. In the absence of BMPR1a signaling, the balance shifts toward BMPR1b (dotted lines in the middle panel), which causes defective astrocytic hypertrophy and increased infiltration in the BMPR1a CKO animals. In contrast, in the BMPR1b KO mice, there is more pronounced BMPR1a signaling (dotted blue arrows) which causes a smaller lesion size with greater compaction of the inflammatory cells.
BMPR1a CKO mice have reduced axon density in the lesion area
Since the functional recovery following severe injuries is limited, we looked at axonal labeling in the lesion as a more direct readout of the signaling effects of BMPR1 signaling on gliosis. We therefore examined the WT, BMPR1a CKO and double KO spinal cords with SMI-312, a pan-axonal neurofilament marker at 1 week post-SCI. In contrast to individual antiphosphoneurofilaments that identify different subsets of neurofilament phosphoepitopes, SMI-312 is a mixture of high-titer monoclonals against the heavily phosphorylated forms of heavy and medium molecular weight neurofilaments (Ulfig et al., 1998) and therefore serves as a convenient marker for axons in general and especially suitable for defining functional and regional differences in normal and pathologic axons. It has been used to visualize axons in an area-specific maturation pattern in human fetal brain and also to visualize aberrantly sprouting axons in neuritic plaques derived from corticocortical fibers in Alzheimer's disease (Masliah et al., 1993).
Sections immediately adjacent to the ones used to measure CD11b distribution were used to look at axon density in the lesion area (see Materials and Methods). Even with severe compression injuries there are still some spared axons that are normally seen in the lesion which were observed in the WT animals (Fig. 10A,B). The BMPR1a CKO mice had an obvious disruption of these spared axons (Fig. 10A,B). Quantification of the axon density in the core of the lesion area showed that BMPR1a CKO animals showed significantly less axonal density compared with the WT animals (Fig. 10C, p < 0.01). Further, this reduction in axon labeling was reversed in the double knock-out animals that were statistically indistinguishable from the WT animals (Fig. 10C). We also normalized the axon density in the lesion area for each section to the density in an uninjured part of the cord >2 mm away from the lesion and there was still a significant reduction in the density observed in BMPR1a CKO animals (Fig. 10D, p < 0.01). This therefore indicates that astrocytic BMPR1a signaling in the acute stages following injury is critical for maintaining the integrity of spared/sprouting axons.
Figure 10.
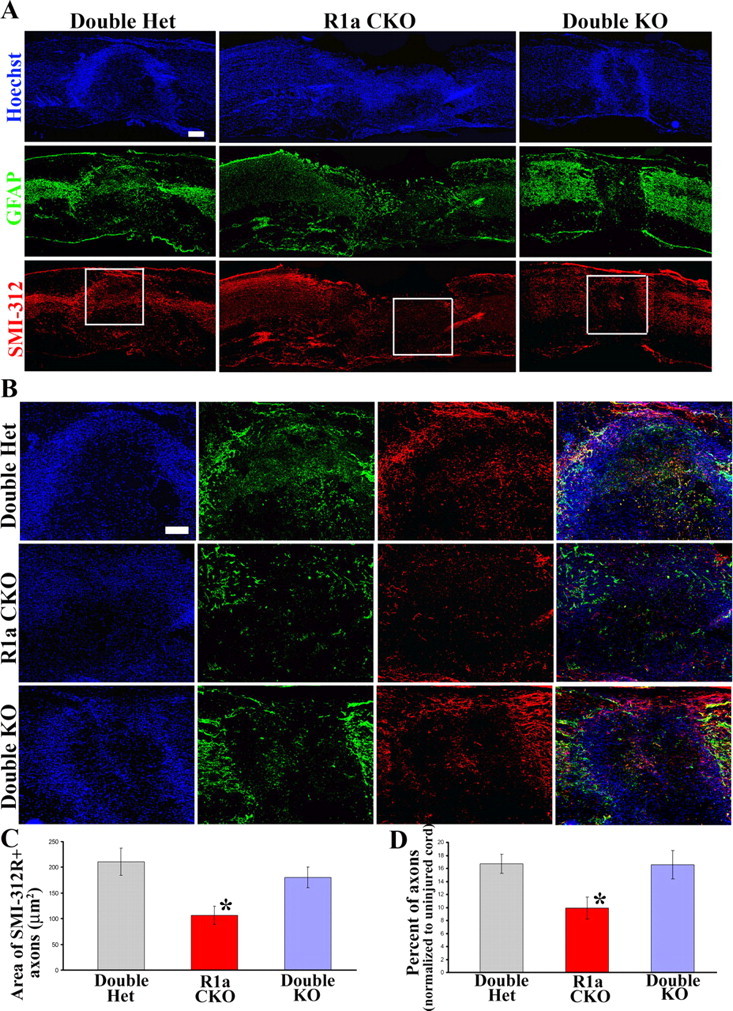
BMPR1a CKO mice have reduced axon density in the lesion area, which is rescued in the double KO mice. A, Low-magnification images of injured spinal cords at 1 week postinjury from WT, BMPR1a CKO and Double KO mice stained with Hoechst nuclear stain (blue), GFAP (green) and SMI-312R (red). (Scale bar: 200 μm). B, Boxed areas in A are shown in higher magnification. Note that there are significantly fewer axons in the lesion area in the BMPR1a KO animals, but a significant number in the double KO animals that is comparable to the WT. Scale bar, 100 μm. C, D, Quantification of total area of SMI-312+ axons in the lesion area (C) as well as normalized to the axon density in the uninjured part of each section (see Materials and Methods) and expressed as a percentage of the same (D).
We also examined the axon density in the lesion area in BMPR1b KO animals (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). SMI-312R immunostaining revealed that while the axon ends did come closer in the BMPR1b KO animals (supplemental Fig. 3G, available at www.jneurosci.org as supplemental material), presumably due to the smaller lesion size, there was not any notable difference in the density of axons in the lesion area compared with WT littermate controls. Quantification of the axon density in the lesion area revealed no difference between the groups (supplemental Fig. 3H, available at www.jneurosci.org as supplemental material).
BMPR1a knock-out mice show deficits in locomotor recovery after severe SCI
The BBB locomotor scale modified for the mouse (Basso et al., 1996; Joshi and Fehlings, 2002a) was used to assess behavioral recovery after SCI. BMPR1a CKO mice showed impaired locomotor recovery compared with WT littermates as early as 1 week after SCI, and this difference persisted through 5 weeks postinjury (Fig. 6D; average BBB scores at 5 weeks in WT, 5.89 ± 0.4 vs BMPR1a CKO, 4.0 ± 0.7, p < 0.05). This suggests that BMPR1a signaling in astrocytes facilitates functional recovery following SCI.
In contrast, BBB scoring showed no differences between BMPR1b WT and KO mice following SCI (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Thus, while loss of BMPR1a signaling impaired locomotor recovery, a null mutation of BMPR1b did not alter recovery. Since the injury model that was used in this study was particularly severe, assessment of functional recovery becomes difficult as the animals do not recover much hindlimb movement. Hence it is possible that the smaller lesion size in the BMPR1b KO could potentially have resulted in functional improvement, had a milder injury been used.
BMPR1a CKO astrocytes have higher levels of miR-21
The above experiments suggested that BMPR1a and BMPR1b signaling exert opposing effects on downstream targets that could, in turn, account for their opposing effects on gliosis. However, in an injury model in vivo, it becomes difficult to tease out such molecular differences, mainly due to the heterogeneity of cell types that are present in the injury site. Hence to elucidate possible downstream differences between the two receptor signaling cascades, we used an in vitro system to specifically address these differences in astrocytes. For this, we cultured serum-derived astrocytes from both KO animals. We examined levels of GFAP by quantitative real-time PCR in these cultures and found that GFAP transcript levels were lower in BMPR1a CKO astrocytes when compared with WT littermate controls (Fig. 11B). BMPR1b mutant astrocytes did not differ in their levels of GFAP (Fig. 11D). We found no difference in the levels of the Id proteins that are known to mediate the effects of BMP4 on GFAP transcription (data not shown). We therefore sought to look for other potential mechanisms that could account for the reduction in GFAP levels.
Figure 11.

Opposing effects of BMPR1a and BMPR1b signaling on posttranscriptional regulation of microRNA-21. A, Real-time Taqman-PCR amplification curves for mature miR-21 in astrocytes derived from WT (blue traces) and BMPR1a CKO (red traces) pups snoR202 serves as a loading control. There is a ∼2 cycle increase in miR-21 levels in BMPR1a CKO astrocytes. B, Normalized transcript levels of pri- and pre-miR-21 in astrocytes derived from WT and BMPR1a CKO pups showing no significant difference between the groups. The KO astrocytes, however, have a significant reduction in GFAP transcript levels. C, Levels of mature miR-21 (normalized to snoR202) in astrocytes derived from WT (Cre−; BMPR1a flox/+) and BMPR1a CKO pups showing a significant increase in the KO. The increase is reversed, and levels are comparable to WT in double KO astrocytes. D, Normalized transcript levels of pri-, pre (normalized to GAPDH)-, and mature miR-21 (normalized to snoR202) in astrocytes derived from either WT (black bars) or BMPR1b KO (blue bars) pups. There is no difference between the groups. E, Normalized transcript levels of genes shown on the x-axis, in astrocytes derived from BMPR1a flox/flox pups and infected with either control GFP-expressing adenovirus (adeno-GFP, green bars) or Cre-expressing adenovirus (adeno-Cre, brown bars). Levels of pri- and pre-miR-21 as well as GFAP were normalized to GAPDH, miR-21 levels were normalized to snoR202. Note that the Cre infected astrocytes have a similar increase in the levels of miR-21 and significant reduction in GFAP transcript levels.
MicroRNAs are endogenous ∼22-nucleotide-long RNAs that have been identified to play important gene regulatory roles by pairing to the messages of protein-coding genes to specify mRNA cleavage or repression of productive translation (Bartel, 2004). MicroRNAs are first transcribed by RNA polymerase II as a primary miRNA (pri-miRNA) transcript that contains an inverted repeat that folds into a short hairpin. The hairpin is cleaved from the rest of the transcript into a pre-miRNA by a nuclear microprocessor complex, into a pre-miRNA hairpin product. This is then exported from the nucleus to the cytoplasm where Dicer processes the pre-miRNA hairpin into a 21–24 nucleotide duplex RNA and subsequently aids in the assembly of the miRNA effector complex, the miRNA-induced silencing complex (miRISC), which can then interact with target messenger RNAs via complementary base pairing (Reinke and Carthew, 2008).
Davis et al. have recently shown that SMAD proteins control the posttranscriptional processing of microRNA-21 (miR-21) (Davis et al., 2008). We therefore, evaluated whether BMPR1a and BMPR1b signaling have any effects on levels of miR-21 in our cultured astrocytes. We found that BMPR1a CKO astrocytes had significantly higher levels of miR-21 (Fig. 11A). However, there was no difference in the levels of pri- or pre-miR-21 (Fig. 11B), suggesting that BMPR1a signaling was negatively regulating the final cytoplasmic processing of pre-miR-21 to mature miR-21. We next examined the levels of miR-21 in BMPR1b KO and found no difference in the levels of miR-21 in BMPR1b WT versus KO astrocytes (Fig. 11D). It was possible, however, that the effects of BMPR1b signaling, were being obscured by BMPR1a, since the cultured astrocytes express very high levels of BMPR1a compared with BMPR1b (data not shown). We hence evaluated the levels of miR-21 in the double KO astrocytes. In these astrocytes, the levels of miR-21 were the same as the wild-type littermate controls (Fig. 11C). Hence the increase in miR-21 in the BMPR1a CKO animals was due to an increase in BMPR1b signaling.
In these studies described above, it was possible that the original progenitor cells from the BMPR1a CKO pups were different when originally plated and hence the increase in miR-21 levels could simply reflect differences in their developmental origin rather than a specific role for BMPR1a in astrocytes themselves. To rule out this possibility as well as the effect of the GFAP Cre line, we cultured astrocytes from BMPR1a flox/flox pups and then infected them with an adenovirus expressing either GFP (control; adeno-GFP) or Cre (adeno-Cre). Ablation of the receptor was confirmed using real-time PCR (Fig. 11E). Once again the adeno-Cre astrocytes had higher levels of miR-21 (p < 0.01) with no effect on pri- or pre-mi-R21 (Fig. 11E) when compared with control adeno-GFP astrocytes. Also, the BMPR1a KO astrocytes had reduced levels of GFAP (Fig. 11E), mimicking the effects observed in the BMPR1a CKO astrocytes. Hence BMPR1a signaling negatively regulates miR-21 levels in astrocytes posttranscriptionally.
miR-21 negatively regulates GFAP in astrocytes
Since BMPR1a ablation caused an increase in miR-21 levels and reduced levels of GFAP, we sought to determine whether miR-21 could affect GFAP levels in astrocytes. For this, we overexpressed miR-21 using a lentiviral overexpression system. The mature miR-21 sequence has been previously described (Lagos-Quintana et al., 2002). The primary mouse miR-21 transcript sequence was identified using the Sanger database and cloned into the lentiviral vector plemiR (see Materials and Methods). Normal astrocytes were infected with either a control RFP-expressing lentivirus (plemiR) or with virus overexpressing miR-21 (plenti-miR-21). Infected cells were selected using puromycin for 4 d at a concentration of 2 μg/ml (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). The RFP fluorescence showed that control astrocytes exhibited the normal morphology of serum-derived astrocytes with a large cell soma and spread-out processes. On the other hand, astrocytes overexpressing miR-21 had smaller processes and reduced cell size (Fig. 12E). We confirmed the overexpression of miR-21 in the astrocytes after selection, using real-time PCR (Fig. 12A,B). Interestingly, we found that the astrocytes overexpressing miR-21 had a reduction (∼50%/1 cycle) in the levels of endogenous miR-21 (Fig. 12A). The miR-21 overexpressing astrocytes had a dramatic reduction in GFAP levels, both at the transcriptional (Fig. 12C) and protein level (Fig. 12D). However, there was no significant effect on the levels of BMPR1a transcript levels, suggesting that the effect on the astrocytes was not directly the result of an opposing effect on the receptor (Fig. 12C). Hence miR-21 negatively regulates GFAP and cell size in astrocytes.
Figure 12.

MicroRNA-21 is a negative regulator of astrocytic size and GFAP levels. A, Real-time SYBR-green PCR amplification plots from astrocytes infected with lentiviruses expressing either RFP (plenti-RFP, blue traces) or miR-21 (plenti-miR-21, red traces). RNA was extracted 4 d after selection (see Materials and Methods). Note that there is a 1-cycle reduction in the levels of pri-miR-21 in the astrocytes overexpressing miR-21. Also there is a significant reduction in the levels of GFAP transcripts in astrocytes overexpressing mir-21. B, Real-time Taqman PCR amplification plots for miR-21 in the same astrocytes as in A showing the increase in miR-21. C, GFAP and BMPR1a transcript levels (normalized to GAPDH) in astrocytes described above. Graphs represent an average of three separate astrocyte cultures ± SEM. D, Western blot analysis showing the significant reduction in GFAP protein levels in astrocytes overexpressing miR-21. GAPDH serves as a loading control. E, Bright field (left columns) and fluorescence (right columns) images for RFP showing the significant reduction in cell size in astrocytes overexpressing miR-21 compared with control RFP. Scale bar: 20 μm.
Discussion
Since BMP signaling plays a fundamental role in generation of astrocytes (Gross et al., 1996; Mabie et al., 1997; Gomes et al., 2003) several groups studied the effects of globally inhibiting BMP signaling after SCI with mixed results (Setoguchi et al., 2004; Enzmann et al., 2005; Matsuura et al., 2008). Astrogliosis has both beneficial as well as detrimental effects (Barnabé-Heider and Frisén, 2008), and global inhibition of BMP signaling does not specifically target the detrimental aspects of gliosis. We show here that BMPR1a signaling promotes reactive gliosis and wound closure whereas BMPR1b signaling inhibits these beneficial processes and increases formation of the glial scar.
BMPR1a and BMPR1b signaling may not serve identical biological roles
Both BMPR1a and BMPR1b are expressed in the nervous system (Zhang et al., 1998), where they may serve overlapping functions (Qin et al., 2006) but BMPR1a and BMPR1b cannot be viewed as being biologically interchangeable under all circumstances (Araya et al., 2008) (Panchision et al., 2001; Gulacsi and Lillien, 2003; Brederlau et al., 2004).
We find that BMPR1a and BMPR1b not only serve different roles, but exert opposing effects. Ablation of astrocytic BMPR1a led to poor reactive gliosis with increased infiltration. BMPR1b KO mice showed “hyperreactive” astrocytes and significantly reduced lesion areas at 1 week postinjury. These effects disappeared in double knock-out mice, suggesting that the acute effects in the BMPR1b KO were dependent upon increased BMPR1a signaling. This suggests that there is a tight balance between the signaling from these two receptors. Following injury, this balance shifts toward BMPR1a signaling to promote the beneficial effects in wound repair. Further, the two type I receptors exert opposing effects on levels of astrocytic miR-21. BMPR1a decreases levels of miR-21 and BMPR1b opposes this effect.
Although we find that both type 1 receptors are expressed by virtually all astrocytes, differences in functions of BMPR1a and BMPR1b could also conceivably reflect differences in the spatiotemporal distribution of the receptors. It is possible that there are subpopulations of astrocytes that preferentially express one receptor subtype that respond differently to BMP signaling because of other unidentified phenotypic differences. In any case, our findings demonstrate that the two different type I receptors serve strikingly different roles in the injured spinal cord and that the final outcome results from a competition between the two receptor subunits.
BMPR1a positively and BMPR1b negatively regulates wound closure following SCI
BMPR1a CKO mice had increased lesion volumes because of increased inflammatory cell infiltration. This parallels the ablation of reactive astrocytes in the initial stages of injury (Faulkner et al., 2004) indicating the vital role of BMPR1a signaling in this process. BMPR1b KO mice conversely had smaller lesion volumes accompanied with increase GFAP levels. Both effects require BMPR1a and occur only after injury (GFAP levels are normal in the uninjured spinal cord). They temporally coincide with the highest levels of BMPR1a levels in the injured cord. At 1 week post-SCI, when the positive effect on lesion volume is most pronounced, levels of BMPR1a are at their peak (Fig. 2).
Infiltration by inflammatory cells occurs in several waves, with a peak of infiltration that occurs initially after injury and one that occurs several weeks after the assault (Kigerl et al., 2006). The second phase is believed to be responsible for secondary pathogenesis (Trivedi et al., 2006). Astrocytes in BMPR1b KO mice are better able to condense the initial infiltrating cells, but in chronic stages, as levels of BMPR1a fall, this ability is lost as more cells arrive in the lesion. This suggests that therapeutic interventions might not only seek to limit BMPR1b signaling but also to extend the period of elevated BMPR1a signaling.
BMPR1a signaling in astrocytes has a role in the formation of the blood–brain-barrier (Araya et al., 2008) and the increased infiltration in BMPR1a CKO mice could reflect delayed closure of the damaged blood–brain-barrier. STAT3 is a critical regulator of this process (Okada et al., 2006; Herrmann et al., 2008), but the levels of activated STAT3 in the BMPR1a CKO mice after SCI were not reduced. Hence, the downstream effectors of BMPR1a signaling are distinct from this pathway.
BMPR1b plays a more significant role in glial scar progression
There was an attenuation of the glial scar at 5 weeks after the injury in the BMPR1b KO. In contrast, BMPR1a CKO animals had greater deficits in the initial reactive hypertrophy, but by 5 weeks postinjury, the difference in gliosis between the groups was less significant. This suggests that BMPR1b signaling regulates glial scar progression. In turn this suggests that the two distinct phases of gliosis, hypertrophy followed by hyperplasia, are regulated by different mechanisms. These observations indicate the duality of signaling pathways that regulate astrocytic hypertrophy versus hyperplasia and suggest that it may be possible therapeutically to reduce gliosis without affecting the initial hypertrophic response or wound healing.
It is possible that the reduced gliosis in the BMPR1b KO is secondary to the earlier effects that limit inflammatory cell infiltration. Infiltrating leukocytes produce mediators that can increase gliosis (Yong et al., 1991; Balasingam et al., 1994), and disruption of signaling mechanisms that perturb this response in astrocytes reduces gliosis (Goldshmit et al., 2004). Hence, the reduced gliosis in the BMPR1b KO could secondarily reflect the reduced cell infiltration in the more acute stages of the injury.
Astrocytic BMPR1a and not BMPR1b signaling is essential for functional recovery after SCI
BMPR1a CKO mice showed poor locomotor recovery beginning in the first week, which persisted in the chronic stages. This is consistent with prior findings where disruption of reactive astrocytes caused worsened functional outcomes (Faulkner et al., 2004; Okada et al., 2006; Herrmann et al., 2008). This was also consistent with the fact that BMPR1a ablation caused a significant reduction in axon density in the lesion area. This further suggests that reactive gliosis in the acute stages following injury is critical for maintaining spared axons and that defects in gliosis compromise the integrity of these axons that has clear implications for functional recovery.
BMPR1b KO mice exhibited normal but not enhanced locomotor recovery despite the accelerated wound closure in these animals. This contrasts the report that ablation of Socs3, which results in similar anatomical findings of accelerated wound closure, is associated with improved BBB scores (Okada et al., 2006). This could reflect a difference in the severity of the injuries that were used in these two studies since the injury in the current study was more severe than the one used by Okada et al. However, there could be other reasons for the lack of locomotor improvement in the BMPR1b KO animals. First, due to the severity of the injury, any subtle improvement would not be manifest, since these animals have extremely limited locomotor performance. Second, we also did not find any difference in SMI-312 density in the lesion area, which would be consistent with this finding. The lack of change in axon density, in turn, could reflect the fact that the BMPR1b KO animals had an increase in levels of GFAP indicating an increase in gliosis, which could, possibly inhibit axonal sprouting. Third, the smaller lesion size in these animals, appears only transiently, since by 5 weeks post-SCI, the lesion size is indistinguishable from the WT littermates. This is in direct contrast to the BMPR1a CKO mice, where the defect in wound closure is even apparent in the chronically injured cord. It is also possible that Socs3 is involved in other components of functional recovery that are unaffected by BMPR1b signaling.
Recent evidence has shown an astrocytic role in the development of neuropathic pain after SCI (Davies et al., 2008). However, severe cases of SCI exhibit a lesser incidence of neuropathic pain compared with milder incomplete lesions (Siddall et al., 1995). Hence, future studies to evaluate such modalities in these knock-out animals, with milder lesions, would be useful not only in correlating it to gliosis, but also if these pathways are the targets of future therapies after SCI.
miR-21 is a regulator of astrocytic GFAP
MicroRNAs have been identified as important regulators of many biological processes (Stefani and Slack, 2008). We find that BMPR1a signaling downregulates miR-21 levels in astrocytes. BMPR1b ablation alone did not affect miR-21. However, BMPR1b ablation rescued the effect of increased miR-21 in BMPR1a KO astrocytes. This shows that the two receptors have opposing effects in regulating miR-21.
miR-21 overexpression mimics the effects of BMPR1a ablation on GFAP. However, the effects with miR-21 overexpression were more pronounced. This is probably because the increase in miR-21 in the BMPR1a KO astrocytes is more modest compared with the levels obtained with lentiviral overexpression. BMPR1a ablation affected posttranscriptional processing of miR-21 without affecting the primary transcript or pre-miR21 levels. This suggests that the effects occur after the pre-micro RNA is exported into the cytoplasm. This is consistent with the finding that the BMPR1a CKO astrocytes, exhibited no difference in the presence of npSMAD.
miR-21 overexpression in astrocytes caused a reduction in GFAP levels as well as in astrocytic size suggesting that miR-21 negatively regulates reactive astrocytic hypertrophy. It remains to be determined whether GFAP is a direct target of miR-21 and whether the effect of cell size is due to an effect on GFAP or vice-versa. Given the more global effect on astrocytic cell size, it seems that the latter possibility is more likely. Further, the algorithm “Targetscan” typically used to predict microRNA targets, does not predict GFAP as a target of miR-21. Hence, miR-21 must affect other targets that in turn affect astrocytic size. Future studies that identify such targets could potentially yield novel designs for therapeutic manipulation of reactive gliosis.
In summary, these findings indicate that BMP signaling regulates multiple stages of gliosis with BMPR1a and BMPR1b exerting opposing effects on the initial reactive hypertrophy. Specific inhibition of BMPR1b signaling for therapeutic purposes, possibly coupled with enhanced BMPR1a signaling, may provide an approach for manipulating gliosis and enhancing functional outcomes after SCI.
Footnotes
This project was supported by a grant from the Morton Cure Paralysis Fund and by National Institutes of Health Grants R01 NS20778 and NS20013 to J.A.K. We thank Yuji Mishina for the BMPR1a mouse lines. Karen Lyons provided us with the BMPR1b KO mouse line. We thank Michael Sofroniew for providing the GFAP Cre mouse line as well as helpful discussions and critical comments on the data.
References
- Araya R, Kudo M, Kawano M, Ishii K, Hashikawa T, Iwasato T, Itohara S, Terasaki T, Oohira A, Mishina Y, Yamada M. BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol Cell Neurosci. 2008;38:417–430. doi: 10.1016/j.mcn.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasingam V, Tejada-Berges T, Wright E, Bouckova R, Yong VW. Reactive astrogliosis in the neonatal mouse brain and its modulation by cytokines. J Neurosci. 1994;14:846–856. doi: 10.1523/JNEUROSCI.14-02-00846.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin SA, Scheff SW. Intermediate filament change in astrocytes following mild cortical contusion. Glia. 1996;16:266–275. doi: 10.1002/(SICI)1098-1136(199603)16:3<266::AID-GLIA9>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Barnabé-Heider F, Frisén J. Stem cells for spinal cord repair. Cell Stem Cell. 2008;3:16–24. doi: 10.1016/j.stem.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W. MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter Animal Spinal Cord Injury Study. J Neurotrauma. 1996;13:343–359. doi: 10.1089/neu.1996.13.343. [DOI] [PubMed] [Google Scholar]
- Brederlau A, Faigle R, Elmi M, Zarebski A, Sjöberg S, Fujii M, Miyazono K, Funa K. The bone morphogenetic protein type Ib receptor is a major mediator of glial differentiation and cell survival in adult hippocampal progenitor cell culture. Mol Biol Cell. 2004;15:3863–3875. doi: 10.1091/mbc.E03-08-0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Leong SY, Schachner M. Differential expression of cell fate determinants in neurons and glial cells of adult mouse spinal cord after compression injury. Eur J Neurosci. 2005;22:1895–1906. doi: 10.1111/j.1460-9568.2005.04348.x. [DOI] [PubMed] [Google Scholar]
- Cheng X, Wang Y, He Q, Qiu M, Whittemore SR, Cao Q. Bone morphogenetic protein signaling and olig1/2 interact to regulate the differentiation and maturation of adult oligodendrocyte precursor cells. Stem Cells. 2007;25:3204–3214. doi: 10.1634/stemcells.2007-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JE, Pröschel C, Zhang N, Noble M, Mayer-Pröschel M, Davies SJ. Transplanted astrocytes derived from BMP- or CNTF-treated glial-restricted precursors have opposite effects on recovery and allodynia after spinal cord injury. J Biol. 2008;7:24. doi: 10.1186/jbiol85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, Hata A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008;454:56–61. doi: 10.1038/nature07086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebendal T, Bengtsson H, Söderström S. Bone morphogenetic proteins and their receptors: potential functions in the brain. J Neurosci Res. 1998;51:139–146. doi: 10.1002/(SICI)1097-4547(19980115)51:2<139::AID-JNR2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Enzmann GU, Benton RL, Woock JP, Howard RM, Tsoulfas P, Whittemore SR. Consequences of noggin expression by neural stem, glial, and neuronal precursor cells engrafted into the injured spinal cord. Exp Neurol. 2005;195:293–304. doi: 10.1016/j.expneurol.2005.04.021. [DOI] [PubMed] [Google Scholar]
- Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Abematsu M, Mori H, Yanagisawa M, Kagawa T, Nakashima K, Yoshimura A, Taga T. Potentiation of astrogliogenesis by STAT3-mediated activation of bone morphogenetic protein-Smad signaling in neural stem cells. Mol Cell Biol. 2007;27:4931–4937. doi: 10.1128/MCB.02435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Goldshmit Y, Galea MP, Wise G, Bartlett PF, Turnley AM. Axonal regeneration and lack of astrocytic gliosis in EphA4-deficient mice. J Neurosci. 2004;24:10064–10073. doi: 10.1523/JNEUROSCI.2981-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 2003;255:164–177. doi: 10.1016/s0012-1606(02)00037-4. [DOI] [PubMed] [Google Scholar]
- Grider MH, Chen Q, Shine HD. Semi-automated quantification of axonal densities in labeled CNS tissue. J Neurosci Methods. 2006;155:172–179. doi: 10.1016/j.jneumeth.2005.12.021. [DOI] [PubMed] [Google Scholar]
- Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 1996;17:595–606. doi: 10.1016/s0896-6273(00)80193-2. [DOI] [PubMed] [Google Scholar]
- Gulacsi A, Lillien L. Sonic hedgehog and bone morphogenetic protein regulate interneuron development from dorsal telencephalic progenitors in vitro. J Neurosci. 2003;23:9862–9872. doi: 10.1523/JNEUROSCI.23-30-09862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjertner O, Hjorth-Hansen H, Börset M, Seidel C, Waage A, Sundan A. Bone morphogenetic protein-4 inhibits proliferation and induces apoptosis of multiple myeloma cells. Blood. 2001;97:516–522. doi: 10.1182/blood.v97.2.516. [DOI] [PubMed] [Google Scholar]
- Joshi M, Fehlings MG. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the mouse: Part 1. Clip design, behavioral outcomes, and histopathology. J Neurotrauma. 2002a;19:175–190. doi: 10.1089/08977150252806947. [DOI] [PubMed] [Google Scholar]
- Joshi M, Fehlings MG. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the mouse: Part 2. Quantitative neuroanatomical assessment and analysis of the relationships between axonal tracts, residual tissue, and locomotor recovery. J Neurotrauma. 2002b;19:191–203. doi: 10.1089/08977150252806956. [DOI] [PubMed] [Google Scholar]
- Kawamura C, Kizaki M, Yamato K, Uchida H, Fukuchi Y, Hattori Y, Koseki T, Nishihara T, Ikeda Y. Bone morphogenetic protein-2 induces apoptosis in human myeloma cells with modulation of STAT3. Blood. 2000;96:2005–2011. [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig BB, Cook JS, Wolsing DH, Ting J, Tiesman JP, Correa PE, Olson CA, Pecquet AL, Ventura F, Grant RA, Chen GX, Wrana JL, Massagué J, Rosenbaum JS. Characterization and cloning of a receptor for BMP-2 and BMP-4 from NIH 3T3 cells. Mol Cell Biol. 1994;14:5961–5974. doi: 10.1128/mcb.14.9.5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Liu F, Ventura F, Doody J, Massagué J. Human type II receptor for bone morphogenic proteins (BMPs): extension of the two-kinase receptor model to the BMPs. Mol Cell Biol. 1995;15:3479–3486. doi: 10.1128/mcb.15.7.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler JA. Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial-astroglial progenitor cells. J Neurosci. 1997;17:4112–4120. doi: 10.1523/JNEUROSCI.17-11-04112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R. An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer's disease. Am J Pathol. 1993;142:871–882. [PMC free article] [PubMed] [Google Scholar]
- Matsuura I, Taniguchi J, Hata K, Saeki N, Yamashita T. BMP inhibition enhances axonal growth and functional recovery after spinal cord injury. J Neurochem. 2008;105:1471–1479. doi: 10.1111/j.1471-4159.2008.05251.x. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y, Hanks MC, Miura S, Tallquist MD, Behringer RR. Generation of Bmpr/Alk3 conditional knockout mice. Genesis. 2002;32:69–72. doi: 10.1002/gene.10038. [DOI] [PubMed] [Google Scholar]
- Nohno T, Ishikawa T, Saito T, Hosokawa K, Noji S, Wolsing DH, Rosenbaum JS. Identification of a human type II receptor for bone morphogenetic protein-4 that forms differential heteromeric complexes with bone morphogenetic protein type I receptors. J Biol Chem. 1995;270:22522–22526. doi: 10.1074/jbc.270.38.22522. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD. Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev. 2001;15:2094–2110. doi: 10.1101/gad.894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wine-Lee L, Ahn KJ, Crenshaw EB., 3rd Genetic analyses demonstrate that bone morphogenetic protein signaling is required for embryonic cerebellar development. J Neurosci. 2006;26:1896–1905. doi: 10.1523/JNEUROSCI.3202-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan P, Panchision DM, Newell LF, McKay RD. BMPs signal alternately through a SMAD or FRAP-STAT pathway to regulate fate choice in CNS stem cells. J Cell Biol. 2003;161:911–921. doi: 10.1083/jcb.200211021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke CA, Carthew RW. BMP signaling goes posttranscriptional in a microRNA sort of way. Dev Cell. 2008;15:174–175. doi: 10.1016/j.devcel.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig BL, Imamura T, Okadome T, Cox GN, Yamashita H, ten Dijke P, Heldin CH, Miyazono K. Cloning and characterization of a human type II receptor for bone morphogenetic proteins. Proc Natl Acad Sci U S A. 1995;92:7632–7636. doi: 10.1073/pnas.92.17.7632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Burke GM, McGuire T, Pisarek AJ, Mukhopadhyay A, Mishina Y, Kessler JA. BMPR1a signaling determines numbers of oligodendrocytes and calbindin-expressing interneurons in the cortex. J Neurosci. 2007;27:7397–7407. doi: 10.1523/JNEUROSCI.1434-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See J, Mamontov P, Ahn K, Wine-Lee L, Crenshaw EB, 3rd, Grinspan JB. BMP signaling mutant mice exhibit glial cell maturation defects. Mol Cell Neurosci. 2007;35:171–182. doi: 10.1016/j.mcn.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoguchi T, Yone K, Matsuoka E, Takenouchi H, Nakashima K, Sakou T, Komiya S, Izumo S. Traumatic injury-induced BMP7 expression in the adult rat spinal cord. Brain Res. 2001;921:219–225. doi: 10.1016/s0006-8993(01)03123-7. [DOI] [PubMed] [Google Scholar]
- Setoguchi T, Nakashima K, Takizawa T, Yanagisawa M, Ochiai W, Okabe M, Yone K, Komiya S, Taga T. Treatment of spinal cord injury by transplantation of fetal neural precursor cells engineered to express BMP inhibitor. Exp Neurol. 2004;189:33–44. doi: 10.1016/j.expneurol.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Siddall P, Xu CL, Cousins M. Allodynia following traumatic spinal cord injury in the rat. Neuroreport. 1995;6:1241–1244. doi: 10.1097/00001756-199506090-00003. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230. doi: 10.1038/nrm2347. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Yamashita H, Sampath TK, Reddi AH, Estevez M, Riddle DL, Ichijo H, Heldin CH, Miyazono K. Identification of type I receptors for osteogenic protein-1 and bone morphogenetic protein-4. J Biol Chem. 1994;269:16985–16988. [PubMed] [Google Scholar]
- Trivedi A, Olivas AD, Noble-Haeusslein LJ. Inflammation and spinal cord injury: infiltrating leukocytes as determinants of injury and repair processes. Clin Neurosci Res. 2006;6:283–292. doi: 10.1016/j.cnr.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulfig N, Nickel J, Bohl J. Monoclonal antibodies SMI 311 and SMI 312 as tools to investigate the maturation of nerve cells and axonal patterns in human fetal brain. Cell Tissue Res. 1998;291:433–443. doi: 10.1007/s004410051013. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Phan KD, Butler SJ. BMP type I receptor complexes have distinct activities mediating cell fate and axon guidance decisions. Development. 2008;135:1119–1128. doi: 10.1242/dev.012989. [DOI] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self-renewal in collaboration with STAT3. Cell. 2003;115:281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- Yi SE, Daluiski A, Pederson R, Rosen V, Lyons KM. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- Yong VW, Moumdjian R, Yong FP, Ruijs TC, Freedman MS, Cashman N, Antel JP. Gamma-interferon promotes proliferation of adult human astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc Natl Acad Sci U S A. 1991;88:7016–7020. doi: 10.1073/pnas.88.16.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mehler MF, Song Q, Kessler JA. Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. J Neurosci. 1998;18:3314–3326. doi: 10.1523/JNEUROSCI.18-09-03314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]