

Abstract
Hemizygous Lis1 mutations cause type 1 lissencephaly, a neuronal migration disorder in humans. The Lis1+/− mouse is a model for lissencephaly; mice exhibit neuronal migration defects but are viable and fertile. On an inbred genetic background, 20% of Lis1+/− mice develop hydrocephalus and die prematurely. Lis1 functions with the microtubule motor cytoplasmic dynein. Because dynactin, a dynein regulator, interacts with end-binding protein 1 (EB1) and β-catenin, two known binding partners of the adenomatous polyposis coli (APC) protein, we looked for a genetic interaction between Lis1 and APC. Mice with a heterozygous truncating mutation in APC (Min mutation) do not exhibit neuronal migration defects or develop hydrocephalus. However, the presence of the APC mutation increases the migration deficit and the incidence of hydrocephalus in Lis1+/− animals. Lis1 and dynein distribution is altered in cells derived from Min mice, and both Lis1 and dynein interact with the C terminus of APC in vitro. Together, our findings point to a previously unknown interaction between APC and Lis1 during mammalian brain development.
Keywords: Cytoplasmic dynein, End-binding protein 1, Neuronal migration, Hydrocephalus
Introduction
Identifying genes mutated in human developmental disorders and the analysis of mouse models for the disorders are important steps in understanding the events governing the development of the mammalian neocortex [1–6]. For example, hemizygous mutations in Lis1/PAFah1b cause type 1 lissencephaly (smooth brain) in humans [7]. This disorder of neuronal precursor migration causes varying degrees of agyria or pachygyria of the cerebral cortex. Lissencephalic brains have thick cortical regions with poorly defined cortical layers. Some patients have enlarged lateral ventricles [1]. Cognitive and motor functions are impaired and increasingly debilitating seizures are associated with the disorder. Six grades of lissencephaly are recognized, with 1 being the severest and 6 being the mildest form. Although the causes of the phenotypic variability are unknown, it has been suggested that some mutant Lis1 alleles retain residual function, while others exhibit dominant negative activity. Very severe lissencephaly such as that associated with Miller-Dieker syndrome may be polygenic [8].
In mice, homozygous deletion or insertion mutations in Lis1 cause early embryonic lethality regardless of the mouse strain [2]. On the other hand, heterozygosity produces various effects dependent on the genetic background. In a mixed genetic background (129SvEv × NIH Black Swiss), the phenotype of both heterozygous mutations is relatively mild; adult mice are viable and fertile. However, attempts at obtaining the mutation on an inbred genetic background were not successful [9], suggesting that genetic modifiers exist.
In the mixed background, ventricles are somewhat enlarged in mutant animals. Distinct, vertically aligned, trabeculated neuronal columns are present, and cortical layers are less well organized. Bromodeoxyuridine assays indicate that migration in the cortex is delayed, but that neurons eventually migrate to the appropriate location [2, 10]. In the insertion mutant, enhanced excitability and morphologically altered neurons are detected in the hippocampus [11]. The mice express approximately 50% of the Lis1 of wild-type littermates. A compound heterozygote with further reduced Lis1 expression (approx. 35% of wild type) exhibits a severer phenotype. In the developing mammalian brain, Lis1 deficiency produced by RNA interference in utero impedes a variety of processes. These include regulation of centrosome/nucleus interactions, interkinetic nuclear oscillations in the ventricular zone, the establishment of a bipolar morphology in pre-migratory cells and their subsequent migration to form the cortical plate [4, 12]. Lis1 is highly conserved, and there is a large body of work in a range of organisms that point to a role for Lis1 in regulating a microtubule motor, cytoplasmic dynein [3, 13]. Dynein carries out intracellular transport of a variety of organelles, vesicles and cytoskeletal components, and contributes to mitosis and the establishment of polarity in many cell types. We reported that Lis1 modestly stimulates dynein's ATPase activity [14], and many of the cellular effects of Lis1 perturbation can be linked to altered dynein function [15]. Lis1 has also been linked to controlling the interaction of dynein with microtubule plus ends.
The list of dynein-associated proteins is growing. This complexity of interactions may contribute to fine control of dynein mechanochemical activity or may regulate switching between dynein's diverse activities, but our understanding of how interacting proteins regulate dynein is minimal, at best [15]. Other proteins that interact with dynein include dynactin, a large multisubunit complex involved in tethering dynein to microtubules during its power stroke to control processivity [16]. Dynactin may also play a role in cargo selection [17]. Lis1, dynein and dynactin are considered microtubule-plus-end-tracking proteins (+TIPS) [18–20] and are thought to play a role in microtubule interactions with the cell cortex. Force generation at the interface between microtubule ends and the cell cortex, and the delivery of signals to the cortex that govern actin assembly and cell polarity, may be dynein dependent [21]. Another plus end protein is the product of the adenomatous polyposis coli protein (APC) gene [22]. APC is generally linked to colon cancer as mutations cause familial adenomatous polyposis [23]. In its tumor suppressor function, APC prevents β-catenin-dependent transcriptional upregulation of a cell cycle program [24]. Like Lis1 and dynein, APC functions in mitosis, migration and establishment of polarity [25]. There is also evidence that APC functions during axon growth. An APC-interacting protein (EB1), binds to p150glued, a subunit of dynactin forming a potential link between APC and dynein [26]. Another partner of APC, β-catenin, targets APC to membrane protrusions in cultured cells [27]. β-Catenin reportedly interacts with dynein and may regulate dynein's ability to capture microtubules at sites of nascent cell contacts [6].
We have now crossed two strains of mice with mutations in Lis1 and APC to determine whether APC and Lis1 interact genetically. A mouse model for lissencephaly (PAFah1bneo, hereafter referred to as Lis1+/− mice) has mild neuronal positioning defects [2]. When the Lis1 mutation is bred into the C57BL/6 mouse strain, 20% of animals develop a severer phenotype, pointing to the presence of genetic modifiers. The mice develop hydrocephalus and altered behavior, and die in the first month. The APC mutant (the APC+/min or Min mouse) is a model for familial colon cancer and develops hundreds of intestinal adenomas, but has no detectable brain abnormalities. Analysis of double mutants indicates that the Min mutation is a genetic enhancer of the Lis1+/− brain phenotype. We provide evidence for an interaction between the C terminus of APC and both Lis1 and dynein. We propose that Lis1 and APC target dynein function such that compounding the mutations results in greater dynein deregulation and severer brain abnormalities.
Methods
Animals
Lis1 interacts with brain-specific PAF1b acetylhydrolases, so Lis1 is also referred to as the noncatalytic or β-subunit of PAFah1b. A heterozygous Lis1 mouse mutant (PAFah1bneo) was originally established in a mixed genetic background (129SvEv × NIH Black Swiss) [2]. These were inbred with the NIH Black Swiss strain over many generations to increase litter size [9]. Thus, NIH Black Swiss is the predominant background strain we began with for our crossbreeding experiments. The Lis1 mutation in PAFah1bneo (Lis1+/−) mice replaces part of exon 6 and all of exon 7 with PKGneo in the opposite transcriptional orientation to Lis1. It is thought to be a true null mutation [2]. The APC+/min (Min) mice used in our experiments were bred and maintained by the Center for Colon Cancer Research at the University of South Carolina. The Min mouse has a mutation at codon 850 in one allele of the APC gene and encodes a truncated version of APC. Because they develop intestinal adenomas, Min mice are a good model for human familial adenomatous polyposis, a hereditary predisposition to colorectal tumors caused by APC mutations that often result in protein truncations [28]. The C57BL/6 background of the Min mouse contributes to the development of intestinal adenomas, as fewer tumors are reported in a mixed background [24, 29]. To generate compound heterozygotes, Lis1+/− mice were backcrossed into the Min strain. Compound ‘LisMin’ heterozygotes were obtained for 5 generations, but because of reduced fertility, 5th- and 6th-generation Lis1+/− mice were crossed with wild-type C57BL/6 mice to generate 7th-generation Lis1+/− animals. These were then crossed with the Min mouse to obtain Lis1+/−, Min, LisMin (double heterozygotes) and wild-type littermates with a greater than 99% C57BL/6 genetic background.
Genotyping
Mice were genotyped at 21 days old by established methods using standardized primers for each mutation. When embryos or perinatal animals were used, genotyping was performed at the time of harvesting.
Histology
Adult mice were perfused with 4% paraformaldehyde. Brains were removed and placed in 4% paraformaldehyde for 24–48 h. Fixed brains were dehydrated, embedded in paraffin and sectioned into 8-μm-thick coronal sections. Cytoarchitecture was visualized by cresyl violet or hematoxylin and eosin staining. Images were captured using either a Nikon SMZ1500 stereomicroscope and Q Capture software (Quantitative Imaging Corporation, Burnaby, B.C., Canada), or with a Zeiss Axiovert microscope and Zeiss Axiovision software.
Cluster Migration Assay
This assay was performed as described elsewhere [5]. Briefly, postnatal day 8 mice were anesthetized, and cerebellar tissue was excised, digested with 2.5% trypsin for 5 min and triturated to obtain a single-cell suspension. Cells were resuspended in 3 ml of Dulbecco's modified Eagle's medium containing 2 mM l-glutamine, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 10% fetal bovine serum, 10% horse serum, 6 mM glucose, 10 μg/ml human transferrin, 10 μg/ml insulin and 10 ng/ml selenium. Cultures were placed in loosely capped 15-ml tubes and incubated at 37°C in an incubator for 24 h to form clusters. These were transferred to 6-well plates coated with 2 ml of poly-d-lysine (100 μg/ml) and laminin (25 μg/ml). After 2 h at 37° C with 5% CO2 to allow clusters to adhere to the plate, the medium was washed off and clusters were incubated in serum-free medium. After 48 h, cells were fixed in 4% paraformaldehyde. Phase contrast images of 25 equal-sized clusters (approx. 100 μm diameter) from 3 cultures were analyzed for cell migration. The number of neurons that had migrated at least 3 μm away from the cluster was determined for each cluster, and the mean for each genotype was calculated. In addition, the distance traveled was recorded for each migrating granule cell. The latter was used to determine whether the distance traveled was affected by the compound mutation.
Western Blot Analysis
Brains were Dounce homogenized in lysis buffer. Lysates were cleared by centrifugation and protein concentrations determined. Twenty, 30 or 50 μg of brain lysate from each genotype was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. Blots were probed with antibodies to mouse polyclonal Lis1 [3] and Ndel1 [12] antibodies, DIC monoclonal antibody (mAb) 74.1 (Santa Cruz), β-catenin mAb (BD Biosciences) and EB1 mAb (BD Biosciences). Primary antibodies were incubated 1 h to overnight, and then blots were exposed to the appropriate horseradish-peroxidase-conjugated secondary antibodies. Labeled proteins were detected using a Western Lightning ECL Plus reagent (Perkin Elmer).
Measuring EB1 Distribution in Neurites
Neuronal cultures were prepared essentially as described elsewhere [30]. On the day of birth, cortices from offspring of Lis1+/− and APC+/min crosses were dissected and pial membranes removed. Cortices were dissociated and plated separately, so genotyping could be determined by PCR of tail digests. Dissociated neurons were plated onto laminin-coated glass coverslips. At 2–3 days after plating, cells were fixed in 100% cold methanol. After blocking in normal goat serum and bovine serum albumin, cells were incubated with a mouse mAb to EB1 (BD Biosciences) for 1 h at room temperature, then exposed to fluorescein-isothiocyanate-conjugated secondary antibodies (Cappel). Nuclei were labeled with bizbenzamide. Coverslips were mounted on glass slides using ProLong Gold mounting medium (Molecular Probes). Regions of 400 μm2 that contained approximately equal numbers of cell bodies (around 10) were selected for analysis. Two areas from 2 cultures derived from 2 different animals per genotype were analyzed (total of 4 areas/genotype). Digital images of these areas were acquired using a high-resolution Axiocam and Axiovert 200 inverted microscope linked to Axiovision data acquisition software. Each image was exposed for the identical amount of time. Images were processed with Image J software so that pixels with intensities above an arbitrarily chosen threshold were labeled red. The number of red pixels within neurites (not in cell bodies) was determined for each image. In addition, the number of high-intensity pixels present in 20 axon segments of 20 μm in length was determined.
Cloning and Protein Expression
A 747-base-pair fragment of the 3′-end of APC (cAPC) was generated by PCR amplification from a full-length human APC construct. This was subcloned into a pET 30 EK/LIC vector (Novagen) for expression of a histidine-tagged peptide in Escherichia coli. Bacteria were exposed to isopropyl-β-d-thiogalactopyranoside for 0 or 3 h and then lysed in B-PER buffer (Pierce) containing lysozyme and a protease inhibitor cocktail. A 30-kDa peptide that was labeled by probing Western blots with a Histag mAb (Novagen) or APC C-20 rabbit polyclonal antibody (Santa Cruz) was present following induction. The recombinant murine Lis1 protein was isolated from Sf9 insect cells following infection with recombinant baculovirus and did not contain detectable dynactin, dynein, Ndel1 or tubulin [14]. This purified Lis1 was able to interact with purified PAFah1b α2-dimers and Ndel1 in brain extracts, increasing confidence that the protein was properly folded.
Dynein Purification
Cytoplasmic dynein was purified from bovine brain as described previously [14]. The dynein contains all subunits in the motor holoenzyme, and contains no detectable Lis1, p150glued, Ndel1, EB1 or tubulin [14]. The dynein purified in this manner is enzymatically active based on its capacity to hydrolyze ATP [14] and its ability to undergo processive movement along microtubules [31].
Immunoprecipitation
Then, 500 μl of a bacterial lysate containing expressed cAPC was coincubated with 6.0 pmol purified brain dynein for 30 min at room temperature, with or without Lis1. To precipitate Lis1, 1 μg of the Lis1 antibody was added to reaction. After rocking for 1 h at 4°C, Lis1 complex was precipitated using protein A Sepharose beads at 4°C. Proteins were eluted from washed beads in sodium dodecyl sulfate sample buffer and analyzed by Western blotting with the anti-Histag and Lis1 antibodies. Blots were also probed for DIC. To control for nonspecific binding to beads or antibodies, identical reactions were carried out in the absence of Lis1 or dynein.
Results
The Incidence of Hydrocephalus in Lis1+/− Mice Is Exacerbated by a Heterozygous APC Min Mutation
Because the Lis1+/− mutation is maintained in a different mouse strain (Black Swiss) than the Min mouse (C57BL/6), Lis1+/− animals were successively backcrossed with the Min mouse for 8 generations to obtain both mutants and double heterozygotes on the BL/6 background. During this period of backcrossing, we observed growth retardation and the appearance of a domed cranium typical of progressive hydrocephalus in some animals (fig. 1A). The affected animals were at first hyperactive but became progressively inanimate and died within the first month after birth. Genotype/phenotype analysis indicated that hydrocephalus was occurring in both Lis1+/− and LisMin mice but at different frequencies. During the course of the crosses, 8 of 71 Lis1+/− mice developed hydrocephalus, while 32 of 88 LisMin double heterozygotes exhibited the phenotype (fig. 1B). The phenotype was never observed in the Min mouse or in wild-type animals, nor was it found in Lis1 mutants in the Black Swiss background. Thus, the BL/6 genetic background produces a proclivity towards hydrocephalus in Lis1+/− animals that is exacerbated by the Min mutation. The nature of the BL/6 genetic modifier(s) of the Lis1 phenotype is not known.
Fig. 1.
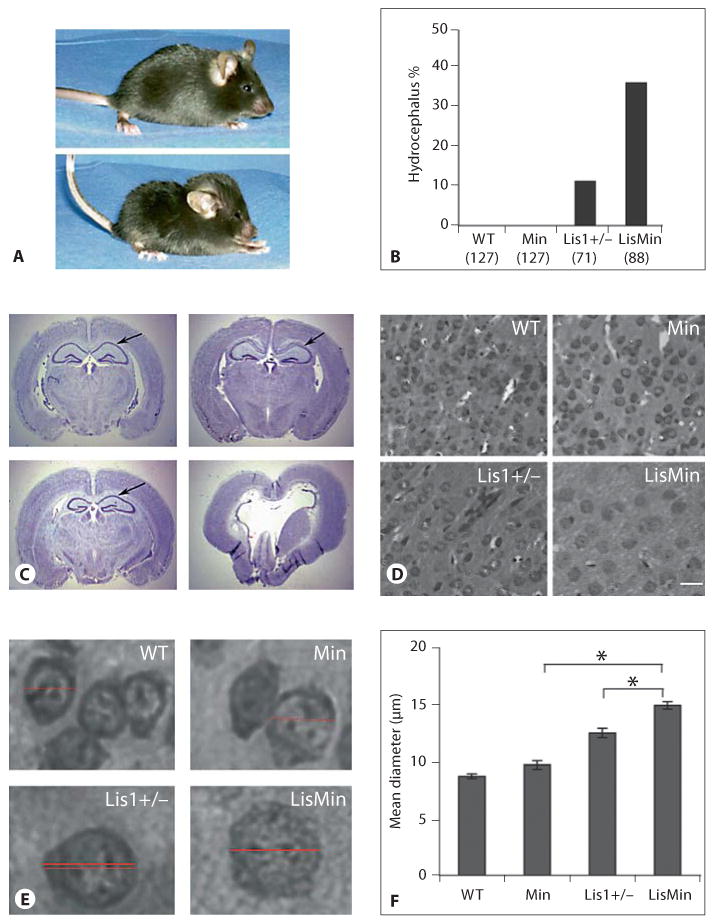
The incidence of hydrocephalus in Lis1+/− mice is increased by a BL/6 genetic background and further by a heterozygous Min mutation. WT = Wild type. A Top panel: a wild-type mouse from a second-generation Lis1+/−/Min backcross. Bottom panel: a compound heterozygote (LisMin) littermate with hydrocephalus. Note the dome-shaped skull and altered posture. B The percentage of offspring with the hydrocephalus phenotype is shown for all offspring obtained during these crosses. The numbers and genotypes are indicated. Parental Black Swiss Lis1+/− mice did not develop hydrocephalus. C Cresyl violet staining of coronal sections of adult brains from littermates (95% BL/6) shows severe disturbances of brain architecture in the LisMin animal (lower right panel). Arrows point out the CA1 region of the hippocampus. D, E Cell bodies in LisMin cortex in these sections appear enlarged. Scale bar = 10 μm. F The somal diameter was determined for 100 cells in digital images (at the same magnification) in the cortex of 3 adjacent brain sections. The diameter was determined by measuring a line through the center of the cells using Axiovision image analysis software (horizontal lines in E). The average somal diameter of neurons in Min mice was not different from that of control mice. Lis1+/− soma were swollen relative to the wild type. This was significantly exacerbated in LisMin brains (* p < 0.00003, Student's t test).
Figure 1C shows coronal brain sections of adult offspring from a cross in which the parental Lis1+/− animal was >95% BL/6. Wild-type, Min, Lis1+/− and LisMin animals were examined (the LisMin littermate was visibly hydrocephalic in this group). Min brains were indistinguishable from wild-type brains, while the Lis1+/− mice had severe neuronal positioning defects in the CA1 region of the hippocampus. The LisMin brain was severely affected. Ventricles were grossly enlarged, and degeneration of white matter tracts and parts of the thalamus, hippocampus and other subcortical structures was apparent. Cortical neurons displayed chromatolysis (swollen cell bodies, reduced Nissl substance and acentric nuclei, fig. 1D–F). Less severe chromatolysis was observed in the Lis1 cortex but was not observed in wild-type or Min cortices.
The Enhancement of the Lis1+/− Phenotype by the Min Mutation Occurs in a Mixed Background in the Absence of Hydrocephalus
We next analyzed coronal sections of mixed background littermates that did not show signs of hydrocephalus to determine if the Min mutation acted as a genetic enhancer in the mixed background. The brains of Min animals were very similar to those of wild-type littermates (fig. 2A). Neurons in CA1, CA2, CA3 and the dentate gyrus of the Lis1+/− mouse were disorganized, but less so than in the BL/6 background. However, the same regions in the compound mutant were severely disturbed (fig. 2B). In addition, the organization of neurons in the cerebral cortex of LisMin mice was altered relative to Lis1 mice (fig. 2C–E). Neurons in the Lis1+/− animal were organized in vertical trabeculae as described previously [2]. In the compound mutant, neurons were more densely packed in deeper layers and were aligned in horizontal bands as well as vertical trabeculae (arrows, fig. 2C, D). Neurons were not obviously chromatolytic but were more likely to be in physical contact with neighboring neurons (fig. 2E).
Fig. 2.
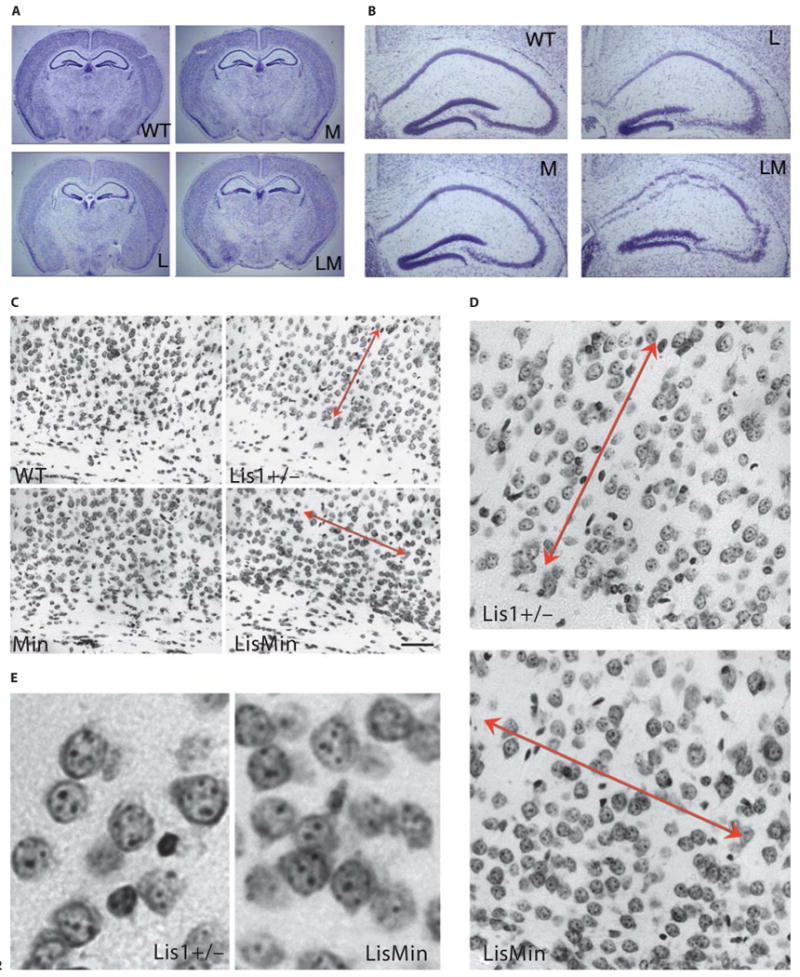
The Min mutation enhances neuronal positioning defects in mixed background Lis1+/− mice. WT = Wild type; M = Min; L = Lis1+/−; LM = LisMin. A Cresyl-violet-stained coronal brain sections from littermates of all 4 genotypes are grossly similar (hydrocephalus was not present in this earlier backcross). B Closer inspection of the hippocampus reveals that neuronal positioning defects are severer in the double heterozygote (LM). The Min hippocampus was similar to its wild-type littermate. C, D Positioning defects in the cortex of Lis1+/− mice include vertical trabeculation of neurons (double-headed arrows, compare to wild-type and Min cortices). In compound heterozygotes, organization is more ‘regimented’, with horizontal as well as vertical trabeculation. Neurons near the ventricular region (towards the bottom) appear more closely packed in the double heterozygote. E Enlargements showing neurons in deep layers of the Lis1+/− and LisMin cortex. In the LisMin cortex, neurons are more tightly packed.
Histopathological Features Are Already Present in Neonatal LisMin Animals
Embryonic day 19 littermates were examined to determine if a severer neuronal positioning defect was detectable in LisMin BL/6 mice prior to the onset of hydrocephalus. Inspection of whole embryos showed that the 4 genotypes were outwardly indistinguishable from one another (fig. 3A–D). Moreover, on gross inspection of brains, all regions were present and of a similar size (not shown). However, histological examination of coronal sections revealed enlarged ventricles in the compound mutant mice accompanied by a reduced septum and striatum (fig. 3H). The granule cells of the dentate gyrus and the pyramidal cells of the CA1, CA2 and CA3 regions of the stratum pyramidale were disorganized in postnatal day 2 Lis1+/− mice (fig. 3K, O) as was reported earlier for adult mice [2]. This disorganization was increased in the compound mutant mice (fig. 3L, P) indicating that Lis1 mutations sensitize mice to the effects of APC truncations on the establishment of hippocampal cytoarchitecture. The ventricles and hippocampus in Min mice (fig. 3F, J, N) were normal compared to that of wild-type littermates (fig. 3E, I, M). This suggests that the APC mutation exacerbates the Lis1 phenotype prior to the onset of obvious clinical signs of hydrocephalus.
Fig. 3.
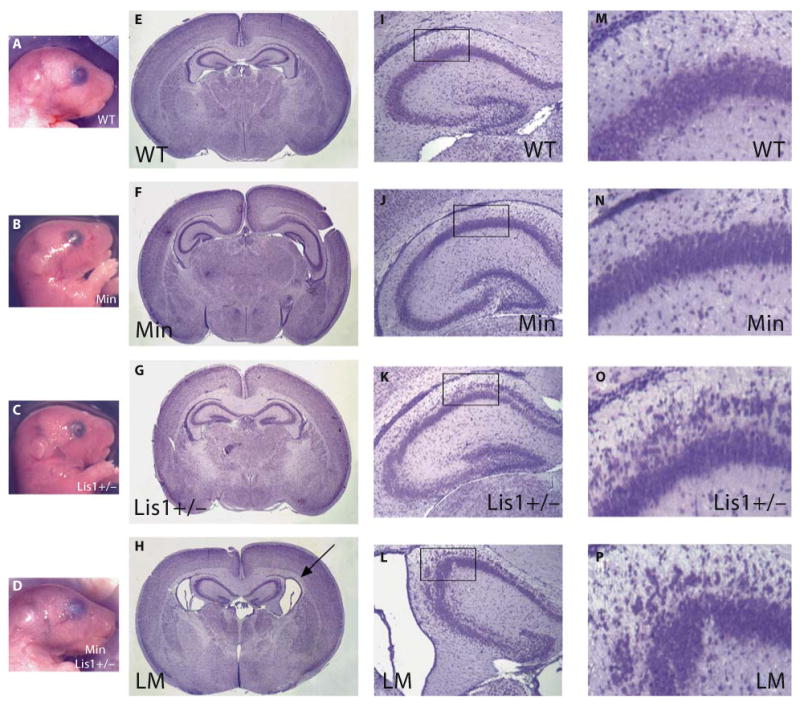
The Min mutation exacerbates hippocampal abnormalities in Lis1+/− BL/6 embryos and results in enlarged ventricles. WT = Wild type; LM = LisMin. A–D Embryonic day 19 embryos from a late-generation cross do not exhibit outward signs of hydrocephalus. E–H Hematoxylin-eosin staining of coronal brain sections of all 4 genotypes revealed that double heterozygotes, but not other genotypes, had enlarged ventricles at this embryonic stage. I–P Compound heterozygotes also had severer neuronal positioning defects in the hippocampus. M–P Enlargements of the regions indicated by the boxes in panels I–L.
Neurons Derived from LisMin Mice Are Less Migration Competent than Neurons from Lis1+/− Animals
To determine if the severer phenotypes observed in the compound mutants are related to further impairment of neuronal migration, we turned to an in vitro assay that was used in the initial characterization of the Lis1+/− mouse (fig. 4). In this assay, unidirectional migration of granule cells from cerebellar reaggregate clusters is measured [2]. The average number of migrating neurons and their distance traveled was analyzed for aggregates from wild-type, Min, Lis1+/− and LisMin animals. Representative clusters from each genotype are shown (fig. 4A). At least 2 animals from each genotype were included in the analysis. Migration from clusters derived from Min animals was indistinguishable from wild-type migration (fig. 4B, C). As expected from previous studies [2], migration from Lis1+/− clusters was compromised. On average, fewer neurons had migrated away from the clusters at different distances from the clusters. The average number of migrating neurons was further reduced by a compounding Min mutation (fig. 4B). Interestingly, fewer LisMin neurons (relative to Lis1+/− neurons) were found within 20 μm from their cluster of origin, while similar numbers were found between 20 and 60 μm from the cluster. This suggests that the Min mutation exacerbates a defect in the initiation of migration caused by Lis1 haploinsufficiency.
Fig. 4.
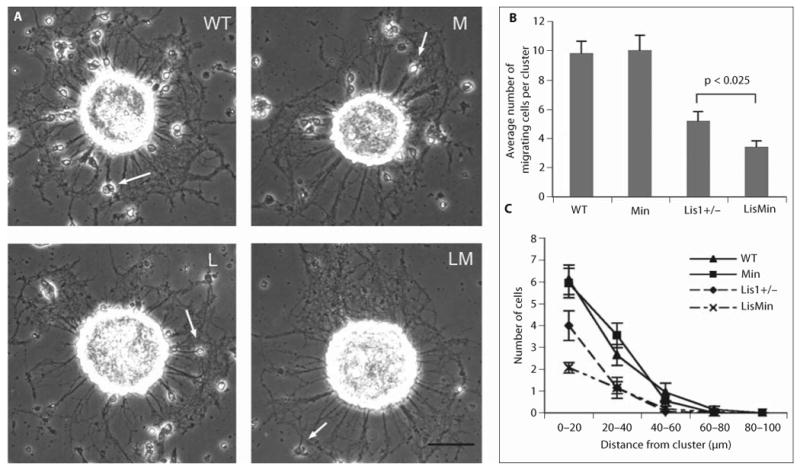
The Min mutation exacerbates the migration defect exhibited by Lis1+/− neurons in a cluster migration assay. This assay uses clusters of cerebellar granule cells to characterize the migration of neonatal neurons along axons. Neurons obtained from neonatal offspring of Lis1+/− Min cross (both BL/6) were dissociated and allowed to form clusters. Clusters were plated onto poly-lysine-coated coverslips and examined by phase contrast microscopy. Twenty-four clusters from 3 cultures were analyzed for each genotype. A Typical clusters from the indicated genotypes. WT = Wild type; M = Min; L = Lis1+/−; LM = LisMin. Arrows point to neurons that migrated away from the cluster along neurites. Scale bar = 50 μm. B For each cluster the number of neurons that had migrated at least 3 μm away from the cluster was determined. The difference between the Lis1+/− clusters and the clusters derived from double mutants is statistically significant (p < 0.025, Student's t test). C The distance that cells had migrated from the clusters is plotted against the number of cells that migrated that distance.
Lis1 Is Reduced and p150glued Is Increased in Adult LisMin Brains
To determine if phenotypic changes in compound heterozygotes correlate with a change in expression of key proteins, we analyzed brain lysates prepared from adult, mixed background littermates (fig. 5D, E). The relative protein levels on Western blots were estimated by densitometry. Values were normalized to extracts stained with Coomassie blue and to α-tubulin probed on the same blots. Both methods yielded similar results. As expected, the level of Lis1 was reduced by approximately 50% in Lis1+/− animals. Two differences in protein expression correlated with phenotype severity (wild type, Min = no phenotype, LisMin severer than Lis1+/−). Lis1 expression was reduced and the dynactin subunit, p150glued, was elevated in LisMin brains. The reduced Lis1 expression in this sample is not likely due to loss of Lis1-expressing neurons, as Ndel1 and dynein are not affected. The higher level of p150glued in the LisMin brain could exacerbate the effects of Lis1 haploinsufficiency on dynein function. Alternatively, p150glued expression may be elevated as a compensatory response to defects in dynein function caused by the Lis1+/− mutation. Other differences in adult expression patterns between genotypes did not correlate with the severity of the brain phenotype.
Fig. 5.
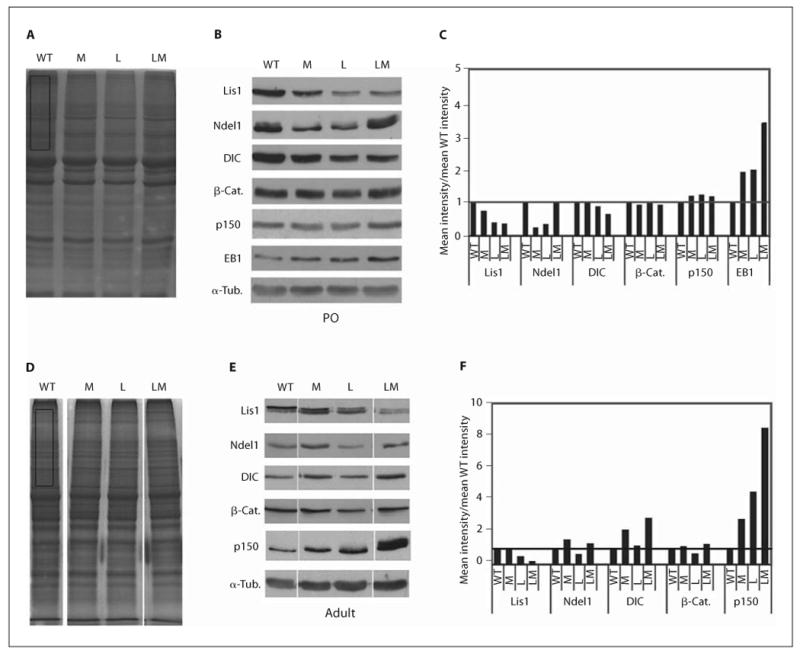
Elevated EB1 in neonatal brains, and reduced Lis1 and elevated p150glued in adult brains of double heterozygotes. A Coomassie-stained gels of 30 μg of lysate prepared from postnatal day 0 (P0) wild-type (WT), Min (M), Lis1+/− (L) and LisMin (LM) cortices. B Typical results from Western blots probed for Lis1-associated proteins (Ndel1, DIC, p150glued), APC-associated proteins (β-catenin (β-Cat.), and EB1) and α-tubulin (α-Tub.). C Densitometric signals were normalized to Coomassie gels (rectangle in A) or to α-tubulin with similar results. The data are expressed relative to wild-type extracts. As expected, extracts from mice with Lis1 mutations contained approximately 50% of the Lis1 of mice without the mutation. Lis1 expression is further reduced in the adult compound heterozygote. EB1 expression is increased in single mutants and further increased in compound mutants. D–F Similar data from brain lysates prepared from adult mice. p150glued expression is increased in single mutants and further increased in the compound heterozygote.
We compared brain extracts prepared from neonatal pups of all 4 genotypes (fig. 5A–C). Lis1 expression was not reduced in neonatal LisMin brains, so a further reduction in Lis1 levels cannot account for the severer neuronal positioning defects that occur during development. However, EB1 expression was increased in both single mutants and further increased in the double mutant, suggesting that defects in EB1 regulation or interactions could contribute to the enhanced phenotype.
The Effect of Lis1+/− and Min Mutations on EB1 Distribution in Neurons Is Additive
Cortical cultures were prepared from cortices of 2 sets of neonatal littermates. Animals were genotyped at the time of harvesting. Three days after plating, cultures were fixed and processed for EB1 immunofluorescence. As described by others, EB1 immunofluorescence labeled comet-shaped structures in cell bodies and neuritic processes. The signal intensity and overall number of these structures appeared to be increased in mutant neurons. Digital images were acquired from all cultures using identical acquisition parameters. The intensities and numbers of comet-shaped structures were analyzed. An arbitrarily chosen threshold was used to isolate bright comets which were pseudocolored red. The average number of above-threshold comets was compared in four 400-μm2 areas per genotype, using cultures derived from at least 2 animals per genotype (fig. 6A). The areas were chosen to be of similar cell and neurite density. Only comets in neurites were counted, as cell bodies were too bright to distinguish individual comets. Lis1+/− and Min neurons had more of these above-threshold comets than wild-type neurons. This was further increased in LisMin neurons.
Fig. 6.
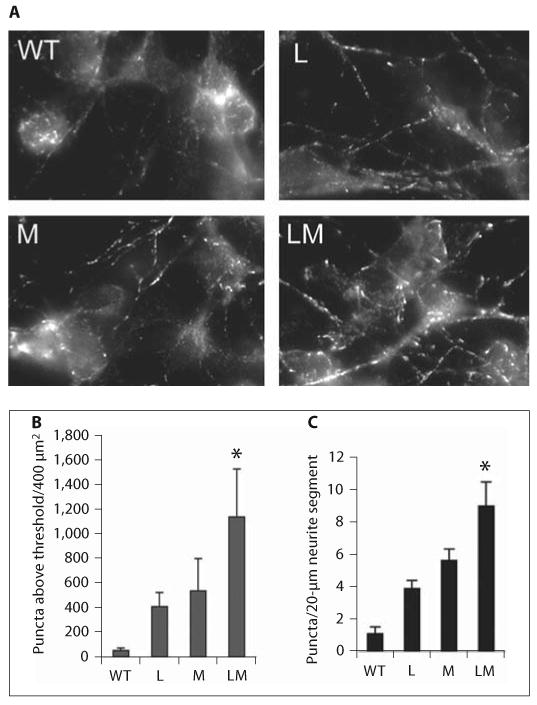
Additive effects of Lis1 haploinsufficiency and APC truncation on EB1 distribution. A EB1 immunolocalization in postnatal day 0 cortical neurons derived from wild-type (WT), Min (M), Lis1+/− (L) and compound mutants (LM) stained for EB1. The average number of pixels within a 400-μm2 region of comet-shaped EB1-labeled structures in neurites above an arbitrary threshold of intensity was determined for cultures of all 4 genotypes. Pixels in or near cell bodies were not included. Four regions were analyzed for each genotype. These were derived from 2 animals per genotype. B The data are presented as the mean ± SD. The difference between each single mutant and the compound mutant was significant by Student's t test (* p < 0.05). C The average number of high-intensity EB1 pixels was determined in 20 neurite segments (20 μm in length), 5 segments from each of the 4 images. The data are presented as the mean ± SE. The difference between each single mutant and the compound mutant was significant by Student's t test (* p < 0.05).
To control for regional or culture-to-culture variation in plating density, we next determined the number of above-threshold comets in 20-μm segments of nonover-lapping neurites (20 segments per genotype). Similar results were obtained (fig. 6B), indicating that the increase in bright comets is not due to the overall number of neurites. Although the significance of the brighter comets is not known, it provides an example of a molecular change caused by each mutation that is, to some extent, additive in the LisMin animals.
A C-Terminal APC Fragment Binds to Purified Lis1 in vitro
Given the genetic interaction, we were interested in determining whether Lis1 and APC interact in the brain. APC staining was observed in cultured cortical neurons where it colocalized extensively with Lis1 (fig. 7A). In Cos7 cells, APC and Lis1 overlap was restricted to membrane protrusions (fig. 7B, C), suggesting that Lis1 and APC interact at protrusions during migration.
Fig. 7.
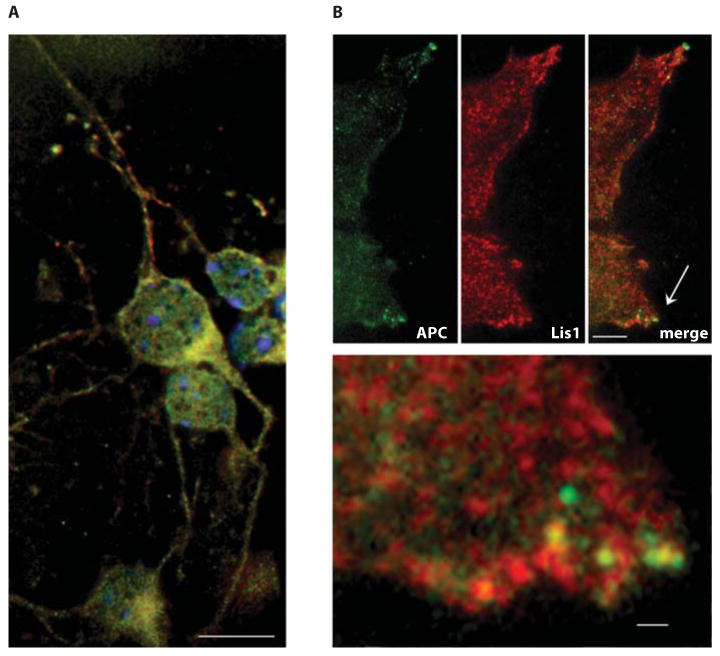
Lis1 colocalizes with APC in neurons and in Cos7 cells. A Cultured cortical neurons from newborn mice express Lis1 (red) and APC (green) that shows substantial overlap in cell bodies. B Less overlap is observed in Cos7 cells, but the signals merge in cortical protrusions, ostensibly at sites of microtubule contact (arrow). The lower panel shows an enlargement of the area near the arrow. A, B, upper panels, scale bars = 5 μm; B, lower panel, scale bar = 1 μm.
In support of the cell and genetic data, purified Lis1 was able to interact with a C-terminal APC fragment in vitro. Full-length recombinant mouse Lis1 was produced in insect cells using a baculovirus expression system. Proper folding of the purified Lis1 was indicated by the observation that it bound with high affinity to the known interacting proteins, Ndel1 and αPAFAH1b [14]. The purified protein appears as a single band on a Coomassie gel. The histidine tag was removed during purification. The C terminus of APC containing the EB1-binding domain (fig. 8A, cAPC, aa 7731–8532) was expressed in E. coli. The histidine-tagged cAPC was easily detected on Western blots with a Histag antibody (fig. 8B). However, the recombinant cAPC did not bind to nickel resin and was not precipitated with the Histag antibody, indicating that the tag was not exposed in the native protein. Therefore, to examine Lis1 binding, bacterial lysates expressing cAPC were incubated with or without Lis1 and potential complexes immunoprecipitated with a Lis1 antibody (fig. 8C). cAPC coprecipitated only when Lis1 was present in the reaction. Bovine brain dynein also coprecipitated with Lis1 if included in the reaction. All subunits of the dynein holoenzyme are present in this preparation, which is free of dynactin, Lis1, Ndel1 and tubulin [14]. The same amount of cAPC coprecipitated with Lis1 even when dynein was included, suggesting that dynein does not compete with APC for Lis1 binding.
Fig. 8.
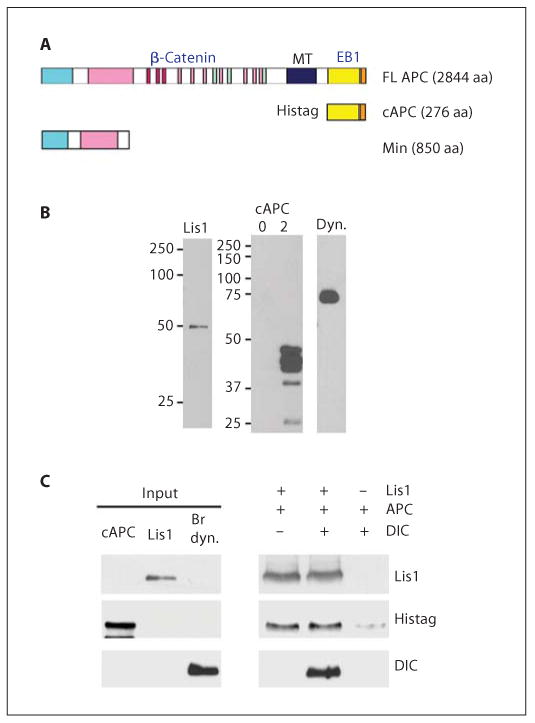
The C terminus of APC binds to purified Lis1 and dynein in vitro. A Schematic of APC forms showing protein interaction domains: FL APC = full-length APC; cAPC = histidine-tagged, C-terminal domain (aa 2577–2844); Min = N-terminal domain expressed in Min mice (aa 1–850); MT = microtubule-interacting domain. B Western blots of proteins used in these studies: left panel = Lis1, expressed in insect cells and purified by nickel chromatography is recognized by a Lis1 antibody; right panel = cAPC expressed in bacteria induced with isopropyl-β-d-thiogalactopyranoside for 2 h was labeled with a Histag antibody. No cAPC was detected in uninduced extracts (0 h, lane 1). GSTEB1 produced in bacteria is labeled with an EB1 antibody (lane 3). Dynein (dyn.) purified from bovine brains is labeled with a DIC antibody. C Lis1 at 10 pmol was added to 500 μl of the 2-hour cAPC extracts, in the presence or absence of 6 pmol dynein. Lis1 was immunoprecipitated. Coprecipitating proteins were analyzed by Western blotting. cAPC coprecipitates with Lis1, independent of dynein.
Discussion
Our data show that a truncating mutation in one allele of the APC gene enhances the effect of Lis1 haploinsufficiency in mice. The APC mutation alone has no effect on neuronal positioning, but increases positioning defects in Lis1+/− mice. This suggests that APC functions in brain development during neuronal migration, but that a Min mutation alone is not sufficient to cause detectable changes. Others have shown that loss of APC reduces the migratory capacity of cultured epithelial cells, fibroblasts and osteosarcoma cells [32, 33]. In the Min mouse, neurons express less wild-type APC but also contain truncated protein. The Min mutation has not been characterized as extensively but was reported to interfere with migration in intestinal enterocytes in vivo [34]. We now find that the presence of the Min allele exacerbated the migration defect exhibited by Lis1+/− cerebellar granule cells in a cluster migration assay. Our findings uncover a novel link between APC function and neuronal migration in the developing brain.
The synthetic phenotype caused by compounding heterozygous Lis1 and APC mutations could arise indirectly if Lis1 and APC participate in unrelated pathways that both impact neuronal migration. However, some evidence points to a more direct interaction between APC and Lis1. For example, the Min mutation causes a fairly dramatic redistribution of Lis1 to microtubule-organizing centers in colon cell lines. This might occur even if Lis1 and APC are not functionally related if the Min protein alters microtubule organization/dynamics. However, APC is reportedly a functional component of centrosomes in Madin-Darby canine kidney cells [35] and may influence Lis1 directly. In support of this, we find that purified Lis1 can interact directly with the C terminus of APC in vitro. We do not know if Lis1 can interact with the C terminus of full-length APC in the brain, but if so, the presence of the Min protein, which would not bind Lis1 normally, could sensitize neurons to reduced Lis1 dosage.
Interestingly, we find that cAPC also interacts with purified dynein motors. Others have shown that dynein associates indirectly with the APC partner, EB1 [5]. In our binding assay, EB1 appears to compete with dynein for binding to this region of APC (data not shown). Given its status as a microtubule-plus-end-tracking protein, our data suggest that a relevant interaction between APC and Lis1/dynein may occur at the microtubule plus end. EB1 distribution and expression are altered in cortical neurons from Lis1+/− and Min mice, and even more so in the double mutant. Since EB1 immunofluorescence appears as comet-shaped structures at microtubule plus ends, our finding further argues that plus ends may be important sites of APC/Lis1 interactions. The p150glued subunit of the dynein regulatory complex, dynactin, binds directly to EB1. It is interesting that p150glued expression is up-regulated in adult brains from both single mutants and even more so in double mutants. However, further experiments will be required to determine if the upregulation contributes to the aggravated phenotype or is a compensatory reaction.
Backcrossing the Lis1 mutation into a C57BL/6 inbred mouse strain resulted in an increased incidence of hydrocephalus. Although this phenotype was not observed in Min mutants in any background, the incidence of hydrocephalus in compound mutants was dramatically increased. Signs of severe chromatolysis in cortical neurons in hydrocephalic LisMin mice also suggest that axonal or synaptic function is perturbed. Mice with compound mutations in both Lis1+/− and reelin pathway members (reelin, Vldlr, Lrp8, Dab1) exhibit progressive hydrocephalus caused by obstruction of the ventricular drainage system caused by defects in the ependymal lining of the third ventricle and the aqueduct of Sylvius [36]. Interestingly, an axonemal dynein heavy-chain gene, Mdnah5, is specifically expressed in ependymal cells, and is critical for the functional integrity of ependymal cilia. In Mdnah5-mutant mice, lack of ependymal flow causes closure of the aqueduct and subsequent formation of tri-ventricular hydrocephalus during early postnatal brain development. Lis1 is localized to some motile cilia in mammals [37] but it is not clear if and how Lis1 functions in these cells. No role has been described for APC in ependymal cells. APC clearly functions in intestinal epithelium and may have similar functions in brain epithelial cells. If so, defects in Lis1 and APC may indirectly converge in ependymal cells to produce the higher incidence of hydrocephalus.
There are clearly genetic modifiers of the Lis1 phenotype in the BL/6 background. One possible candidate is the MOM1 locus, discovered because this locus influences intestinal tumor formation in Min animals [38]. The relevant gene is thought to encode a phospholipase A protein (PLA2IIA) that is inactivated in BL/6 mice. Although there is not a clear link between PLA2IIA function and Lis1, phospholipase A can contribute to the synthesis of platelet-activating factor (PAF) [39]. Lis1 forms a tripartite complex with PAF acetylhydrolases, which can compete with a potential Lis1-activating protein for binding to Lis1 [40]. Future studies may be able to determine if PAF synthesis is altered in BL/6 mice, and if PAF can influence Lis1 function.
Lissencephaly in humans is quite variable, both at the level of brain morphology and at the level of clinical manifestations, so uncovering a genetic interaction between Lis1 and APC is interesting from a disease standpoint. Our work suggests that the APC and MOM1 genes are candidates for potential sites of lissencephaly modifiers.
References
- 1.Kato M, Dobyns WB. Lissencephaly and the molecular basis of neuronal migration. Hum Mol Genet. 2003;12:R89–R96. doi: 10.1093/hmg/ddg086. [DOI] [PubMed] [Google Scholar]
- 2.Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 3.Smith DS, Niethammer M, Ayala R, Zhou Y, Gambello MJ, Wynshaw-Boris A, Tsai LH. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 4.Tsai JW, Chen Y, Kriegstein AR, Vallee RB. LIS1 RNA interference blocks neural stem cell division, morphogenesis, and motility at multiple stages. J Cell Biol. 2005;170:935–945. doi: 10.1083/jcb.200505166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berrueta L, Tirnauer JS, Schuyler SC, Pellman D, Bierer BE. The APC-associated protein EB1 associates with components of the dynactin complex and cytoplasmic dynein intermediate chain. Curr Biol. 1999;9:425–428. doi: 10.1016/s0960-9822(99)80190-0. [DOI] [PubMed] [Google Scholar]
- 6.Ligon LA, Karki S, Tokito M, Holzbaur EL. Dynein binds to β-catenin and may tether microtubules at adherens junctions. Nat Cell Biol. 2001;3:913–917. doi: 10.1038/ncb1001-913. [DOI] [PubMed] [Google Scholar]
- 7.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein β-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 8.Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14–3–3ε is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- 9.Paylor R, Hirotsune S, Gambello MJ, Yuva-Paylor L, Crawley JN, Wynshaw-Boris A. Impaired learning and motor behavior in heterozygous Pafah1b1 (Lis1) mutant mice. Learn Mem. 1999;6:521–537. doi: 10.1101/lm.6.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McManus MF, Nasrallah IM, Pancoast MM, Wynshaw-Boris A, Golden JA. Lis1 is necessary for normal non-radial migration of inhibitory interneurons. Am J Pathol. 2004;165:775–784. doi: 10.1016/S0002-9440(10)63340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, McBain CJ. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 13.Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O'Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 14.Mesngon MT, Tarricone C, Hebbar S, Guillotte AM, Schmitt EW, Lanier L, Musacchio A, King SJ, Smith DS. Regulation of cytoplasmic dynein ATPase by Lis1. J Neurosci. 2006;26:2132–2139. doi: 10.1523/JNEUROSCI.5095-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1, and what they tell us about brain development. Genes Dev. 2006;20:1384–1393. doi: 10.1101/gad.1417206. [DOI] [PubMed] [Google Scholar]
- 16.Kincaid MM, King SJ. Motors and their tethers: the role of secondary binding sites in processive motility. Cell Cycle. 2006;5:2733–2737. doi: 10.4161/cc.5.23.3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004;20:759–779. doi: 10.1146/annurev.cellbio.20.012103.094623. [DOI] [PubMed] [Google Scholar]
- 18.Mimori-Kiyosue Y, Tsukita S. ‘Search-and-capture’ of microtubules through plus-end-binding proteins (+TIPs) J Biochem (Tokyo) 2003;134:321–326. doi: 10.1093/jb/mvg148. [DOI] [PubMed] [Google Scholar]
- 19.Carvalho P, Tirnauer JS, Pellman D. Surfing on microtubule ends. Trends Cell Biol. 2003;13:229–237. doi: 10.1016/s0962-8924(03)00074-6. [DOI] [PubMed] [Google Scholar]
- 20.Wu X, Xiang X, Hammer JA., 3rd Motor proteins at the microtubule plus-end. Trends Cell Biol. 2006;16:135–143. doi: 10.1016/j.tcb.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Dujardin DL, Vallee RB. Dynein at the cortex. Curr Opin Cell Biol. 2002;14:44–49. doi: 10.1016/s0955-0674(01)00292-7. [DOI] [PubMed] [Google Scholar]
- 22.Nathke IS, Adams CL, Polakis P, Sellin JH, Nelson WJ. The adenomatous polyposis coli tumor suppressor protein localizes to plasma membrane sites involved in active cell migration. J Cell Biol. 1996;134:165–179. doi: 10.1083/jcb.134.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senda T, Shimomura A, Iizuka-Kogo A. Adenomatous polyposis coli (Apc) tumor suppressor gene as a multifunctional gene. Anat Sci Int. 2005;80:121–131. doi: 10.1111/j.1447-073x.2005.00106.x. [DOI] [PubMed] [Google Scholar]
- 24.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 25.Hanson CA, Miller JR. Non-traditional roles for the Adenomatous polyposis coli (APC) tumor suppressor protein. Gene. 2005;361:1–12. doi: 10.1016/j.gene.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 26.Strickland LI, Wen Y, Gundersen GG, Burgess DR. Interaction between EB1 and p150glued is required for anaphase astral microtubule elongation and stimulation of cytokinesis. Curr Biol. 2005;15:2249–2255. doi: 10.1016/j.cub.2005.10.073. [DOI] [PubMed] [Google Scholar]
- 27.Sharma M, Leung L, Brocardo M, Henderson J, Flegg C, Henderson BR. Membrane localization of adenomatous polyposis coli protein at cellular protrusions: targeting sequences and regulation by β-catenin. J Biol Chem. 2006;281:17140–17149. doi: 10.1074/jbc.M513027200. [DOI] [PubMed] [Google Scholar]
- 28.Pellman D. Cancer: a CINtillating new job for the APC tumor suppressor. Science. 2001;291:2555–2556. doi: 10.1126/science.1057337. [DOI] [PubMed] [Google Scholar]
- 29.Moser AR, Luongo C, Gould KA, McNeley MK, Shoemaker AR, Dove WF. ApcMin: a mouse model for intestinal and mammary tumorigenesis. Eur J Cancer. 1995;31A:1061–1064. doi: 10.1016/0959-8049(95)00181-h. [DOI] [PubMed] [Google Scholar]
- 30.Meberg PJ, Miller MW. Culturing hippocampal and cortical neurons. Methods Cell Biol. 2003;71:111–127. doi: 10.1016/s0091-679x(03)01007-0. [DOI] [PubMed] [Google Scholar]
- 31.King SJ, Schroer TA. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol. 2000;2:20–24. doi: 10.1038/71338. [DOI] [PubMed] [Google Scholar]
- 32.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batile E, Simon-Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kroboth K, Newton IP, Kita K, Dikovskaya D, Zumbrunn J, Waterman-Storer CM, Nathke IS. Lack of adenomatous polyposis coli protein correlates with a decrease in cell migration and overall changes in microtubule stability. Mol Biol Cell. 2007;18:910–918. doi: 10.1091/mbc.E06-03-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahmoud NN, Boolbol SK, Bilinski RT, Martucci C, Chadburn A, Bertagnolli MM. Apc gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 1997;57:5045–5050. [PubMed] [Google Scholar]
- 35.Louie RK, Bahmanyar S, Siemers KA, Votin V, Chang P, Stearns T, Nelson WJ, Barth AI. Adenomatous polyposis coli and EB1 localize in close proximity of the mother centriole and EB1 is a functional component of centrosomes. J Cell Sci. 2004;117:1117–1128. doi: 10.1242/jcs.00939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Assadi AH, Zhang G, Beffert U, McNeil RS, Renfro AL, Niu S, Quattrocchi CC, Antalffy BA, Sheldon M, Armstrong DD, Wynshaw-Boris A, Herz J, D'Arcangelo G, Clark GD. Interaction of reelin signaling and Lis1 in brain development. Nat Genet. 2003;35:270–276. doi: 10.1038/ng1257. [DOI] [PubMed] [Google Scholar]
- 37.Pedersen LB, Rompolas P, Christensen ST, Rosenbaum JL, King SM. The lissencephaly protein Lis1 is present in motile mammalian cilia and requires outer arm dynein for targeting to Chlamydomonas flagella. J Cell Sci. 2007;120:858–867. doi: 10.1242/jcs.03374. [DOI] [PubMed] [Google Scholar]
- 38.Markova M, Koratkar RA, Silverman KA, Sollars VE, MacPhee-Pellini M, Walters R, Palazzo JP, Buchberg AM, Siracusa LD, Farber SA. Diversity in secreted PLA2-IIA activity among inbred mouse strains that are resistant or susceptible to Apc Min/+ tumorigenesis. Oncogene. 2005;24:6450–6458. doi: 10.1038/sj.onc.1208791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem. 2000;69:419–445. doi: 10.1146/annurev.biochem.69.1.419. [DOI] [PubMed] [Google Scholar]
- 40.Tarricone C, Perrina F, Monzani S, Massi-miliano L, Kim MH, Derewenda ZS, Knapp S, Tsai LH, Musacchio A. Coupling PAF signaling to dynein regulation: structure of LIS1 in complex with PAF-acetylhydrolase. Neuron. 2004;44:809–821. doi: 10.1016/j.neuron.2004.11.019. [DOI] [PubMed] [Google Scholar]