

Abstract
Methionine sulfoxide reductase maintains adhesin function during oxidative stress. Using Streptococcus gordonii as a model, we now show the mechanistic basis of adhesin maintenance provided by MsrA. In biofilms, S. gordonii selectively expresses the msrA gene. When the wild-type strain was grown with exogenous hydrogen peroxide (H2O2), msrA-specific mRNA expression significantly increased, while acid production was unaffected. In the presence of H2O2, an msrA-deletion mutant (ΔMsrA) showed a 6 h delay in lag phase growth, a 30% lower yield of H2O2, significantly greater inhibition by H2O2 on agar plates (reversed by complementation), 30% less adhesion to saliva-coated hydroxyapatite, 87% less biofilm formation, and an altered electrophoretic pattern of SspAB protein adhesins. Using mass spectrometry, methionine residues in the Met-rich central region of SspB were shown to be oxidized by H2O2 and reduced by MsrA. In intact wild-type cells, MsrA co-localized with a cell wall-staining dye, and MsrA was detected in both cell wall and cytosolic fractions. To maintain normal adhesion and biofilm function of S. gordonii in response to exogenous oxidants, therefore, msrA is up-regulated, methionine oxidation of adhesins and perhaps other proteins is reversed, and adhesion and biofilm formation is maintained.
Keywords: Streptococcus gordonii, methionine sulfoxide reductase, oxidation, adhesins, adhesion, biofilms
Introduction
Methionine sulfoxide reductase (MsrA) is a highly conserved enzyme among eukaryotes and prokaryotes (Cabreiro et al., 2006, Moskovitz, 2005). In bacteria, methionine oxidation is cytotoxic and a major result of myeloperoxidase-mediated killing by neutrophils (Rosen et al., 2009). MsrA confers protection against methionine oxidation, making bacterial cells more resistant to killing or damage. In sublethal oxidative conditions, bacteria utilize MsrA to reverse the adverse affects of oxidation on intra- (Ezraty et al., 2004) and extracellular surface proteins (Kiliç et al., 1999, Vriesema et al., 2000). MsrA-mediated protection against oxidation of extracellular proteins appears to enable several species of bacteria to maintain adhesion function (Wizemann et al., 1996), which is crucial to survival of most species. For most species of bacteria, adhesion precedes biofilm formation (Nobbs et al., 2009) (Kline et al., 2009). Preservation of adhesion function is, therefore, crucial to biofilm formation.
Streptococcus gordonii is a pioneer colonizer of dental plaque in the oral cavity, which must adhere successfully in fluctuating environmental conditions in the mouth to avoid clearance by swallowing and aid survival (Schachtele et al., 2007, Nobbs et al., 2009). Oral streptococcal adhesion depends on macromolecular assemblies selected from more than 20 adhesins; the P1 (antigen I/II) adhesin family is among the best characterized. While in the oral cavity, S. gordonii is a harmless commensal species, whereas gaining access into the blood circulation provides opportunity for pathogenic traits to emerge, particularly when heart valves become infected (Kiliç et al., 1999) (Meyer et al., 1998). Hence, S. gordonii, like the other viridans streptococci, are frequent causative agents of infective endocarditis. Pathogenesis depends on the ability of the streptococci to adhere to injured heart valves and initiate formation of sessile biofilms.
During experimental S. gordonii endocarditis in rabbits, biofilms form on the heart valves and msrA expression is induced (Kiliç et al., 1999). The heart valve infection is characterized by aggregated masses of platelets and some neutrophils and monocytes that form a vegetative fibrin-encased mass, trapping the bacterial cells within (Herzberg, 1996). This environment exposes the infecting S. gordonii to oxidative stress from reactive oxygen species produced by the aggregated platelets and white blood cells (Ostrowski et al., 2009, Presterl et al., 2001). Furthermore, S. gordonii (and S. sanguinis) pyruvate oxidase yields H2O2 in the presence of oxygen (Kreth et al., 2008). H2O2 is freely diffusible and can oxidize intra- and extracellular macromolecules (Ma and Eaton, 1992). It is unclear whether oxidative stress directly induces MsrA as a protective mechanism. Hence, MsrA is important during the two different life styles of S. gordonii, in the oral biofilm and during infective endocarditis.
As the pathogenesis of infective endocarditis is modeled, several environmental conditions could induce expression of MsrA. A shift in pH from slightly acidic in dental plaque to neutral in the blood induces S. gordonii MsrA, affecting growth in aerobic and anaerobic conditions (Vriesema et al., 2000). As in infective endocarditis, polymicrobial infection may be sufficient to induce MsrA. MsrA is expressed by S. gordonii during growth with Porphyromonas gingivalis in mixed species biofilms in vitro (Kuboniwa et al., 2006), presumably a consequence of oxidative stress in this biofilm community. Loss of MsrA increases the sensitivity of cells to H2O2 as shown for E. coli (Rosen et al., 2009). We now show that S. gordonii induces expression of MsrA in response to hydrogen peroxide, maintaining adhesion and biofilm formation. Cell surface MsrA facilitates adhesion maintenance. MsrA targets methionine residues of adhesins to preserve functional conformation as we modeled using the methionine-rich central region of SspB.
Results
Morphology of the wild-type S. gordonii V288 and ΔmsrA mutant
After overnight culture with or without added H2O2, Gram stained V288 and ΔmsrA mutant cells were indistinguishable under the light microscope (image not shown). When observed by field emission scanning electron microscopy (FESEM), wild-type S. gordonii showed finely textured surfaces and septae (Fig. 1A). The ΔmsrA mutant also showed surface blebs (Fig. 1B). In the presence of H2O2, surface blebs were also visible in wild-type cells (Fig. 1C) and increased in size and frequency on the ΔmsrA mutants (Fig. 1D). These results indicate that H2O2 causes severe envelope damage, which can be counteracted by functional expression of msrA.
FIG. 1. Ultrastructural morphology of the wild-type and msrA mutant strains.
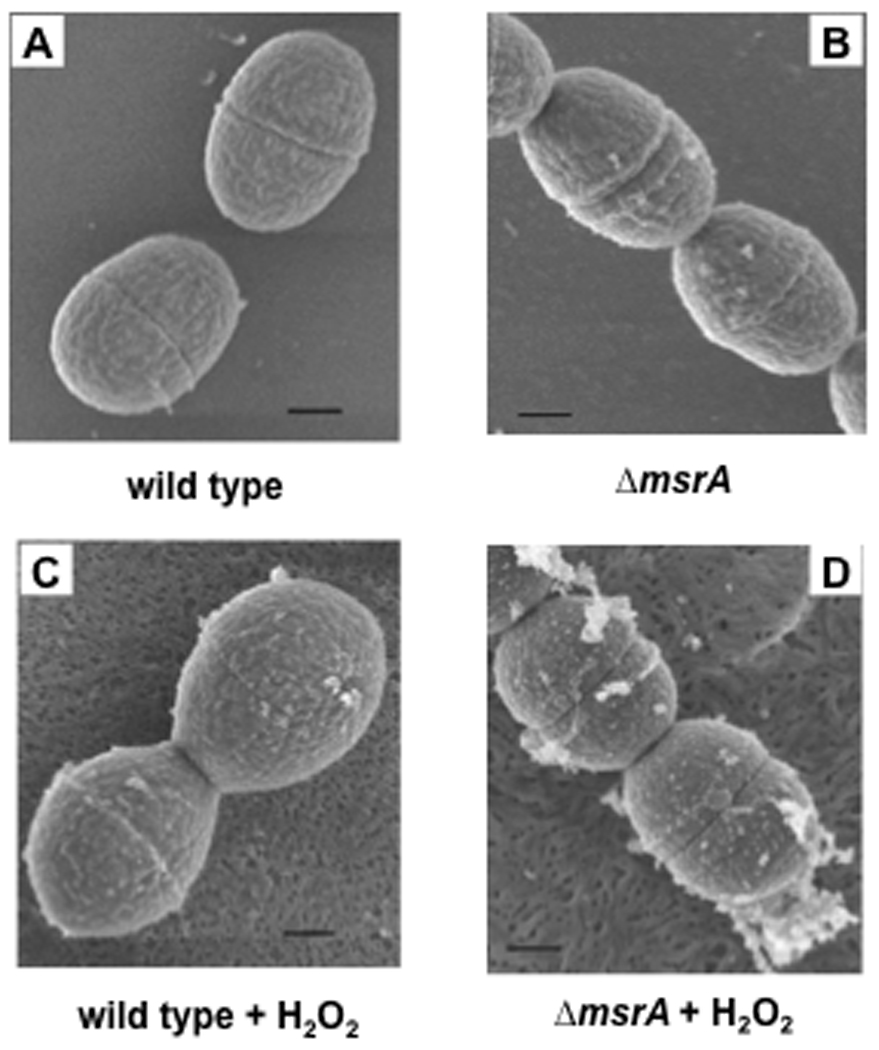
Bacteria were cultured overnight in FMC medium with or without H2O2. After fixation and coating, the cells were observed under a Hitachi S-900 field emission scanning microscope. The bar in each panel represents 200 nm. A. Wild-type cells. B. msrA mutant. C. Wild-type cells with H2O2. D. msrA mutant with H2O2.
MsrA protects growth during exogenous H2O2 stress
S. gordonii V288, the ΔmsrA mutant, and the complemented mutant grew similarly in FMC medium with 5% CO2 (Fig. 2; open symbols). When the medium was supplemented with 0.5 mM H2O2 (exogenous; filled symbols), the lag phase of growth was prolonged for all strains. In the presence of exogenous H2O2, the ΔmsrA mutant (filled circles) required about 6 h longer than the wild-type cells (filled triangles) to enter exponential growth. When msrA was restored by complementation (filled diamond), peroxide-resistance (decreased lag phase) was restored to greater levels than the wild-type cells, probably reflecting over-abundance of MsrA.
Fig. 2. Growth of wild-type, msrA mutant, and complemented mutant strains with and without H2O2.
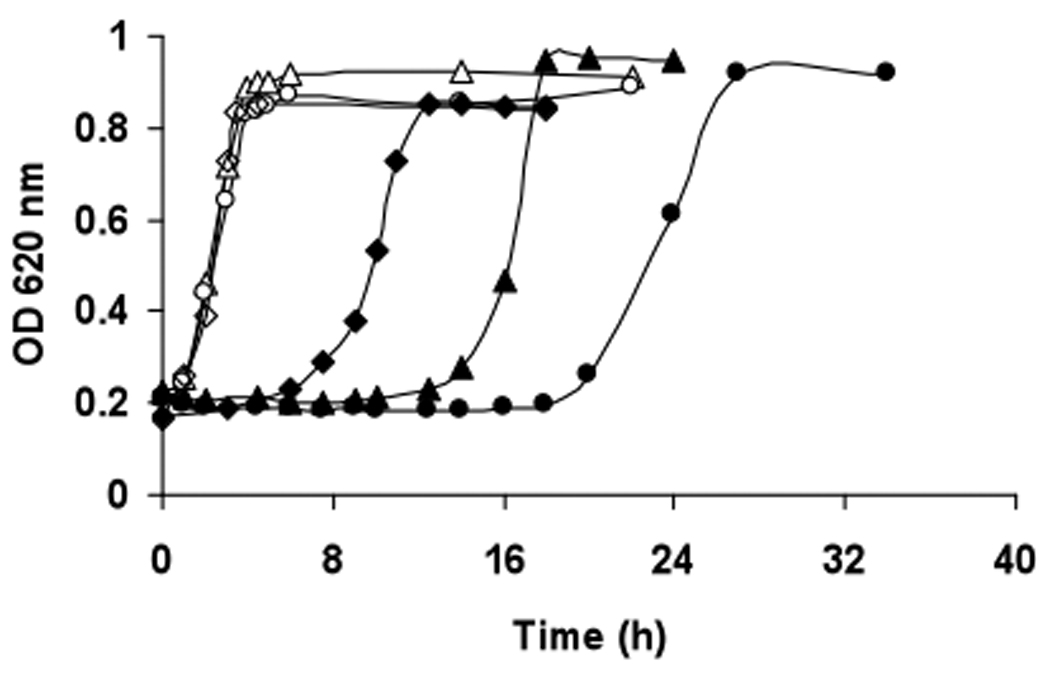
Strains were pre-cultured overnight in FMC media, and diluted with fresh media until a starting OD620 nm = 0.2 was obtained. As indicated, H2O2 was added to the cultures to a final concentration of 0.5 mM. Symbols: Δ, wild-type strain; ○, msrA mutant; ◊, complemented mutant; filled symbols represent the same strains cultured with H2O2.
The wild-type, ΔmsrA mutant, and complemented strains were also analyzed for growth inhibition on THB agar. In the presence of H2O2, the wild-type cells showed an area of inhibition of 14.0 ± 0.7 cm2 (mean ± SD, N = 4) and the ΔmsrA mutant, 19.3 ± 1.0 cm2 (mean ± SD, n = 4), a 35% increase in sensitivity attributable to the mutation (p < 0.01). The MsrA-complemented strain showed an area of inhibition of 11.7 ± 0.9 cm2 (mean ± SD, n = 4), suggesting that sensitivity to H2O2 was attributed to the deletion of MsrA. The strains grew similarly when distilled H2O was added to the disk, rather than H2O2 (data not shown).
msrA-dependent production of H2O2 (endogenous) and pyruvate oxidase-specific mRNA
Spent FMC medium from stationary phase cells was analyzed for released H2O2 as a function of cell (optical) density. The wild-type cells produced about 30% more H2O2 than the ΔmsrA mutant (p < 0.01) (Fig. 3A). The RNA was then isolated from both strains and analyzed for expression of pyruvate oxidase-specific mRNA. Pyruvate oxidase catalyzes the conversion of pyruvate to acetyl-phosphate and generates H2O2. Consistent with the H2O2 yield, the ΔmsrA mutant expressed about 30% less pyruvate oxidase-specific mRNA than the wild-type cells (p < 0.01) (Fig. 3B).
Fig. 3. H2O2 yield and pyruvate oxidase mRNA expression by wild-type and msrA mutant strains.
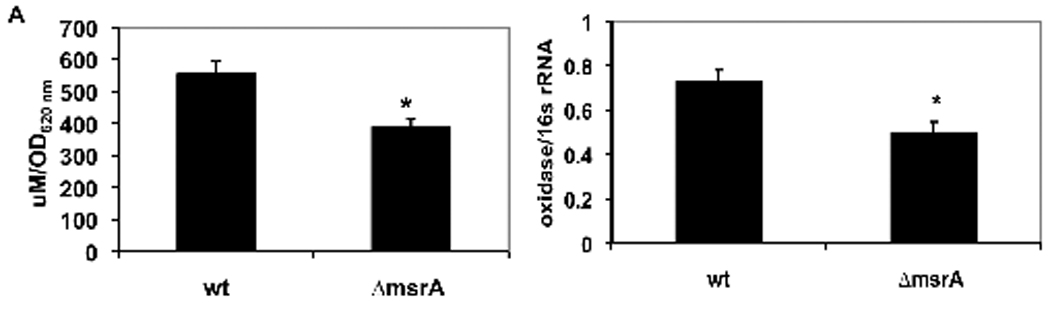
Bacteria were cultured in FMC medium at 37°C with 5% CO2 until stationary phase. A. The concentration of H2O2 released into the cell-free spent medium was detected by the o-dianisidine/horseradish peroxidase (Nunez de Kairuz et al., 1988) and expressed relative to the OD620 nm of cells. B. The mRNA expression level was detected by real-time PCR and reported relative to 16s rRNA expression. The data shown are the means ± SEM for 3 experiments. The means were significantly different, *p < 0.01.
Exogenous H2O2 up-regulates expression of msrA
To determine whether expression of msrA is a response to oxidative stress, S. gordonii V288 cells were cultured overnight in FMC media with or without 0.5 mM added H2O2. In the continuous presence of H2O2, msrA-specific mRNA expression was 2.2 ± 0.04 fold (mean ± SEM, n = 3 experiments) greater than in the absence. When H2O2 was added to mid-logarithmic phase cells, msrA-specific mRNA expression was 2.3 ± 0.4 fold greater one hour later than in the absence of oxidative stress.
Since shifts in pH may influence the expression of msrA (Vriesema et al., 2000), we considered the possibility that exogenous H2O2 modulates acid production, altering msrA expression and growth of S. gordonii. Cells were harvested during log growth at increasing cell densities (A620 nm = 0.4 to 1.2). During log phase growth, acidification of the media was similar in the presence and absence of 0.5 mM exogenous H2O2 (data not shown).
Adhesion and biofilm formation by the ΔmsrA mutant
To learn whether MsrA influences the ability of S. gordonii to adhere, wild-type and ΔmsrA mutant cells were grown in the presence and absence of H2O2. When grown without H2O2, wild-type and ΔmsrA mutant strains adhered similarly to sHA (Fig. 4A) and formed equivalent in vitro biofilms (Fig. 4B). After growth in exogenous H2O2, fewer mutant cells adhered (30% less) (p < 0.01) (Fig. 4A) and biofilms formed with 87% less mass (p < 0.01) than wild-type cells when compared to the absence of H2O2 (Fig. 4B). Adhesion and biofilm formation by the wild-type strain was not significantly affected by exogenous H2O2 and complementation of the ΔmsrA mutant strain restored adhesion and biofilm formation to wild-type strain levels.
Fig. 4. Effect of H2O2 on adhesion and biofilm formation by wild-type and msrA mutant strains.
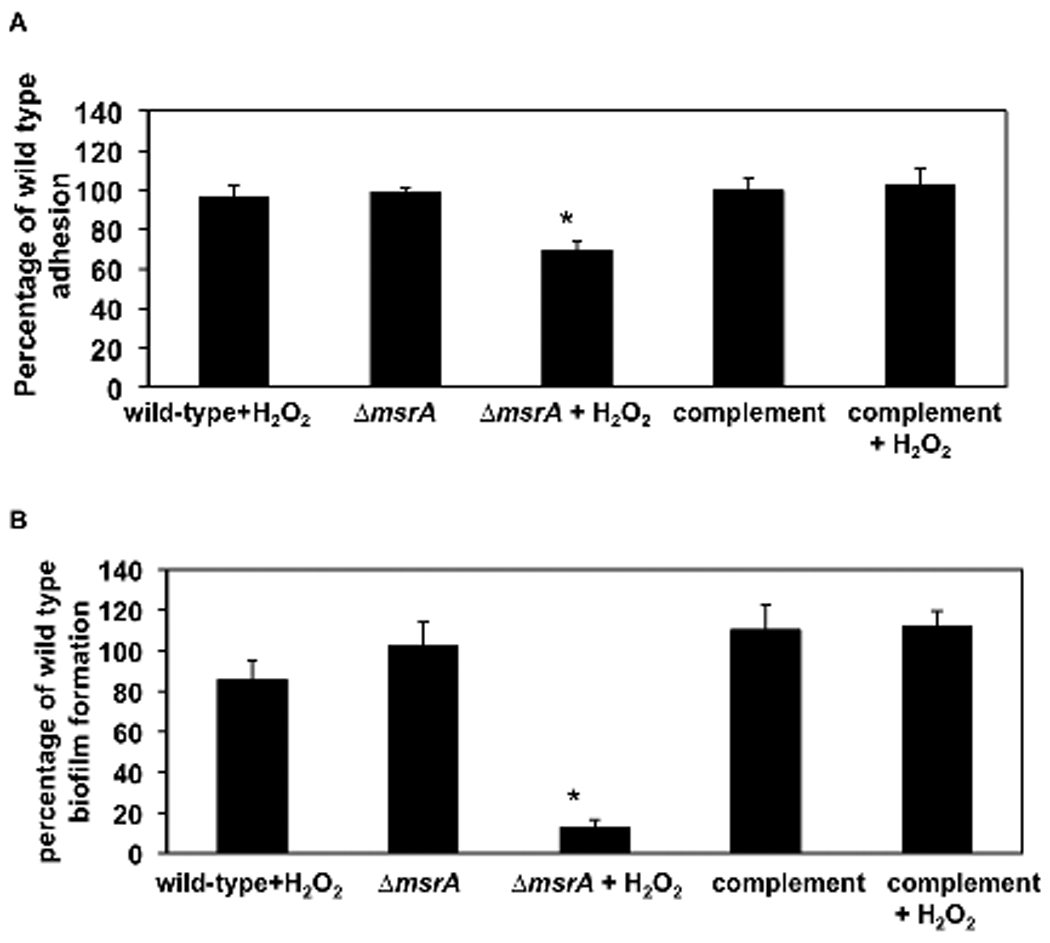
A. Adhesion. Cells were cultured with 3H-thymidine overnight in FMC medium with or without 0.5 mM H2O2, harvested, washed, mixed with sHA, and incubated with continuous inversion at ambient temperature for 1 h. The adhesion percentage of wild-type cells was normalized to 100%. The data shown are the means ± SEM for 4 experiments. B. Biofilm formation. Cells were cultured in 96-well plates at 37°C for 24 h in FMC with or without 0.5 mM H2O2. Biofilm formation by the wild-type cells was normalized to 100%. The data shown are the means ± SD of one triplicate experiment, representative of 4 experiments. *p < 0.01.
Effects of H2O2 on SspA/B (P1 antigen) on S. gordonii
Adhesion of S. gordonii is strongly associated with expression of adhesins. We determined the effect of exogenous H2O2 on the structural integrity of a model adhesin, SspAB. Wild-type and ΔmsrA cells were grown with or without H2O2, cell wall protein preparations were isolated, separated in native gels, and probed with P1 (SspAB) antigen-specific antibodies in Western blots. Oxidation by H2O2 did not appear to affect SspAB in wild-type cells (Fig. 5A). In ΔmsrA, oxidation by H2O2 caused a shift in native gel mobility and apparent loss of antigenicity of SspA/SspB, which was not seen in the absence of added H2O2 (Fig. 5A). The overall profiles of surface proteins from wild-type and ΔmsrA cells were similar when grown in the presence or absence of exogenous H2O2 (Fig. 5B).
Fig. 5. Western blot analysis of the effect of H2O2 on SspA/B.
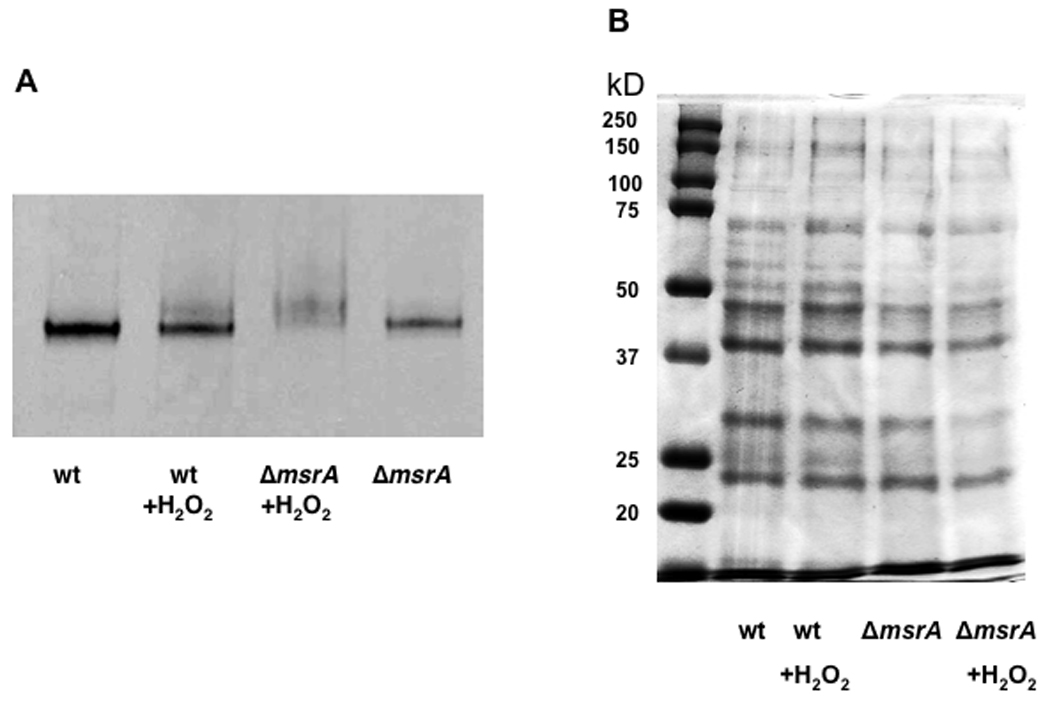
The MsrA− strain and the S. gordonii wild-type cells were grown overnight in FMC with or without H2O2 (0.5 mM final concentration). Cells were digested with mutanolysin, cell wall macromolecules were isolated, and concentrated by ultrafiltration. A. Western blot. Prepared proteins (10 µg) were electrophoresed on a non-denaturing 4–20% native gradient polyacrylamide gel. Proteins were transferred to nitrocellulose membrane and reacted with anti-P1 (anti-SspAB). Lanes: 1, S. gordonii V288; 2, S. gordonii V288 with H2O2; 3, ΔmsrA strain with H2O2; 4, ΔmsrA strain. B. Profiles of prepared proteins. Prepared proteins (16 µg) were electrophoresed on a 10% SDS-PAGE and stained with Coomassie Blue.
Oxidation of met-rich central region of SspB and reduction by MsrA
To serve as target substrates for oxidation and MsrA reduction, the Met-rich central region of SspB (residues 594 to 774) and an SspB protein fragment of the same length without Met (control) (residues 336 to 517) were cloned and expressed in E. coli with an N-terminal His-tag. MsrA from S. gordonii wild-type cells was also cloned, expressed and purified to apparent homogeneity (Fig. 6A, right lane). Untreated, H2O2-oxidized and MsrA-reduced Met-rich and control protein fragments were resolved using 4–15% native PAGE (Fig. 6B) and detected in Western blots using anti-tetra-His (Fig. 6C) or anti-P1 antibodies (Fig. 6D). In the presence of H2O2, only the Met-rich protein fragment (Fig. 6C, D, lane 2) replicated the native gel mobility shift shown by native P1 protein. MsrA restored the band to the untreated position (Fig. 6C, D, lane 3). Synthesized without Met-residues, the control protein fragment was unaffected by H2O2 treatment (Fig. 6E).
Fig. 6. H2O2 oxidation and reduction of Met-rich protein fragments.
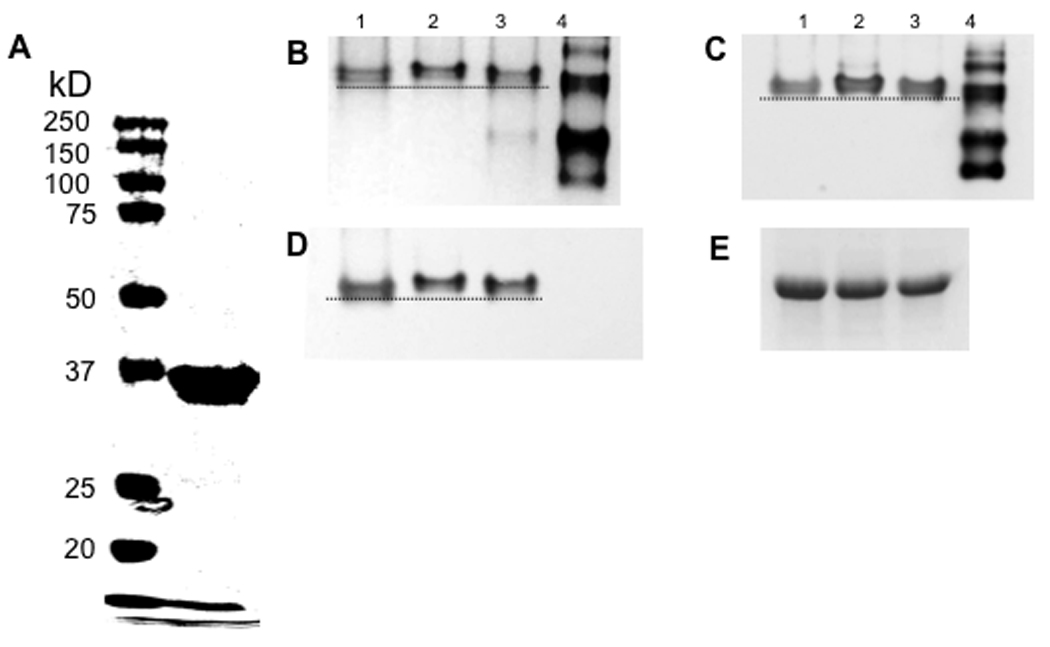
A. Purified recombinant MsrA. Purified MsrA (19 µg; right lane) was electrophoresed on 10% SDS-PAGE and stained with Coomassie Blue. Molecular weight markers are shown in left lane. B–D. Electrophoresis of target substrate protein fragments on native 4–15% polyacrylamide gels. As described in the Materials and Methods, purified Met-rich protein fragments (lane 1) were incubated with 50 mM H2O2 (lane 2), followed by incubation with purified MsrA (lane 3). Used to reduce protein fragments in lane 3, cloned and expressed his-tagged MsrA is resolved on native gel (lane 4). B. 4–15% PAGE stained with Coomassie Blue; C. Western blot using anti-tetra-His antibody; D. Western blot using rabbit anti-P1 antibody; and E. Control protein fragments were treated as in lanes 1 to 3 above and detected in Western blot using rabbit anti-P1 antibody. Dotted lines highlight the band shift.
The Met-rich protein fragments, including untreated, H2O2-treated, and MsrA-reduced samples, were extracted from the polyacrylamide gels, digested with trypsin, and analyzed by mass spectrometry for all oxidation forms of Met. After digestion, four tryptic peptides resolved (Fig. 7, sequences in pink, blue, black and green); one peptide was not detectable (gray). The mass ratios represent the relative amount of oxidized and reduced methionine-containing peptides. Three met-containing peptides showed met oxidation after H2O2 treatment and reduction of Met[O] by MsrA (Table 2; Fig. S1).
Fig. 7. MASS-SPEC analysis of oxidation and reduction of SspB met-rich peptides.
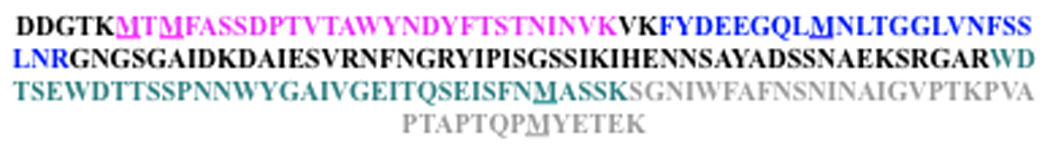
Cloned met-rich protein fragments were separated by PAGE, digested with trypsin, and analyzed by MASS-SPEC. Amino acid sequence of met-rich segment of SspB, with colors identifying the fragments obtained after trypsin digestion.
Table 2.
Mass ratios of oxidized to reduced met-containing peptides
| Sequence | oxidized/reduced (± SE) | ||
|---|---|---|---|
| untreated | oxidized | reduced | |
| FYDEEGQLMNLTGGLVNFSSLNR | 0.1 ± 0.04 | 60.8 ± 53.7 | 0.1 ± 0.01 |
| WDTSEWDTTSSPNNWYGAIVGEITQSEISFNMASSK | 0.03 ± 0.02 | 17.7 ± 4.5 | 1.0 ± 0.5 |
| MTMFASSDPTVTAWYNDYFTSTNINVK | 0.1 ± 0.01 | 628.5 ± 78.7 | 0.1 ± 0.04 |
Methionine sulfoxide reductase activity associated with S. gordonii
To confirm MsrA activity under the experimental conditions used in this study, whole cell Msr activity was determined. The wild-type strain showed a specific activity of 38.1 ± 1.4 nmol NADPH/mg protein/min, the ΔmsrA mutant 19.4 ± 7.1 nmol/mg/min, and the complemented strain 46.4 ± 7.6 nmol/mg/min for (n = 3, p ≤ 0.05). The results further corroborate an active role of MsrA during growth and correlate with the repairing function of MsrA.
MsrA localization in vivo
To rescue oxidized surface-proteins, MsrA needs to localize in proximity to the substrate cellular proteins. To localize MsrA under in vivo conditions, a fluorescent-tagged MsrA was constructed. The FLAsH tag was localized to cell fractions and also visualized in vivo using fluorescent microscopy. msrA-FLAsH complemented mutant cells were harvested during mid-logarithmic phase growth, fractionated, and fluorescent bands were observed in SDS-PAGE in both cell wall and cytosolic fractions, but not in the spent media (Fig. 8A). To determine cellular localization in vivo, whole bacterial cells were incubated with FLAsH-EDT2, and counterstained with the cell wall dye WGA Alexa Fluor 555 (Fig. 8B). MsrA fluorescence was associated with the cell wall and cytosol.
Fig. 8. Cellular localization of MsrA.
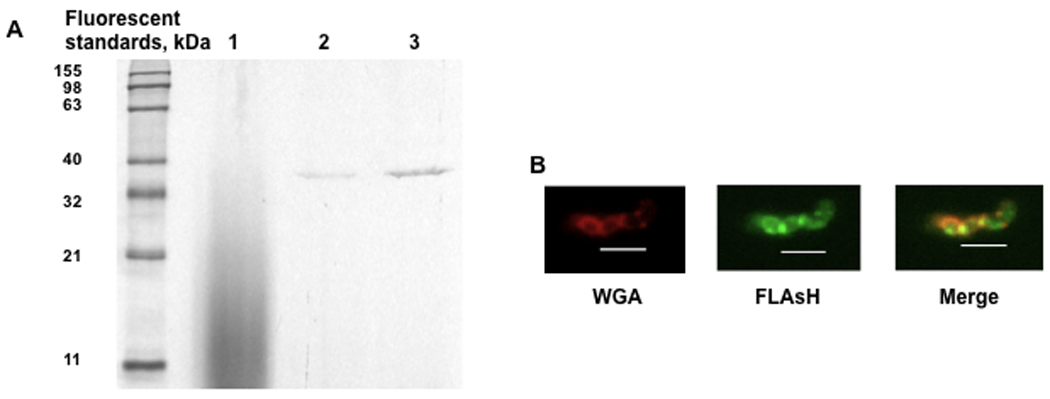
The ΔmsrA strain was complemented with a msrA-FlAsH construct. The msrA-FlAsH complemented mutant cells were harvested at mid-logarithmic phase, spent medium was concentrated by ultrafiltration, and cell wall and cytosolic fractions were isolated. A. Resolution of msrA-FlAsH fractions (5 µg protein/ lane) using 10% SDS-PAGE, a Lumio Green Detection kit (invitrogen) and UV transilluminator. Lane 1, spent medium; lane 2, cell wall proteins; and lane 3, cytosolic proteins. B. Whole cell localization of MsrA. The msrA-FLAsH complemented mutant was harvested at mid-logarithmic phase, washed with TBST buffer, incubated with 20 µM FLAsH-EDT (green) for 60 min, washed, stained with 1 µg/ml WGA Alexa Fluor 555 (red; cell wall-binding lectin), and observed using a Nikon ECLIPSE E800 fluorescence microscope. Images were obtained at 1000 × magnification with a Spot camera. Scale bars represent 2 µm.
Discussion
S. gordonii and the other oral streptococci are the most prevalent group of bacteria in dental plaque (Schachtele et al., 2007). Dental plaque is a challenging oxidative environment. Indeed, oxidative stress contributes to interspecies competition as S. gordonii and S. sanguinis produce hydrogen peroxide to antagonize growth of one another and the oral pathogen, S. mutans (Kreth et al., 2008). Pyruvate oxidase-dependent production of anti-bacterial H2O2 must be accompanied by a mechanism for anti-oxidant protection of the producer organisms. We now show that expression of msrA by S. gordonii is an integral response to H2O2-mediated stress. Yet, it does not appear to be part of a generalized stress response since expression of msrA is independent of adaptation to changes in pH in vitro. The sensitivity to hydrogen peroxide shown in the ΔmsrA mutant could be reversed by complementation demonstrating that the H2O2-sensitive phenotype was specifically attributable to loss of the msrA gene. Indeed, msrA is now shown to protect several cellular functions that are probably integral to the life cycle of this organism, including growth, adhesion to a biologically relevant surface (sHA), and biofilm formation.
Oxidative stress affects bacterial cell morphology and integrity. For example, S. sanguinis cells cultured in 95% O2 and 5% CO2 were larger and showed more septal notches than cells maintained in air and 5% CO2 (Rosan et al., 1973). Bacillus subtilis and Pseudomonas fluorescens developed rough cell surfaces, indentations, and distorted shapes after treatment with peracetic acid/hydrogen peroxide (Lindsay et al., 1999). At a low H2O2 concentration (< 2.5 mM), E. coli cells expressed extensive cell filaments and released lactate dehydrogenase (Brandi et al., 1989), a marker of membrane damage. At higher concentrations of exogenous H2O2, E. coli lost viability and released large amounts of lactate dehydrogenase into the culture medium (Brandi et al., 1989).
While some morphological changes may reflect cellular damage, the exposure to oxidants may reflect structural adaptations that increase resistance to oxidative stress. For example, after 3 days in culture, an amorphous extracellular substance surrounds the rugose phenotype of Salmonella enterica serovar Typhimurium and the cells form aggregates. These rugose phenotypes more readily form biofilms and are more resistant to H2O2 than the smooth phenotype (Anriany et al., 2001). In the conditions of our experiments, S. gordonii expressed discrete ultrastructural blebs on the surface in the presence of H2O2, which were exaggerated in the ΔmsrA mutant and not seen under the light microscope. Most effectively produced in the absence of MsrA, the extracellular blebs may contain oxidized surface substances or intracellular substances that were released secondary to hydrogen peroxide-mediated membrane damage or by a H2O2-dependent release mechanism such as we have reported for DNA (Kreth et al., 2009). The composition and function of the blebs remain to be determined.
S. gordonii production of H2O2 (Barnard et al., 1999) promotes endogenous oxidation, but damage to streptococcal cells is noticeably affected by deletion of MsrA, which results in increased surface blebs. To cope with endogenous H2O2, the ΔmsrA mutant appeared to down-regulate pyruvate oxidase, decreasing H2O2 production. The ΔmsrA mutant and the wild-type strain grew similarly in liquid medium, formed biofilms of similar mass, and adhered indistinguishably to sHA. Since the production of H2O2 was only partially lost, S. gordonii must express an anti-oxidant system other than MsrA to prevent self-inflicted H2O2-mediated injury. One possible explanation is that another methionine sulfoxide reductase may exist in S. gordonii, as the Msr activity was not totally lost in the msrA mutant. S. gordonii genome data (Genbank) indicated that another Msr sequence may exist and microorganisms such as Pseudomonas aeruginosa, E. coli, Staphylococcus aureus, like mammalian cells, express multiple Msr enzymes (Hansel et al., 2005, Ezraty et al., 2005). Future studies may address alternative anti-oxidation systems in S. gordonii that provide protection against endogenous H2O2.
In response to exogenous H2O2, S. gordonii V288 requires MsrA for anti-oxidant protection to achieve optimal growth. When challenged with H2O2, the wild-type and ΔmsrA mutant strains showed delayed onset of log-phase growth in liquid medium of about 14 and 20 hours, respectively; the ΔmsrA mutant also showed significantly greater growth sensitivity to exogenous H2O2 on agar plates. In the ΔmsrA strain, complementation with the msrA gene restored growth in the presence of oxidative stress. To accommodate to exogenous hydrogen peroxide-mediated stress, we show that S. gordonii up-regulates expression of msrA-specific mRNA. The msrA gene can also be up-regulated by a slight increase in pH (Vriesema et al., 2000). We considered the possibility that exogenous hydrogen peroxide increased the pH of strain V288 cultures, which could be more directly responsible for the increase in msrA expression. As expected, pH decreased during growth and was unaffected by added hydrogen peroxide. These data suggest strongly that hydrogen peroxide is an environmental factor that regulates the expression of the msrA gene independently of pH to protect growing cells against exogenous oxidative stress.
Whereas MsrA maintains adhesion function in the presence of oxidative stress (Wizemann et al., 1996), we now show that MsrA protects the structural integrity of surface adhesins. Purified MsrA reduced H2O2-oxidized, met-rich SspB protein fragments to restore apparent antigenicity. The restoration of antigenicity with the modeled protein fragments was indistinguishable from SspAB isolated from ΔMsrA cells in the presence of H2O2 when compared to wild-type cells. The wild-type and ΔMsrA cells expressed similar levels of sspA and sspB B genes as tested by qPCR (data not shown). Furthermore, H2O2 caused no obvious changes in the overall pattern or intensity of protein expression suggesting that SspAB were expressed but when oxidized were not recognized by our antibodies. Hence, SspB (SspAB), on the surface of the bacteria, is oxidized when the ΔMsrA mutant is exposed to H2O2 and is protected by MsrA when the wild-type bacteria are exposed to H2O2.
The wild-type strain of S. gordonii showed little functional impairment in adhesion to sHA or formation of in vitro biofilms in the presence of H2O2. After growth in H2O2-containing media, the harvested cells were resuspended in physiological buffer without H2O2 to determine residual damage to the cell surface. When compared to the wild-type cells, the ΔmsrA mutant cells in the presence of H2O2 showed significant loss of adhesion to sHA, a model of the saliva-coated tooth surface (Zhang et al., 2005), and altered electrophoretic mobility and antigenicity of a major adhesin SspAB. The overall protein profile was unaffected suggesting that transcription, protein stability or transport was unaffected.
We modeled the altered electrophoretic mobility, antigenicity, and substrate specificity for MsrA using an SspB protein fragment that includes the globular hinge region. The refolded met-rich fragment was soluble to 5 mg/ml as used in our experiments in non-denaturing conditions. At best, this protein fragment offers us an approximation of the conformation of the native met-rich domain in the intact protein. The protein will change in conformation when oxidized as our data suggest. We also know little of the conformation of SspB as it is exported in an unfolded state and susceptible to oxidation. To represent the entire sequence of the met-rich protein fragment, information from both the crystallographic structures of SpaP from S. mutans (Larson et al., 2010) and the variable domain of SspB from S. gordonii (Forsgren et al., 2009) were utilized to create the in silico model for our analysis. The static conformation of our cloned SspB met-rich fragment is predicted to be similar to the same region in the native protein (data not shown). In the intact protein, the central met-rich region is located in the globular variable domain, which is presented to external substrates at the end of a long unique, alpha-helix fibrillar stalk. Hence, this globular domain appears to have little interaction with other domains of the intact protein, and is poised to interact with adhesion targets. The met-rich protein fragment appears, therefore, to reasonably simulate oxidation of the met-rich central region in SspB (and the homologous region in SspA by inference), which was reversed or prevented by MsrA. Indeed, the met-rich variable region in SspAB appears to be important to the overall conformation and function of members of the P1 family of surface adhesins (Brady et al., 1998).
To protect adhesins on the cell surface, MsrA might be effective if expressed on the cell wall but a cytoplasmic or membrane-associated enzyme could protect against oxidation of proteins before or during export. Before or during export, the nascent, unfolded protein might be most vulnerable to oxidation. In some species of bacteria (Skaar et al., 2002, Spector et al., 2003), MsrA is targeted to the plasma membrane and also retained in the cytoplasm when post-translationally modified. Although MsrA in S. gordonii lacks a classical signal-peptide sequence, we found Msr activity to be associated with the cell wall, membrane and cytosolic fractions. Fluorescent-tagged MsrA co-localized with cell wall reactive WGA and was recovered from both cell wall and cytosolic fractions of cells, indicating that MsrA in S. gordonii locates in proximity to protein targets on the cell surface and within the cell. Indeed, finding that MsrA localizes in the three cell fractions suggests a mechanism to protect proteins any virtually any cellular locale against diffusible H2O2 originating from extra- or intracellular sources. Additional studies are needed to precisely co-localize MsrA with substrate surface proteins.
While protecting against exogenous peroxide, MsrA may also protect cells by regulating production of H2O2 by the host strain. The wild-type strain produced more H2O2 than the ΔmsrA mutant. S. gordonii appears to adapt to a deficit in MsrA by reducing production or increasing degradation of H2O2. Since msrA expression in wild-type S. gordonii is up-regulated in response to exogenous H2O2, and pyruvate oxidase–dependent production of endogenous hydrogen peroxide is down-regulated in the ΔmsrA mutant, oxidant protection in S. gordonii cells may reflect coordinately regulated genes. Such a mechanism implies that cells must signal in response to peroxides. Indeed, we have shown that S. gordonii releases DNA in response to a H2O2-dependent signal, without evidence of membrane damage or autolysis (Kreth et al, 2009). The mechanism by which cells sense hydrogen peroxide or other oxidants is not well established. Environmental oxidants are known to regulate some transcriptional regulator and stress genes. For example, during adaptation to H2O2, Salmonella typhimurium induces expression of about 30 proteins (Christman et al., 1985, Morgan et al., 1986). Genes of at least nine of the induced proteins are controlled by a transcriptional factor, OxyR (Morgan et al., 1986, Christman et al., 1985), which protects cells from oxidative stress (Demple, 1998, Kim et al., 2002, Zheng et al., 1998). A similar, specific oxidant-response regulating system may also exist in S. gordonii. Our data predict that this system down-regulates H2O2 production, and increases expression of anti-oxidant and other stress proteins. The oxidant-response regulating system functions to protect against environmental peroxide, facilitate peroxide-mediated anti-bacterial challenges against other bacteria, and maintain essential functions, including adhesion, growth and biofilm formation. The msrA gene is suggested to play a central role in this system.
Experimental procedures
Bacterial strains, oligonucleotides and DNA manipulation
The parent strain in these experiments was Streptococcus gordonii V288, which we (Kiliç et al., 1999) and others (Vriesema et al., 2000) have analyzed previously. In this study, oligonucleotides (listed in Table 1) were synthesized by Integrated DNA Technologies, Inc (Coralville, IA). Chromosomal DNA was prepared from mutanolysin-treated streptococcal cells as we report (Herzberg et al., 1990) (Zhang et al., 2009) using the QIAGEN 100/G Genomic Tip system (QIAGEN; Valencia, CA). DNA restriction and modification enzymes were used under conditions specified by the manufacturer (Promega; Madison, WI). PCR products were purified using the High Pure PCR Product Purification kit (Roche; Indianapolis, IN).
Table 1.
Primers used in this study
| Primer | Sequencea |
|---|---|
| msrAp.F | CGCGGATCCGCTGAAATTTATCTAGCAGGC |
| msrAp.R | CCCAAGCTTTTTCATGTGTTGAAGTAGATA |
| msrAup.F | AAGCTTCATCACCGCAAAACTCAAGA |
| msrAup.R | GAATTCCACCTCATTTCTTTGCGTCTA |
| msrAdn.F | GGATCCGCGTTGGCAAAAGTTTTTAGA |
| msrAdn.R | GAGCTCCCTTACTTGACGACTGGGTCT |
| msrAreal.F | ACGCAAGAGCAATACCAGGTG |
| msrAreal.R | AGACCGCCCGCTGACTC |
| msrAw.F | AACCGGCCGCATCACCGCAAAACTCA |
| msrAw.R | CCCAAGCTTTTATTTCATCCTCTAGAAAGGAGA |
| Flash.F | CGCGGATCCCGGTACCTCGAGCCTTC |
| Flash.R | CCCAAGCTTTTATTTCATCCTCTAGAAAGGAGA |
| sspB.F1 | CGCGGATCCGACGATGGAACCAAAATG |
| sspB.R1 | CCCAAGCTTTTTCTCTGTCTCATACATTGG |
| sspBc.F | CGCGGATCCGCTGTTGAAGAAAACACA |
| sspBc.R | CCCAAGCTTTTCCTGCTGCCCATATTG |
underlined sequences indicate restriction enzyme sites. All primers are original to this study.
Construction of a ΔmsrA mutant
A msrA-deletion mutant of S. gordonii was constructed by allelic exchange of the whole msrA sequence in S. gordonii with the erythromycin-resistance determinant, ermAM (Erm; 10 µg ml−1) as previously described (Nobbs et al., 2007). Briefly, using primers msrAup.F/msrAup.R and msrAdn.F/msrAdn.R, two flanking sequences of the msrA gene were amplified with PCR, incorporating restriction enzyme sites for HindIII/EcoRI for the upstream fragment and BamHI/SacI for the downstream fragment. A DNA fragment containing the ermAM gene was ligated between these two flanking regions. The insert DNA from the plasmid was then PCR amplified, purified, and transformed into S. gordonii, generating the ΔmsrA mutant. Predicted insertions were confirmed by PCR amplification and sequencing.
Complementation of msrA and construction of fluorescent labeled MsrA
To complement msrA, the complete msrA gene plus native promoter was amplified from S. gordonii V288 genomic DNA using primers msrAw.F/msrAw.R. The PCR product was cloned into the integration vector pFW5 encoding a spectinomycin resistance cassette (Podbielski et al., 1996). The confirmed plasmid was subsequently transformed into S. gordonii ΔmsrA. Chromosomal integration was achieved via homologous recombination with the intact msrA promoter sequence in the ΔmsrA mutant strain and selection using 100 µg ml−1 spectinomycin. Correct chromosomal insertion was confirmed by PCR amplification and sequencing.
To label MsrA with a fluorescent tag for cellular localization studies, a similar strategy of chromosomal knock-in was used as described above. Briefly, a tetracysteine-tag (Tsien et al., 1998) was incorporated at the 3’ end of msrA with PCR using primers Flash.F/Flash.R. The amplification strategy replaced the stop-codon of the msrA gene with the tetracysteine tag encoding its own stop codon. The PCR product was cloned into pFW5. The confirmed plasmid was transformed into S. gordonii ΔmsrA to produce an msrA-FLAsH complemented strain using 100 µg ml−1 spectinomycin and confirmed by PCR amplification, sequencing and fluorescent imaging.
RNA isolation
FMC medium (Terleckyj et al., 1975) with and without H2O2 (0.5 mM final concentration) was inoculated with S. gordonii and the cells were harvested after overnight growth. In some experiments, cultures in FMC medium were allowed to grow to mid-logarithmic phase, before H2O2 was added to half of the culture. H2O2-treated and untreated cells were then allowed to grow for one more hour, all cells were harvested, and RNA was isolated using a FastRNA® Pro Blue Kit (MP Biomedicals; Solon, OH) according to the manufacturer’s instructions. The RNA obtained was checked for integrity using agarose gel electrophoresis and then digested with DNase I (Takara; Madison, WI) for 2 h at 37°C and purified with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. The amount of RNA recovered was measured by absorbance at 260 nm in a spectrophotometer.
Reverse transcription
To determine changes in expression of msrA-specific mRNA, reverse transcription was performed to synthesize cDNA as previously described (Zhang et al., 2004). RNA (10 µg) was mixed with 500 pmoles of random hexamer primers (Promega) to a final volume of 68 µl, incubated at 80°C for 5 minutes, and placed on ice for 5 min. A reverse transcriptase enzyme master mixture (32 µl), containing 20 µl of 5× reaction buffer (Promega), 5 µl (100 U) anti-RNase (Ambion), 5 µl of 10 mM dNTP, and 2 µl (100 U) of Maloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT, Promega), was then added to the primer-annealed RNA. The reaction mixture was incubated at 40°C for 1 h and at 92°C for 10 min. As negative controls, all RNA samples were also incubated without reverse transcriptase.
Real-time PCR
The expression of msrA-specific mRNA in S. gordonii V288 was determined by real-time PCR assay using a Mx3000P™ QPCR System following the manufacturer’s instructions (Stratatgene; Cedar Creek, TX). The product of reverse transcription was used as the template for real-time PCR amplification using a mixture containing specific primers msrAreal.F/msrAreal.R. The PCR conditions were: 95°C × 10 min; 40 × [95°C × 30 sec, 55°C × 1 min, 72°C × 1 min]. To estimate the relative amount of specific mRNA, msrA and the 16S rRNA genes were estimated using a serial dilution of 100, 10, 1, 0.1, 0.01, and 0.001 ng purified S. gordonii V288 genomic DNA as the standard curve. The relative amount of msrA was estimated by normalizing to 16S rRNA as a housekeeping gene.
FLAsH labeling and fluorescence microscopy
To localize MsrA, the msrA-FLAsH complemented strain was prepared using the green fluorescent FLAsH-EDT2 labeling reagent following the manufacturer’s instructions (Invitrogen). Briefly, an overnight culture of bacteria was centrifuged, re-inoculated, and grown at 37°C in 5% CO2. When cell density reached A620nm = 0.5, bacteria were centrifuged, washed, and resuspended in Tris-buffered saline with 0.05% Tween-20 (TBST) buffer, supplemented with 1 µM of Tris (2-carboxyethyl) phosphine hydrochloride (TCEP)-HCl and 20 µM FLAsH-EDT2, and incubated at RT for 60 min. The cell wall was stained with 1 µg/ml WGA Alexa Fluor 555 (Invitrogen). The stained cells were placed on microscope slides, sealed with cover slips, and visualized using a Nikon Eclipse E800 microscope and a 100×/1.40 Plan Apo oil-immersion lens. Images were processed with MetaMorph software (Molecular Devices, Sunnyvale, CA). Brightness and contrast were adjusted uniformly for the entire image.
Growth of bacteria
To test the sensitivity to oxidation, S. gordonii V288 wild-type, ΔmsrA mutant and complemented strains were grown in chemically-defined FMC medium as we have reported (Herzberg et al., 1990). Bacteria were pre-cultured overnight at 37°C in 5% CO2 in FMC; an aliquot was diluted to A 620 nm = 0.2 and incubated at 37°C in 5% CO2 in the presence or absence of 0.5 mM H2O2. At indicated times, the OD620 nm was determined. In some experiments, the pH of the culture medium was measured at selected times of growth.
H2O2 inhibition assay
The S. gordonii strains were cultured separately in 1 ml of THB medium (Todd-Hewitt broth, Difco; Sparks, MD). After overnight growth, cells in spent media were mixed with 5 ml handwarmed THB soft-agar (0.7%) and poured onto a Petri dish. A filter paper disk (1 mm diameter) containing 10 µl of distilled H2O or 31% H2O2 (Sigma) was placed in the center of the agar surface. Plates were then incubated at 37°C for 24 h. Inhibition was quantified as the area of clearing between the margins of the filter and the emergent bacterial colonies.
Quantitation of H2O2 production
The H2O2 concentration in the supernatant of FMC stationary phase cultures was measured as described using a colorimetric assay (Nunez de Kairuz et al., 1988). Briefly, bacterial supernatant (0.2 ml) was mixed with reaction buffer (0.8 ml) (10 mM phosphate buffer containing 0.16 mM o-dianisidine, 1.2 µg ml−1 horseradish peroxidase, and 0.02% Triton-X100), incubated at 37°C for 20 minutes, and color development was determined at A570 nm. The concentration of H2O2 was determined from a standard curve prepared with fresh dilutions of hydrogen peroxide solution.
MsrA activity
MsrA activity was analyzed at 25°C by monitoring H-transfer from NADPH to methionine sulfoxide (Met(O)) by recording change in A340 nm as described (Hassouni et al., 1999). Briefly, the supernatant of sonicated whole cell extract in 50 mM Tris·HCl, pH 7.4 was mixed with 0.2 mM NADPH, 78.43 µg/ml thioredoxin, 3.04 µg/ml thioredoxin reductase, and 3.3 mM Met(O). The change in NADPH oxidation was determined by measuring absorbance at 340 nm at minute intervals for 10 min.
Cell wall protein isolation and Western blot
To learn whether MsrA protects surface adhesins, S. gordonii V288 was inoculated into FMC medium. To some cultures, H2O2 was added immediately to a final concentration of 0.5 mM. After overnight growth, cells were harvested and cell wall macromolecules were isolated as described previously (Herzberg et al., 1983). Briefly, bacteria were cultured overnight, washed and resuspended in 20 mM Tris buffer (pH 6.8), 10 mM MgCl2 and 26% raffinose, and mutanolysin (Sigma; Milwaukee, WI) was added at 80 U/ml and incubated at 37°C for 75 min. Protoplasts were centrifuged at 12,400 × g for 20 min, and the supernatant was collected, dialyzed and concentrated by ultrafiltration with a Centricon Plus-20 centrifugal filter unit with PL-30 membrane (Millipore; Billerica, MA). The protein concentration was detected using a BCA Protein Assay Kit (Pierce; Rockford, IL). Proteins were loaded on a 4–20% precast mini polyacrylamide gel under non-denaturing conditions and transferred to a nitrocellulose membrane. The membrane was blocked with 3% BSA in PBS buffer overnight and then incubated with rabbit anti-P1 antibody (Jenkinson et al., 1993), washed and incubated with anti-rabbit IgG HRP conjugate (secondary antibody). Bands were detected using ECL chemiluminescent reagents (Amersham Biosciences). Anti-P1 antibody reacts specifically with SspAB from S. gordonii. To resolve overall profiles, surface proteins were also separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) under denaturing conditions, stained with Coomassie Blue (Bio-Rad, Hercules, CA) and destained in a methanol-acetic solution (40% methanol and 10% acetic acid).
Expression and purification of MsrA and the Met-rich segment of SspB
The msrA gene and the Met-rich segment of sspB (SGO_0211) were each amplified using primers msrAp.F/msrAp.R and sspB.F1/sspB.R1, respectively, cloned into the pQE-80L vector (QIAGEN) and expressed in NEB 5α F'Iq competent E. coli cells (New England Biolabs) with an N-terminal His-tag as we described (Zhang et al., 2009). The cells were grown in LB medium to OD600 ~ 0.5–0.6. Expression of the recombinant protein fragments was then induced by incubation with IPTG (1 mM) for 4 h at 37°C. Cells were lysed by sonication. To ensure proper refolding, the Met-rich protein fragment was solubilized from inclusion bodies using a Novagen protein refolding kit as per the manufacturer’s instructions, refolded by a series of dialysis steps with Slide-A-Lyzer MINI Dialysis Unit (Pierce), and purified by Ni-NTA column chromatography. As a negative control, an SspB protein fragment of the same length without Met was cloned using primers sspBc.F/sspBc.R and prepared similarly. Recombinant MsrA was purified from the soluble fraction of cell lysate by Ni-NTA column chromatography. The purified Met-rich segment was incubated at 2 mg/ml (final cncentration) with 50 mM H2O2, and then incubated with purified MsrA. Untreated, H2O2-oxidized and MsrA-reduced Met-rich protein fragments at 5 µg/12 µl/lane were resolved by native 4–15% PAGE gel and resolved using anti-tetra His antibody (QIAGEN) in Western blot.
MASS-SPEC analysis
Protein fragment samples (5 µg) were separated by 10% PAGE and digested with trypsin using the protocol of Trypsin Profile IGD Kit for In-gel Digests (SIGMA). Digested samples were analyzed in the Center for Mass Spectrometry and Proteomics at the University of Minnesota. In brief, samples were absorbed on a C18 trap (5 mm × 300 µm ID) and separated using a 150 mm × 75 µm ID C18 column, using a linear gradient of 10% to 75% acetonitrile/0.1% formic acid at a flow rate of 0.4 µl/min. Column effluent was introduced to a triple quadrupole instrument (4000 Q TRAP, Applied Biosystems, Foster City, CA) via an ESI source. The mass spectrometer was operated in information-dependent acquisition (IDA) mode with multiple reaction monitoring (MRM). All possible oxidation forms of Met were monitored. For each peptide, MRM transitions were calculated based on in-silico digestion of the Met-rich segment of SspB. Targeted MRM transitions trigger IDA that identifies the peptide. Identified peptides were quantitated using a regular MRM method and the data were processed with Analyst® software (Applied Biosystems).
Saliva-coated hydroxyapatite (sHA) adhesion assay
The adhesion assay was performed with S. gordonii essentially as described previously (Zhang et al., 2004, Gong et al., 2000). In brief, bacterial cells were cultured overnight in FMC medium with 3H-thymidine (10 µCi ml−1), centrifuged and diluted to A620nm = 0.3 with modified Gibbons' buffer (1 mM KH2PO4, K2HPO4 buffer, pH 6.8, with 50 mM KCl, 1 mM CaCl2, 0.1 mM MgCl2). The cells (1 ml; specific activity about 103 cells CPM−1) were then incubated with 10 mg of sHA for 1 h at ambient temperature with continuous inversion on a roto-torque. The unattached cells were removed by aspiration. The sHA with attached bacteria was washed three times to remove additional unattached cells. Radioactivity associated with sHA was counted using a liquid scintillation counter. The percentage of adhesion was calculated as the ratio of counts per minute (CPMs) associated with sHA to the total CPMs in the 1 ml input suspension × 100.
Biofilm formation assay
To screen S. gordonii strains for the ability to form biofilms on polystyrene surfaces, the assay of O' Toole (O'Toole et al., 1998) and Loo (Loo et al., 2000) as modified (Zhang et al., 2004) was used. In brief, bacterial cells were pre-cultured overnight in FMC chemically-defined synthetic medium with or without 0.5 mM H2O2, diluted 200-fold in FMC, and inoculated into wells of duplicate microtitre plates (Costar 3799). The bacteria were then incubated at 37°C until reaching stationary phase growth, unattached cells were removed by aspiration and 25 µl of 1% crystal violet (CV) was added to each well to stain the sessile biofilm. The plates were incubated at room temperature for 15 min and rinsed thoroughly with water. To each well of CV-stained bacterial biofilm, 200 µl of 95% ethanol was added to release the CV and 125 µl was then transferred to a new polystyrene microtitre plate. Biofilm formation was quantified as the absorbance at 568 nm as analyzed with an ELISA plate reader.
Field Emission Scanning Electron Microscopy (FESEM)
An overnight culture was harvested by centrifugation at 700 × g for 20 min, resuspended in PBS, fixed with 0.1 M sodium cacodylate buffer containing 2.5% glutaraldehyde and 7.5% sucrose, and one drop was placed on poly-L-lysine (Sigma)-covered glass chips (5 × 10 mm). The cells were allowed to adhere for 30 min, rinsed with 0.1 M cacodylate buffer, immersed in OsO4 secondary fixative for 30 min, and dehydrated by an ascending series of alcohol solutions. The bacteria were examined by backscatter electron imaging with an Hitachi S-900 field emission scanning microscope using an Autrata-modified YAG detector at 2.5 keV (5 keV for back scatter microscopy) as described (Olmsted et al., 1993).
Statistics
Descriptive statistics, including the mean and standard deviation, were calculated. Statistical analysis of data was performed using the student t-test to compare the means of two groups. Data were considered as significantly different if the two-tailed p-value was ≤ 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant R01 DE08590 to MCH.
References
- Anriany YA, Weiner RM, Johnson JA, De Rezende CE, Joseph SW. Salmonella enterica serovar Typhimurium DT104 displays a rugose phenotype. Appl Environ Microbiol. 2001;67:4048–4056. doi: 10.1128/AEM.67.9.4048-4056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard JP, Stinson MW. Influence of environmental conditions on hydrogen peroxide formation by Streptococcus gordonii. Infect Immun. 1999;67:6558–6564. doi: 10.1128/iai.67.12.6558-6564.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LJ, Cvitkovitch DG, Geric CM, Addison MN, Joyce JC, Crowley PJ, Bleiweis AS. Deletion of the central proline-rich repeat domain results in altered antigenicity and lack of surface expression of the Streptococcus mutans P1 adhesin molecule. Infect Immun. 1998;66:4274–4282. doi: 10.1128/iai.66.9.4274-4282.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandi G, Fiorani M, Pierotti C, Albano A, Cattabeni F, Cantoni O. Morphological changes in Escherichia coli cells exposed to low or high concentrations of hydrogen peroxide. Microbiol Immunol. 1989;33:991–1000. doi: 10.1111/j.1348-0421.1989.tb03157.x. [DOI] [PubMed] [Google Scholar]
- Cabreiro F, Picot CR, Friguet B, Petropoulos I. Methionine sulfoxide reductases: relevance to aging and protection against oxidative stress. Ann N Y Acad Sci. 2006;1067:37–44. doi: 10.1196/annals.1354.006. [DOI] [PubMed] [Google Scholar]
- Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- Demple B. A bridge to control. Science. 1998;279:1655–1656. doi: 10.1126/science.279.5357.1655. [DOI] [PubMed] [Google Scholar]
- Ezraty B, Aussel L, Barras F. Methionine sulfoxide reductases in prokaryotes. Biochim Biophys Acta. 2005;1703:221–229. doi: 10.1016/j.bbapap.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Ezraty B, Grimaud R, El Hassouni M, Moinier D, Barras F. Methionine sulfoxide reductases protect Ffh from oxidative damages in Escherichia coli. EMBO J. 2004;23:1868–1877. doi: 10.1038/sj.emboj.7600172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsgren N, Lamont RJ, Persson K. Crystal structure of the variable domain of the Streptococcus gordonii surface protein SspB. Protein Science. 2009;18:1896–1905. doi: 10.1002/pro.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong K, Mailloux L, Herzberg MC. Salivary film expresses a complex, macromolecular binding site for Streptococcus sanguis. J Biol Chem. 2000;275:8970–8974. doi: 10.1074/jbc.275.12.8970. [DOI] [PubMed] [Google Scholar]
- Hansel A, Heinemann SH, Hoshi T. Heterogeneity and function of mammalian MSRs: enzymes for repair, protection and regulation. Biochim Biophys Acta. 2005;1703:239–247. doi: 10.1016/j.bbapap.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Hassouni ME, Chambost JP, Expert D, Van Gijsegem F, Barras F. The minimal gene set member msrA, encoding peptide methionine sulfoxide reductase, is a virulence determinant of the plant pathogen Erwinia chrysanthemi. Proc Natl Acad Sci U S A. 1999;96:887–892. doi: 10.1073/pnas.96.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg MC. Platelet-streptococcal interactions in endocarditis. Crit Rev Oral Biol Med. 1996;7:222–236. doi: 10.1177/10454411960070030201. [DOI] [PubMed] [Google Scholar]
- Herzberg MC, Brintzenhofe KL, Clawson CC. Cell-free released components of Streptococcus sanguis inhibit human platelet aggregation. Infect Immun. 1983;42:394–401. doi: 10.1128/iai.42.1.394-401.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzberg MC, Erickson PR, Kane PK, Clawson DJ, Clawson CC, Hoff FA. Platelet-interactive products of Streptococcus sanguis protoplasts. Infect Immun. 1990;58:4117–4125. doi: 10.1128/iai.58.12.4117-4125.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson HF, Terry SD, McNab R, Tannock GW. Inactivation of the gene encoding surface protein SspA in Streptococcus gordonii DL1 affects cell interactions with human salivary agglutinin and oral actinomyces. Infect Immun. 1993;61:3199–3208. doi: 10.1128/iai.61.8.3199-3208.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiliç AO, Herzberg MC, Meyer MW, Zhao X, Tao L. Streptococcal reporter gene-fusion vector for identification of in vivo expressed genes. Plasmid. 1999;42:67–72. doi: 10.1006/plas.1999.1408. [DOI] [PubMed] [Google Scholar]
- Kim SO, Merchant K, Nudelman R, Beyer WF, Jr, Keng T, DeAngelo J, et al. OxyR: a molecular code for redox-related signaling. Cell. 2002;109:383–396. doi: 10.1016/s0092-8674(02)00723-7. [DOI] [PubMed] [Google Scholar]
- Kline KA, Falker S, Dahlberg S, Normark S, Henriques-Normark B. Bacterial adhesins in host-microbe interactions. Cell Host Microbe. 2009;5:580–592. doi: 10.1016/j.chom.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Kreth J, Vu H, Zhang Y, Herzberg MC. Characterization of hydrogen peroxide-induced DNA release by Streptococcus sanguinis and Streptococcus gordonii. J Bacteriol. 2009;191:6281–6291. doi: 10.1128/JB.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreth J, Zhang Y, Herzberg MC. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol. 2008;190:4632–4640. doi: 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboniwa M, Tribble GD, James CE, Kilic AO, Tao L, Herzberg MC, et al. Streptococcus gordonii utilizes several distinct gene functions to recruit Porphyromonas gingivalis into a mixed community. Mol Microbiol. 2006;60:121–139. doi: 10.1111/j.1365-2958.2006.05099.x. [DOI] [PubMed] [Google Scholar]
- Larson MR, Rajashankar KR, Patel MH, Robinette RA, Crowley PJ, Michalek S, Brady LJ, Deivanayagam C. Elongated fibrillar structure of a streptococcal adhesin assembled by the high-affinity association of α- and PPII-helices. PNAS. 2010;107:5983–5988. doi: 10.1073/pnas.0912293107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay D, von Holy A. Different responses of planktonic and attached Bacillus subtilis and Pseudomonas fluorescens to sanitizer treatment. J Food Prot. 1999;62:368–379. doi: 10.4315/0362-028x-62.4.368. [DOI] [PubMed] [Google Scholar]
- Loo CY, Corliss DA, Ganeshkumar N. Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol. 2000;182:1374–1382. doi: 10.1128/jb.182.5.1374-1382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Eaton JW. Multicellular oxidant defense in unicellular organisms. Proc Natl Acad Sci U S A. 1992;89:7924–7928. doi: 10.1073/pnas.89.17.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MW, Gong K, Herzberg MC. Streptococcus sanguis-induced platelet clotting in rabbits and hemodynamic and cardiopulmonary consequences. Infect Immun. 1998;66:5906–5914. doi: 10.1128/iai.66.12.5906-5914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RW, Christman MF, Jacobson FS, Storz G, Ames BN. Hydrogen peroxide-inducible proteins in Salmonella typhimurium overlap with heat shock and other stress proteins. Proc Natl Acad Sci U S A. 1986;83:8059–8063. doi: 10.1073/pnas.83.21.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskovitz J. Methionine sulfoxide reductases: ubiquitous enzymes involved in antioxidant defense, protein regulation, and prevention of aging-associated diseases. Biochim Biophys Acta. 2005;1703:213–219. doi: 10.1016/j.bbapap.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Nobbs AH, Lamont RJ, Jenkinson HF. Streptococcus adherence and colonization. Microbiol Mol Biol Rev. 2009;73:407–450. doi: 10.1128/MMBR.00014-09. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobbs AH, Zhang Y, Khammanivong A, Herzberg MC. Streptococcus gordonii Hsa environmentally constrains competitive binding by Streptococcus sanguinis to saliva-coated hydroxyapatite. J Bacteriol. 2007;189:3106–3114. doi: 10.1128/JB.01535-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez de Kairuz MS, Olazabal ME, Oliver G, Pesce de Ruiz Holgado AA, Massa E, Farias RN. Fatty acid dependent hydrogen peroxide production in Lactobacillus. Biochem Biophys Res Commun. 1988;152:113–121. doi: 10.1016/s0006-291x(88)80687-9. [DOI] [PubMed] [Google Scholar]
- O'Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol. 1998;30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- Olmsted SB, Erlandsen SL, Dunny GM, Wells CL. High-resolution visualization by field emission scanning electron microscopy of Enterococcus faecalis surface proteins encoded by the pheromone-inducible conjugative plasmid pCF10. J Bacteriol. 1993;175:6229–6237. doi: 10.1128/jb.175.19.6229-6237.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski S, Kasielski M, Kordiak J, Nowak D. Elevated resting and agonist-induced whole blood chemiluminescence in patients with active infective endocarditis. Interact Cardiovasc Thorac Surg. 2009;8:12–16. doi: 10.1510/icvts.2008.183285. [DOI] [PubMed] [Google Scholar]
- Presterl E, Rokita E, Graninger W, Hirschl AM. Dysregulation of monocyte oxidative burst in streptococcal endocarditis. Eur J Clin Invest. 2001;31:902–906. doi: 10.1046/j.1365-2362.2001.00891.x. [DOI] [PubMed] [Google Scholar]
- Rosan B, Eisenberg RJ. Morphological changes in Streptococcus sanguis associated with growth in the presence of oxygen. Arch Oral Biol. 1973;18:1441–1444. doi: 10.1016/0003-9969(73)90118-0. [DOI] [PubMed] [Google Scholar]
- Rosen H, Klebanoff SJ, Wang Y, Brot N, Heinecke JW, Fu X. Methionine oxidation contributes to bacterial killing by the myeloperoxidase system of neutrophils. Proc Natl Acad Sci U S A. 2009;106:18686–18691. doi: 10.1073/pnas.0909464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtele CF, Nobbs A, Zhang Y, Costalonga M, Herzberg MC. Oral Streptococci: Commensals and Opportunistic Pathogens. In: Hakenbeck R, Chhatwal S, editors. The Molecular Biology of Streptococci. Norfolk, UK: Horizon Scientific Press; 2007. pp. 411–462. [Google Scholar]
- Skaar EP, Tobiason DM, Quick J, Judd RC, Weissbach H, Etienne F, et al. The outer membrane localization of the Neisseria gonorrhoeae MsrA/B is involved in survival against reactive oxygen species. Proc Natl Acad Sci U S A. 2002;99:10108–10113. doi: 10.1073/pnas.152334799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector D, Etienne F, Brot N, Weissbach H. New membrane-associated and soluble peptide methionine sulfoxide reductases in Escherichia coli. Biochem Biophys Res Commun. 2003;302:284–289. doi: 10.1016/s0006-291x(03)00163-3. [DOI] [PubMed] [Google Scholar]
- Terleckyj B, Willett NP, Shockman GD. Growth of several cariogenic strains of oral streptococci in a chemically defined medium. Infect Immun. 1975;11:649–655. doi: 10.1128/iai.11.4.649-655.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriesema AJ, Dankert J, Zaat SA. A shift from oral to blood pH is a stimulus for adaptive gene expression of Streptococcus gordonii CH1 and induces protection against oxidative stress and enhanced bacterial growth by expression of msrA. Infect Immun. 2000;68:1061–1068. doi: 10.1128/iai.68.3.1061-1068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizemann TM, Moskovitz J, Pearce BJ, Cundell D, Arvidson CG, So M, et al. Peptide methionine sulfoxide reductase contributes to the maintenance of adhesins in three major pathogens. Proc Natl Acad Sci U S A. 1996;93:7985–7990. doi: 10.1073/pnas.93.15.7985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lei Y, Khammanivong A, Herzberg MC. Identification of a novel two-component system in Streptococcus gordonii V288 involved in biofilm formation. Infect Immun. 2004;72:3489–3494. doi: 10.1128/IAI.72.6.3489-3494.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lei Y, Nobbs A, Khammanivong A, Herzberg MC. Inactivation of Streptococcus gordonii SspAB alters expression of multiple adhesin genes. Infect Immun. 2005;73:3351–3357. doi: 10.1128/IAI.73.6.3351-3357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Whiteley M, Kreth J, Lei Y, Khammanivong A, Evavold JN, et al. The two-component system BfrAB regulates expression of ABC transporters in Streptococcus gordonii and Streptococcus sanguinis. Microbiology. 2009;155:165–173. doi: 10.1099/mic.0.023168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Aslund F, Storz G. Activation of the OxyR transcription factor by reversible disulfide bond formation. Science. 1998;279:1718–1721. doi: 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.