
Figure 6.
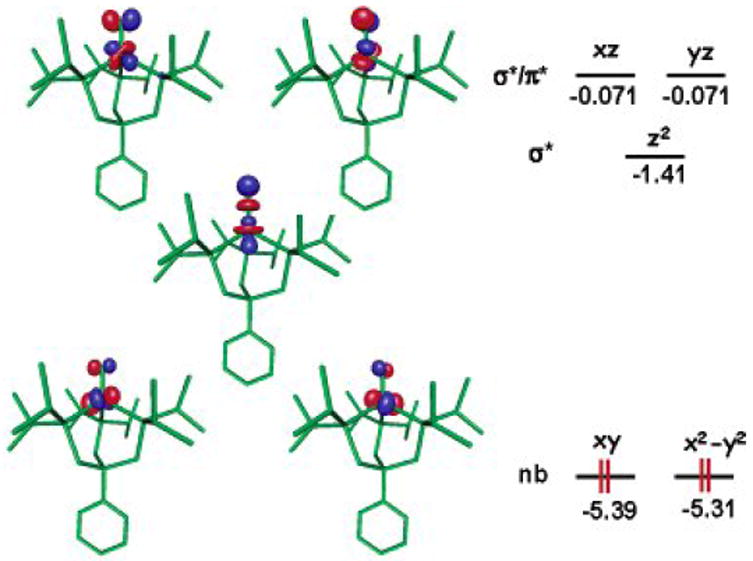
Theoretically predicted geometry and electronic structure (DFT,JAGUAR 5.0, B3LYP/LACVP**) for S = 0 [PhBPiPr3]Fe≡N. Lobal representations correspond to the frontier orbitals (energies in eV). Structural parameters: Fe-P = 2.28, 2.28, 2.29 Å; N-P-Fe = 117, 117, 119°; P-Fe-P = 99, 101, 101°; Fe-N = 1.490 Å. Reprinted with permission from ref[52]. Copyright 2004 American Chemical Society.