

Abstract
Endoplasmic reticulum (ER) stress, often resulting from cellular accumulation of misfolded proteins, occurs in many neurodegenerative disorders, in part because of the relatively long lifetime of neurons. Excessive accumulation of misfolded proteins activates the unfolded protein response (UPR) that dampens protein synthesis and promotes removal of misfolded proteins to support survival of ER-stressed cells. However, the UPR also initiates apoptotic signaling to kill cells if recovery is not achieved. Thus, there is much interest in identifying determinants of the life-death switch and interventions that promote recovery and survival. One intervention that has consistently been shown to protect cells from ER stress-induced apoptosis is application of inhibitors of glycogen synthase kinase-3 (GSK3). Therefore, we examined where in the UPR pathway GSK3 inhibitors intercede to impede signaling towards apoptosis. Apoptosis following UPR activation can be mediated by activation of two transcription factors, ATF4 and ATF6, that activate expression of the death-inducing transcription factor C/EBP homologous protein (CHOP/GADD153) following ER stress. We found that ER stress induced activated ATF6 and ATF4, but these responses were not altered by pretreatment with GSK3 inhibitors. However, inhibition of GSK3 effectively reduced the expression of CHOP, and this was apparent in several types of neural-related cells and was evident after application of several structurally diverse GSK3 inhibitors. Therefore, reduction of CHOP activation provides one mechanism by which inhibitors of GSK3 are capable of shifting cell fate towards survival instead of apoptosis following ER stress.
Keywords: ER stress, CHOP, GADD153, glycogen synthase kinase-3, unfolded protein response
Introduction
The accumulation of misfolded proteins is one of the most prevalent disease-induced causes of cellular dysfunction, and is particularly destructive in diseases afflicting neurons because of their long lifetimes [1]. The unfolded protein response (UPR) is a crucial stress response that evolved in eukaryotes to manage deleterious effects of excessive misfolded proteins caused by disease, environmental, or genetic factors. The UPR is activated in response to endoplasmic reticulum (ER) stress, which can be caused by misfolded protein accumulation in the ER, calcium depletion, inflammation, glucose deprivation, and other conditions [2,3]. The UPR generally represses translation, upregulates production of ER chaperones, and promotes other mechanisms directed towards reducing the accumulation of misfolded proteins to re-establish ER homeostasis. With severe or persistent ER stress, the UPR also initiates apoptotic signaling leading to cell death [4,5]. Thus, the UPR is a signaling program that supports adaptation to stress and recovery, but also terminates highly stressed cells, which has led to much interest in identifying determinants of the life-death switch and interventions that promote recovery and survival.
Three major signaling pathways activated by the UPR have been identified. One of these involves activation of PKR-like ER kinase (PERK), which is released from glucose-regulated protein 78 (GRP78) by ER stress [6]. Activated PERK phosphorylates eukaryotic initiation factor 2α (eIF2α) on serine 51, which represses translation by inhibiting the guanine nucleotide exchange factor eIF2B to block the recycling of eIF2-GDP to eIF2-GTP [7–9]. Although most protein synthesis is repressed by phosphorylated eIF2α, synthesis of activating transcription factor 4 (ATF4) is up-regulated to induce stress responsive genes [8,10]. Among these is the proapoptotic protein C/EBP homologous protein (CHOP/GADD153), which is a key determinate in cellular fate and facilitates ER stress-induced apoptosis [11]. CHOP is a short lived protein, so with mild or transient ER stress, CHOP expression is not prolonged and the cell can adapt and recover. However, if the ER stress is intense or prolonged, CHOP expression is persistent and cell death ensues [12].
Similar to PERK, IRE1 is a trans-ER membrane kinase held inactive by GRP78. ER stress releases IRE1 from GRP78, and IRE1 oligomerizes, autophosphorylates, and becomes active [6]. Active IRE1 splices the mRNA of the transcription factor X box-binding protein 1 (XBP1), removing 26 nucleotides, leading to accumulation of active XBP1, which promotes expression of ER-resident molecular chaperones, including GRP78 [13]. The third arm of the UPR activates ATF6 [14], which is released from GRP78 in the ER and translocates to the golgi where it is cleaved by site 1 and site 2 proteases [14,15]. The ATF6 cleavage product, p50ATF6, translocates to the nucleus and promotes expression of ER stress responsive genes, including CHOP, which can lead to apoptosis [13].
The UPR is a concerted effort by the cell to restore ER homeostasis and survive, but it also leads to cell death by apoptosis if these recovery mechanisms fail. ER stress-induced apoptosis utilizes the intrinsic (mitochondrial) pathway involving activation of caspase-9 and caspase-3 [16]. Regulation of the ER stress-induced execution phase of apoptosis is not completely understood, but clearly involves glycogen synthase kinase-3 (GSK3). GSK3 represents two ser/thr kinase paralogs that are commonly referred to as isoforms, GSK3α and GSK3β, that are the products of distinct genes [17]. GSK3 is constitutively active but is regulated through inhibitory phosphorylation at serine 21 of GSK3α and serine 9 of GSK3β [17]. GSK3 is a critical component in the Wnt signaling pathway where it associates with Axin to phosphorylate β-catenin, marking it for proteasomal mediated degradation [18]. Inhibition of GSK3 suppresses caspase-3 activation and subsequent cell death induced by the ER stress-inducing agents thapsigargin (an ER Ca2+-ATPase inhibitor) and tunicamycin (which inhibits N-linked glycosylation and protein folding in the ER) [19]. Several groups have verified the obligatory role of GSK3 in ER stress-induced apoptosis using a variety of cell types, ER stress models, and GSK3 inhibitors [20–27]. Despite considerable evidence that GSK3 has a crucial role promoting ER stress-induced apoptosis, the mechanism by which GSK3β facilitates apoptosis remains undefined. Here we provide evidence that in cells of neuronal lineage GSK3 regulates the outcome of UPR signaling by promoting CHOP expression.
Materials and methods
Cells and reagents
SH-SY5Y human neuroblastoma cells were grown in RPMI1640 with 10% horse serum and 5% fetal clone. Mouse embryonic fibroblasts (MEFs) were grown in Dulbecco’s modified eagle medium supplemented with 10% fetal bovine serum and 15 mM HEPES (Invitrogen, Carlsbad, CA). RN46a cells were grown in DMEM/F12 (1:1) supplemented with 10% fetal bovine serum. Primary astrocytes were prepared from cerebral cortex of 1-day-old C57Bl/6 mice as described previously [28] and cultured in DMEM/F-12 (1:1) medium supplemented with 10% fetal bovine serum and 0.3% glucose. All media were supplemented with 2 mM L-glutamine, 100 units/ml penicillin, and 100 µg/ml streptomycin (Cellgro, Herndon, VA), and cells were grown in humidified, 37°C chambers with 5% CO2, except the immortalized hippocampal cells which were grown at 33°C. Primary neural precursor cells (NPCs) were prepared at gestational day 12–13 from the telencephalons of C57BL/6 mice, expanded as neurospheres, and experimental procedures were carried out with monolayers of NPCs cultured as described previously [29]. Primary mouse neurons were prepared and cultured as described previously [30]. The 5X-ATF6-luciferase has been described previously [31]. Sources of chemicals were: tunicamycin, thapsigargin (Alexis, San Diego, CA), kenpaullone, lithium chloride (Sigma, St. Louis, MO), salubrinal (Dako Corp., Carpintera, CA), 6-bromoindirubin-3'-oxime (BIO; CalBiochem), CHIR99021 (Dr. Hilary Mclauchlan, University of Dundee), and insulin-like growth factor-1 (IGF-1; Intergen, Purchase, NY).
Immunoblotting
Cells were washed twice with PBS and were lysed with lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 2 mM EGTA, 0.5% NP-40, 1 mM sodium orthovanadate, 100 µM phenylmethanesulfonyl fluoride, 0.1 µM okadaic acid, 50 mM sodium fluoride, and 10 µg/ml each of leupeptin, aprotinin, and pepstatin). The lysates were sonicated and centrifuged at 20,800×g for 15 min. Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL). Samples were mixed with Laemmli sample buffer (2% SDS), placed in a boiling water bath for 5 min, proteins were resolved in SDS-polyacrylamide gels, transferred to nitrocellulose, and incubated with antibodies to CHOP (Santa Cruz Biotechnology, Santa Cruz, CA), phospho-ser51-eIF2α (#3597), cleaved caspase-3, proteolyzed 85 kDa fragment of poly(ADP-ribose) polymerase (PARP), phospho-Ser9-GSK3β (Cell Signaling, Danvers, MA), total GSK3β, (BD-PharMingen/Transduction Laboratories, San Diego, CA), phospho-ser9/21-GSK3 α/β, total GSK3 α/β (Upstate Biotechnology Inc., Lake Placid, NY), or β-actin (Sigma, St. Louis, MO). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse, or goat anti-rabbit IgG, followed by detection with enhanced chemiluminescence, and the protein bands were quantitated with a densitometer.
RT-PCR
For RT-PCR analysis RNA was isolated using Trizol (Invitrogen) according to the manufacturer’s protocol. The RNA was quantified using a Nano-drop spectrophotometer. The RT-PCR reaction was performed using the SuperScript one-step RT-PCR system (Invitrogen) according to the manufacturer’s protocol. To analyze CHOP RNA, 500 ng of RNA was used in the reaction with the following primers: forward (CTGCAAGAGGTCCTGTCTTC) and reverse (CAATCAGAGCTCGGCGAGTC) for 40 cycles. The resulting cDNA was then visualized on a 1.5% agarose gel containing ethidium bromide and imaged on a multi-imager (Bio-Rad).
Real time PCR
RNA was isolated using the RNeasy kit (Qiagen). cDNA synthesis was performed using oligo(dT) and reverse transcriptase Superscript Preamplification System according to manufacturer’s instructions (Invitrogen). Real-time PCR was performed using the Light Cycler 280 (Roche Applied Biosciences) to detect sybr green incorporation, according to the manufacturer’s instructions. The fold-increase of CHOP or ATF4 mRNA accumulation was normalized to the housekeeping gene GAPDH. Primer sequences: CHOP forward (CAGAGGTCACAAGCACCT), CHOP reverse (TCCCTGGTCAGGCGCTC) ATF4 forward (AGTGGCATCTGTATGAGCC) ATF4 reverse (TCTTCTGGCGGTACCTAGT) GAPDH forward (GCTGGGGCTCACCTGAAGGG) GAPDH reverse (GGATGACCTTGCCCACAGCC).
Luciferase assay
Cells were plated and simultaneously transfected using Fugene6 (Roche) with the indicated vector and/or luciferase vector and Renella control vector at a ratio of 10:1 (Luciferase:Renella). 24 h after transfection, cells were treated for the indicated times. Assays were conducted using the dual luciferase assay system (Promega). All luciferase values were normalized to the internal Renella control.
Statistical Analysis
Statistical analysis used was one-way ANOVA with post-hoc Dunnett’s multiple comparison tests.
Results
GSK3 promotes ER stress-induced apoptosis in SH-SY5Y cells
To examine the effects of GSK3 inhibitors on activation of the UPR, we first tested the effects of the selective GSK3 inhibitor lithium [32] on apoptotic signaling activated by tunicamycin-induced ER stress in human SH-SY5Y neuroblastoma cells. Inhibition of GSK3 with lithium reduced tunicamycin (4 µg/ml)-induced caspase-3 activation and reduced cleavage of the caspase-3 substrate PARP (Fig. 1). These results confirm previous reports that apoptosis induced by ER stress is promoted by GSK3 and attenuated by inhibitors of GSK3 [19–27].
Fig. 1. GSK3 inhibition reduces ER stress-induced caspase-3 activation in SH-SY5Y cells.
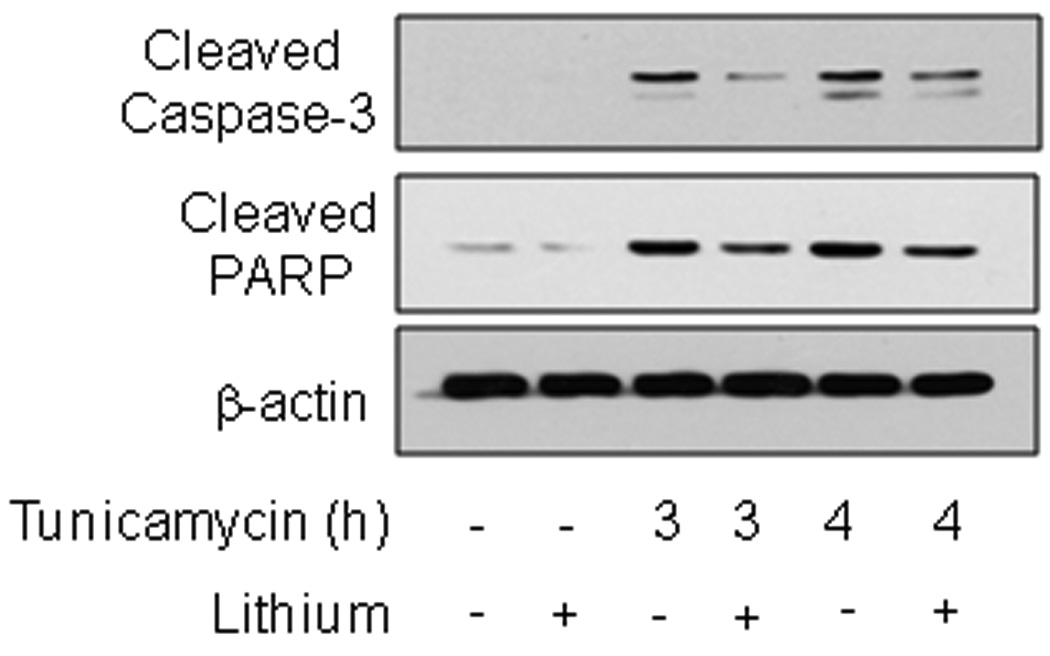
SH-SY5Y cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 3 or 4 h. Cell lysates were immunoblotted for active cleaved caspase-3, cleaved PARP, and β-actin.
Inhibition of GSK3 does not impair ER stress-induced activation of ATF6 or ATF4
Induction of ER stress and the UPR activate ATF6 and ATF4 to up-regulate expression of CHOP, which has been suggested to be critical for regulating the switch from survival to apoptosis [11,12]. Therefore, we tested if inhibition of GSK3 with lithium blocked the activation of either ATF4 or ATF6 after induction of ER stress. The activation of ATF6 was measured using a luciferase construct containing 5 tandem repeats of the ATF6 consensus DNA binding site [31] expressed in SH-SY5Y cells. Treatment with tunicamycin (4 µg/ml) caused a large increase in ATF6 activation and this was not significantly inhibited by lithium treatment (Fig. 2). This result indicated that GSK3 is not required for ATF6 activation to promote CHOP expression to initiate apoptotic signaling. Treatment of SH-SY5Y cells with tunicamycin (4 µg/ml) also increased expression of ATF4 mRNA, and this was not blocked, but instead additively enhanced, by inhibiting GSK3 with lithium (Fig. 3A). This finding indicated that GSK3 is not required for ER stress-induced ATF4 expression. The ATF4 pathway leading to CHOP up-regulation also can be activated by salubrinal, which blocks the eIF2α-linked protein phosphatase 1 (PP1))/GADD34 phosphatase complex [38], leading to increased phosphorylation of eIF2α, followed by activation of ATF4 to increase CHOP expression [8,9]. Therefore, to further test the influence of GSK3 inhibition on ATF4-induced CHOP expression without the influences of other UPR signaling pathways induced by ER stress, we tested the effects of inhibiting GSK3 with lithium on the response to salubrinal. As expected, treatment of SH-SY5Y cells with salubrinal increased phospho-ser51-eIF2α and after 14 h this was associated with a modest increase in CHOP expression (Fig. 3B). Treatment with lithium did not inhibit salubrinal-induced CHOP expression, but instead enhanced it, consistent with lithium-induced enhancement of ATF4 expression (Fig. 3A), confirming that the ATF4 pathway leading to CHOP was not blocked by inhibition of GSK3. However, upon measuring CHOP expression following induction of ER stress with tunicamycin, reduced CHOP was evident in cells co-treated with lithium and tunicamycin. These results indicated that lithium does not block ATF4-induced CHOP expression after salubrinal treatment, but may reduce CHOP expression following ER stress.
Fig. 2. GSK3 inhibition does not influence ATF6 expression induced by ER stress.
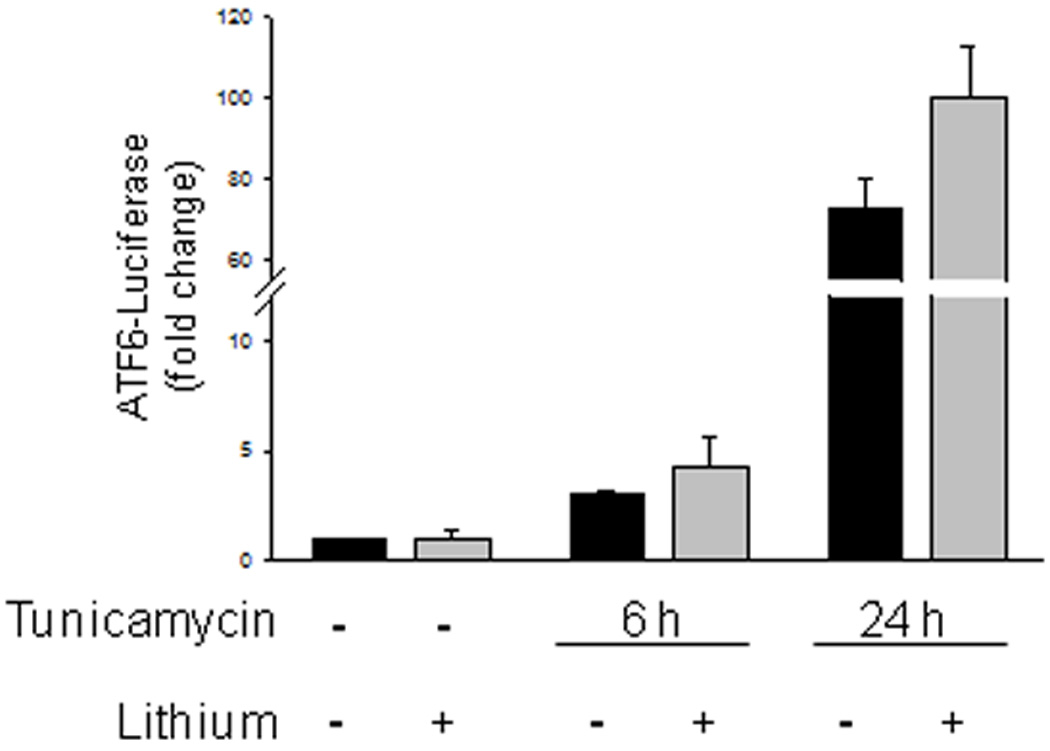
SH-SY5Y cells were transfected with a 5x-ATF6-luciferase construct, and 24 h after transfection cells were treated with 1 µg/ml tunicamycin for 6 or 24 h with or without a 1 h 20 mM lithium chloride pretreatment, and luciferase activity was measured and normalized to Renella expression. Values are means ± SEM, n=3.
Fig. 3. GSK3 inhibition does not impair ATF4 activation.
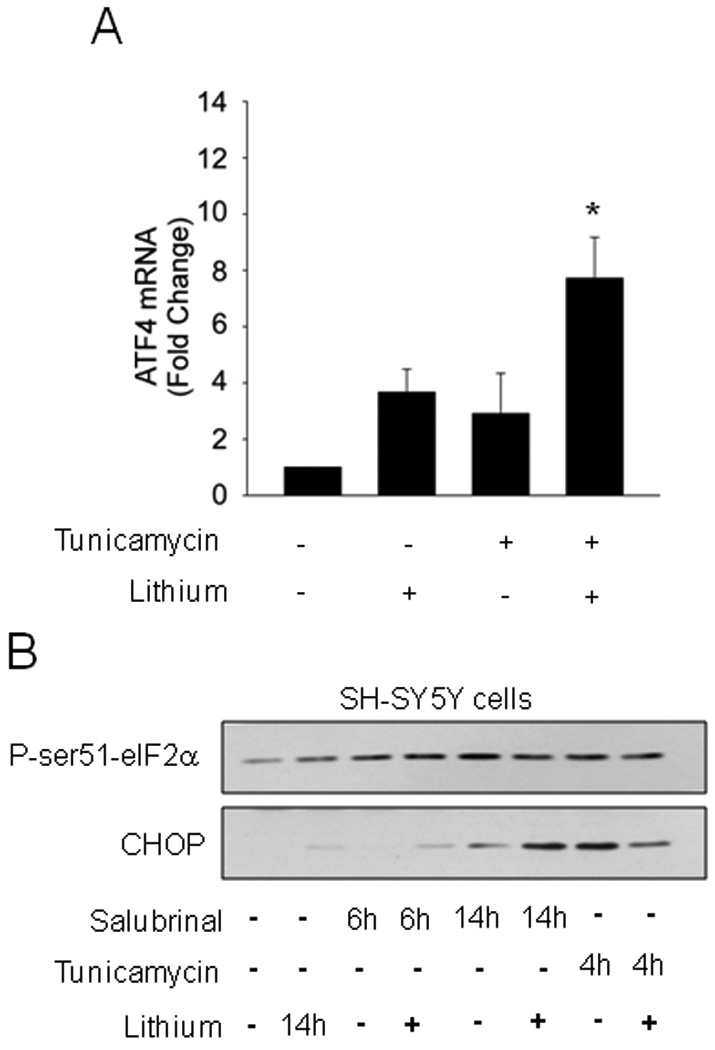
(A) SH-SY5Y cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 2.5 h. ATF4 mRNA was analyzed by qRT-PCR. Values are means ± SEM, n=3, *p< 0.05 compared with control. (B) SH-SY5Y cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 50 µM salubrinal for 6 or 14 h, or with 4 µg/ml tunicamycin for 4 h. Cell lysates were immunoblotted for phospho-ser51-eIF2α and CHOP.
GSK3 promotes ER stress-induced CHOP
CHOP has been reported to be a key determinant of cell fate, such that CHOP expression induced during ER stress promotes apoptosis [11,12]. In agreement with this conclusion, attenuation by lithium treatment of caspase-3 activation 3 and 4 h after tunicamycin treatment (Fig. 1) was associated with reduced CHOP levels (Fig. 4A). To verify that active GSK3 is required for increases in CHOP following ER stress, SH-SY5Y cells were pretreated with three other structurally distinct small molecule selective GSK3 inhibitors. These included the paullone derivative kenpaullone that has been reported to selectively inhibit GSK3 [33], the indirubin derivative selective GSK3 inhibitor 6-bromoindirubin-3'-oxime (BIO) [34], or CHIR99021, a substituted aminopyrimidine derivative reported to be the most selective inhibitor of GSK3 that has been described [35]. Pretreatment with each of the three structurally diverse GSK3 inhibitors reduced tunicamycin-induced increases in CHOP levels by greater than 75% (Fig. 4B). Along with the results with lithium treatment, these findings indicate that GSK3 promotes increases in CHOP following ER stress and this is attenuated by inhibitors of GSK3.
Fig. 4. GSK3 inhibition reduced tunicamycin-induced increased CHOP level.
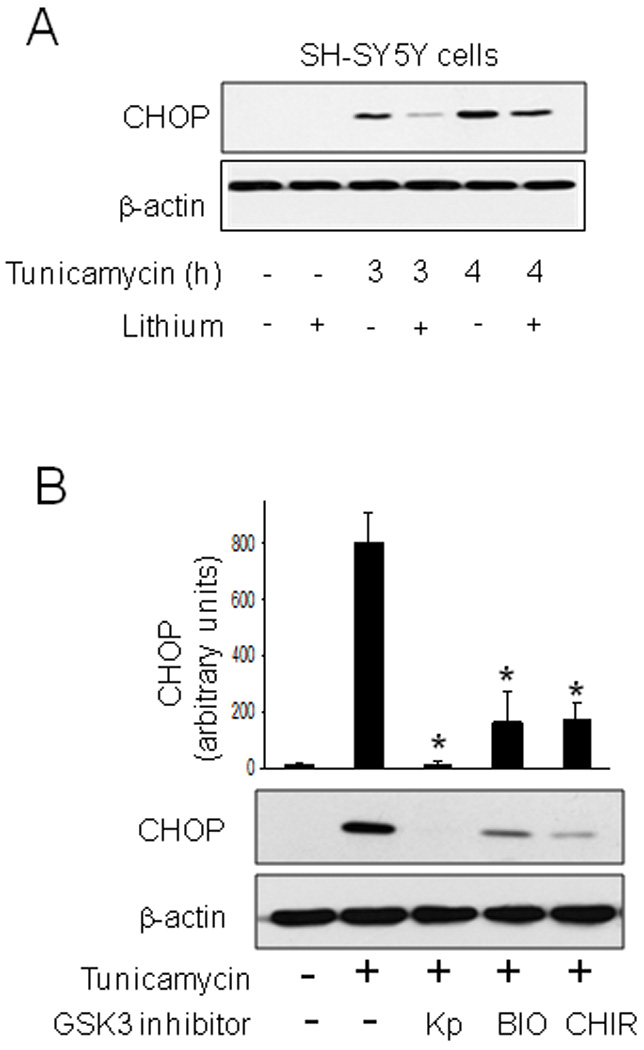
(A) SH-SY5Y cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 3 or 4 h. Cell lysates were immunoblotted for CHOP and β-actin. (B) SH-SY5Y cells were pretreated for 1 h with the GSK3 inhibitors 5 µM kenpaullone (Kp), 5 µM 6-bromoindirubin-3'-oxime (BIO), or 5 µM CHIR99021 (CHIR), followed by treatment with 4 µg/ml tunicamycin for 4 h and immunoblotting for CHOP and β-actin. Quantitative values are means ± SEM, n=3, *p< 0.05 compared with no GSK3 inhibitor.
CHOP mRNA levels were measured to test if reduced CHOP protein levels resulted from reduced CHOP expression by GSK3 inhibition. Pretreatment with lithium effectively reduced the tunicamycin-induced increase in the level of CHOP mRNA (Fig. 5A). qRT-PCR measurements confirmed that lithium treatment attenuated by more than 50% the tunicamycin-induced accumulation of CHOP mRNA (Fig. 5B). These results indicate that the regulation of CHOP protein levels by GSK3 results from its action on CHOP expression
Fig. 5. GSK3 inhibition reduced CHOP expression after tunicamycin treatment.
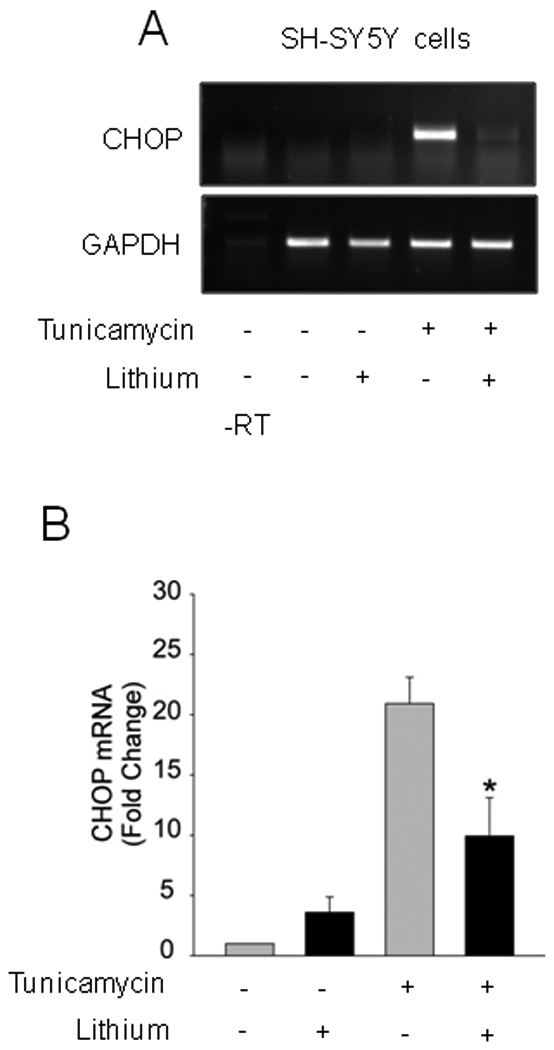
SH-SY5Y cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 2.5 h, and CHOP mRNA was analyzed by (A) RT-PCR or (B) qRT-PCR. Quantitative values are means ± SEM, n=3, *p< 0.05 compared with no lithium.
Comparison of ER stressors and cell types in GSK3 regulation of CHOP
We tested if CHOP expression was dependent on GSK3 in other neuronal-derived cells and after induction of ER stress with another agent, thapsigargin, which inhibits the ER Ca++-ATPase. In immortalized hippocampal neurons, tunicamycin treatment increased CHOP levels and this was diminished by pretreatment with the GSK3 inhibitors lithium and kenpaullone (Fig. 6A), demonstrating that this regulatory effect is not restricted to SH-SY5Y cells. Additionally, the reduction of ER stress-induced CHOP by GSK3 inhibitors was not unique to ER stress induced by tunicamycin, as lithium and BIO also attenuated CHOP expression induced by treatment with thapsigargin in immortalized hippocampal neurons (Fig. 6B). Furthermore, in RN46A cells, developed from the rat raphe nucleus [36], pretreatment with lithium also attenuated tunicamycin-induced CHOP expression (Fig. 7A).
Fig. 6. GSK3 inhibition reduced CHOP levels in hippocampal neurons treated with tunicamycin or thapsigargin.
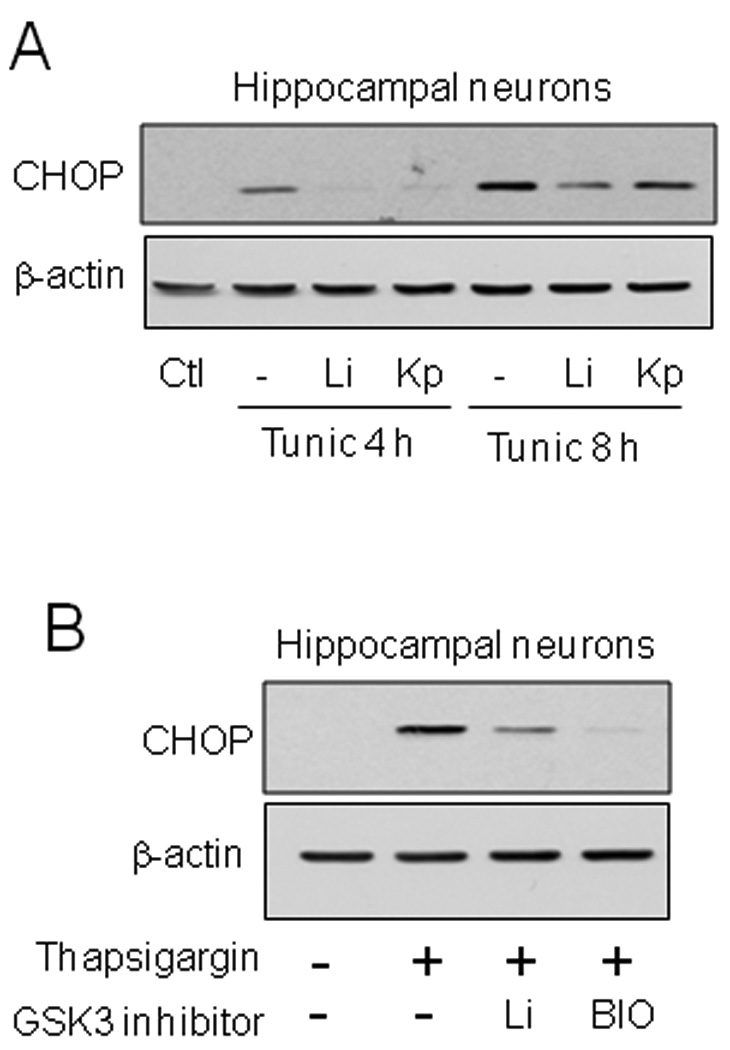
(A) Immortalized hippocampal neurons were pretreated with or without 20 mM lithium chloride (Li) or 5 µM kenpaullone (Kp) for 1 h followed by treatment with 4 µg/ml tunicamycin for 4 or 8 h. (B) Immortalized hippocampal neurons were pretreated with or without 20 mM lithium chloride or 5 µM 6-bromoindirubin-3'-oxime (BIO) for 1 h followed by treatment with 2 µM thapsigargin for 4 h. Cell lysates were immunoblotted for CHOP and β-actin.
Fig. 7. GSK3 inhibition reduced CHOP levels in neuronal lineage cells.
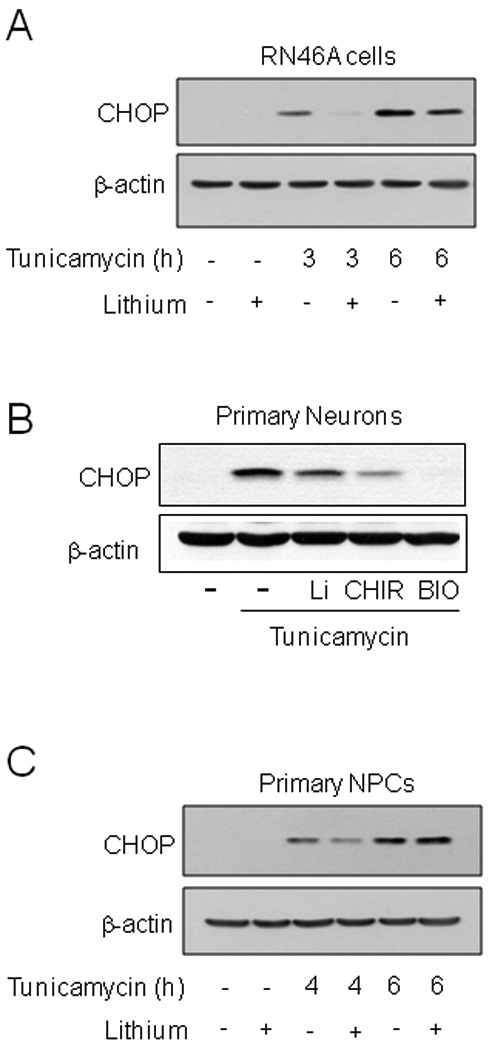
(A) RN46A cells were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 3 or 6 h. (B) Primary cortical neurons were treated with or without 20 mM lithium chloride (Li), 5 µM CHIR99021 (CHIR) or 5 µM BIO for 1 h followed by treatment with 4 µg/ml tunicamycin for 3 h. (C) Primary neural precursor cells (NPCs) were treated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 4 or 6 h. Cell lysates were immunoblotted for CHOP and β-actin.
To test the influence of GSK3 on CHOP expression in primary cells, mouse primary cortical neurons and mouse neural precursor cells were used. In primary cortical neurons the GSK3 inhibitors lithium, CHIR, and BIO strongly attenuated CHOP expression induced by tunicamycin treatment (Fig. 7B). However, treatment of neural precursor cells with lithium only modestly delayed tunicamycin-induced CHOP expression (Fig. 2E).
Considering that NPCs are multipotent and have not developed a neuronal phenotype [37], we tested if the influence of GSK3 on ER-stress induced CHOP expression may be restricted to cells of neuronal lineage. Consistent with this hypothesis, lithium did not attenuate tunicamycin-induced CHOP expression in either MEFs (Fig. 8A) or in mouse primary astrocytes (Fig. 8B). These results demonstrate for the first time that GSK3, in a neuronal selective manner, promotes upregulation of CHOP after ER stress, a mechanism that likely contributes to the facilitation by GSK3 of ER stress-induced apoptosis in neuronal-derived cells.
Fig. 8. GSK3 does not regulate CHOP expression in MEFs and primary astrocytes.
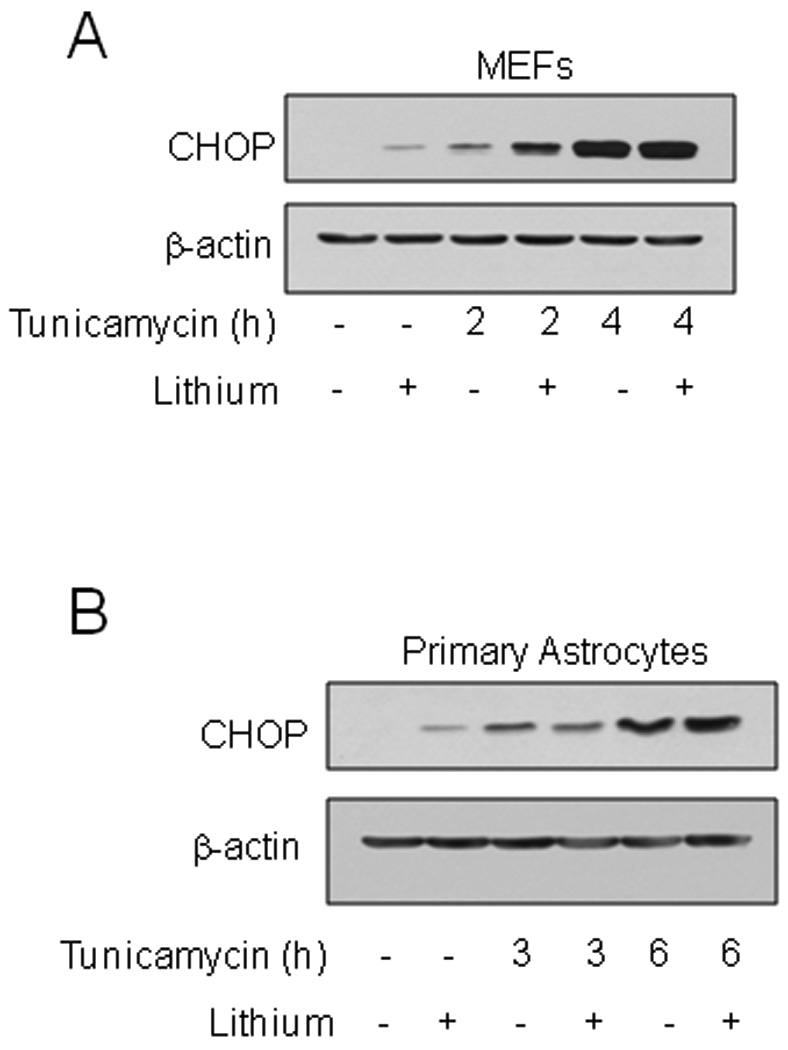
(A) MEFs were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 2 or 4 h. (B) Primary mouse astrocytes were pretreated with or without 20 mM lithium chloride for 1 h followed by treatment with 4 µg/ml tunicamycin for 3 or 6 h. Cell lysates were immunoblotted for CHOP and β-actin.
Discussion
ER stress is a prominent mechanism contributing to neuronal dysfunction, and sometimes death, in a number of prevalent neurodegenerative diseases [1–5]. Activation of the UPR follows the accumulation of misfolded proteins, one cause of ER stress that is associated with many neurodegenerative diseases. The UPR promotes recovery mechanisms, such as a general down-regulation of protein synthesis accompanied by increased production of chaperones. However, along with these recovery mechanisms, an apoptotic signaling pathway is initiated so cells can be eliminated if recovery is not achieved. The balance between recovery and apoptosis appears to be controlled by several regulators, one of which is activation of the death-inducing transcription factor CHOP, which is considered a crucial determinant in promoting apoptosis following UPR activation [12]. Thus, there is much interest in identifying determinants of the life-death switch, particularly focused on CHOP because of its key role, and interventions that promote recovery and survival. One mechanism to enhance cellular recovery and survival from ER stress is the application of inhibitors of GSK3 [19–27]. Upon examining the interactions between GSK3 and ER stress-induced signaling pathways, we found that the expression of CHOP was reduced in neurons in the presence of GSK3 inhibitors, indicating that this action contributes to the neuroprotective actions of GSK3 inhibitors in neurons exposed to ER stress.
Many investigators have established that GSK3 inhibitors provide protection from ER stress-induced apoptosis following exposure of a variety of cell types to several conditions that induce ER stress [19–27], but the underlying mechanisms by which GSK3 promotes apoptotic signaling induced by ER stress remain unclear. We first found that GSK3 inhibitors protected human neuroblastoma SH-SY5Y cells and immortalized mouse hippocampal neurons from ER stress induced by thapsigargin, tunicamycin, or brefeldin A [19]. GSK3 inhibitors also protected cerebellar granule neurons from brefeldin A-induced ER stress and apoptosis [20], SH-SY5Y cells, PC12 cells and cerebellar granule neurons from 6-hydroxydopamine-induced ER stress and apoptosis [21], PC12 cells from thapsigargin-induced ER stress and apoptosis [22], HepG2 cells from ER stress-induced apoptosis after exposure to tunicamycin, A23187, or glucosamine [23], mouse insulinoma cells from ER stress-induced apoptosis after exposure to tunicamycin or thapsigargin [24]. primary rat cortical neurons from ER stress and apoptosis after treatment with thapsigargin [25] or amyloid β-peptide [26], and leukemia cells from tunicamycin-induced ER stress and apoptosis [27]. Although it is clear that GSK3 promotes apoptotic signaling following ER stress, little is known about the influence of GSK3 on the UPR signaling pathway activated by ER stress and or about how inhibitors of GSK3 reduce apoptosis after ER stress. Mechanisms proposed to contribute to protection from ER stress-induced apoptosis by inhibitors of GSK3 include inhibition of Bax activation [20], up-regulation of the expression of GRP78 and bcl-2 and reduced intracellular calcium levels [22], and increased expression of chaperone proteins [23]. Here we found that ER stress-induced upregulation of the proapoptotic transcription factor CHOP is promoted by GSK3. Interestingly, this may be a neuron-selective interaction, as CHOP expression was independent of GSK3 in MEFs and astrocytes, but further studies are necessary to more fully identify the cell-type selectivity of this regulatory mechanism and to determine if the regulation of CHOP is differentially mediated by either GSK3α or GSK3β. Regulating CHOP may be a crucial action during ER stress because activation of CHOP has been established as a key mediator of the switch from recovery mechanisms towards apoptosis following activation of UPR signaling [11,12]. CHOP has been reported to promote apoptosis by inducing the expression of GADD34, an eIF2α phosphatase, to restore protein synthesis [11], increasing the oxidation of proteins [11], and suppressing expression of the anti-apoptotic protein Bcl-2 [38], and CHOP deficiency protects cells from ER stress-induced apoptosis [39]. CHOP is a short-lived protein, so when cells are able to dissipate misfolded proteins the levels of CHOP decrease rapidly to terminate the apoptotic pathway. Since GSK3 inhibitors reduced CHOP activation, this likely raises the threshold for apoptosis after ER stress, perhaps by prolonging the time necessary for CHOP to increase to levels that activate the apoptotic program, thereby contributing to the well-established reduction in apoptosis that occurs in the presence of GSK3 inhibitors in cells experiencing ER stress [19–27]. Surprisingly, the mechanism by which GSK3 regulates CHOP expression did not involve regulation of the two most prominent transcription factors that can induce CHOP, ATF4 and ATF6. Promotion of CHOP expression by GSK3 cannot be the only mechanism by which GSK3 promotes apoptosis during ER stress because GSK3 inhibitors protect many cell types from ER stress-induced apoptosis, but they only reduced CHOP expression in cells of neuronal-lineage. Previous studies have shown that GSK3 targets other proteins involved in apoptotic signaling downstream of CHOP expression (reviewed in [40]). This includes evidence that GSK3 regulates the anti-apoptotic protein Mcl-1 and pro-apoptotic proteins Bim and Bax [41–43], and also phosphorylates the voltage-dependent anion channel, an abundant outer mitochondrial membrane protein implicated in maintaining the mitochondrial membrane potential [44]. Thus, the finding that CHOP is regulated by GSK3 appears to be one of several mechanisms by which GSK3 can promote apoptosis following ER stress and may be a mechanism selective to cells of neuronal-lineage.
In summary, inhibition of GSK3 reduces ER stress-induced CHOP expression and apoptotic signaling. By regulating ER stress-induced CHOP expression, GSK3 provides a fulcrum to balance UPR-associated survival pathways with apoptotic signaling, which is enhanced when GSK3 is activated. These actions of GSK3 regulating CHOP strengthen previous conclusions that GSK3 is an important determinant of the life-death switch mediated by ER stress and the UPR.
Acknowledgements
This research was supported by grants from the NIH (MH092970, MH038752).We thank Dr. Ron Prywes for the 5x-ATF6-luciferase construct.
Abbreviations
- ATF4
activating transcription factor 4
- BIO
6-bromoindirubin-3'-oxime
- CHOP
C/EBP homologous protein
- ER
endoplasmic reticulum
- eIF2α
eukaryotic initiation factor 2α
- GRP78
glucose-regulated protein 78
- GSK3
glycogen synthase kinase-3
- MEFs
mouse embryonic fibroblasts
- NPCs
neural precursor cells
- PARP
poly(ADP-ribose) polymerase
- PERK
PKR-like ER kinase
- PP1
protein phosphatase 1
- UPR
unfolded protein response
- XBP1
X box-binding protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors have no financial conflict of interest.
REFERENCES
- 1.Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–383. doi: 10.1016/s0143-4160(03)00139-8. [DOI] [PubMed] [Google Scholar]
- 2.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev. Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshida H. ER stress and disease. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- 4.Schröder M, Kaufman RJ. The mammalian unfolded protein response. Ann. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 5.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 7.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 8.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 9.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem. Soc. Trans. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 10.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 11.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell Biol. 2000;20:6755–6767. doi: 10.1128/mcb.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 15.Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 16.Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 17.Woodgett JR. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990;9:2431–2438. doi: 10.1002/j.1460-2075.1990.tb07419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ciani L, Salinas PC. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci. 2005;6:351–362. doi: 10.1038/nrn1665. [DOI] [PubMed] [Google Scholar]
- 19.Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase-3β in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002;277:44701–44708. doi: 10.1074/jbc.M206047200. [DOI] [PubMed] [Google Scholar]
- 20.Brewster JL, Linseman DA, Bouchard RJ, Loucks FA, Precht TA, Esch EA, Heidenreich KA. Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase-3β and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol. Cell Neurosci. 2006;32:242–253. doi: 10.1016/j.mcn.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Chen G, Bower KA, Ma C, Fang S, Thiele CJ, Luo J. Glycogen synthase kinase 3β (GSK3β) mediates 6-hydroxydopamine-induced neuronal death. FASEB J. 2004;18:1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- 22.Hiroi T, Wei H, Hough C, Leeds P, Chuang DM. Protracted lithium treatment protects against the ER stress elicited by thapsigargin in rat PC12 cells: roles of intracellular calcium. GRP78 and Bcl-2, Phamacogenomics J. 2005;5:102–111. doi: 10.1038/sj.tpj.6500296. [DOI] [PubMed] [Google Scholar]
- 23.Kim AJ, Shi Y, Austin RC, Werstuck GH. Valproate protects cells from ER stress-induced lipid accumulation and apoptosis by inhibiting glycogen synthase kinase-3. J. Cell Sci. 2005;118:89–99. doi: 10.1242/jcs.01562. [DOI] [PubMed] [Google Scholar]
- 24.Srinivasan S, Ohsugi M, Liu Z, Fatrai S, Bernal-Mizrachi E, Permutt MA. Endoplasmic reticulum stress-induced apoptosis is partly mediated by reduced insulin signaling through phosphatidylinositol 3-kinase/Akt and increased glycogen synthase kinase-3β in mouse insulinoma cells. Diabetes. 2005;54:968–975. doi: 10.2337/diabetes.54.4.968. [DOI] [PubMed] [Google Scholar]
- 25.Takadera T, Fujibayashi M, Kaniyu H, Sakota N, Ohyashiki T. Caspase-dependent apoptosis induced by thapsigargin was prevented by glycogen synthase kinase-3 inhibitors in cultured rat cortical neurons. Neurochem. Res. 2007;32:1336–1342. doi: 10.1007/s11064-007-9310-4. [DOI] [PubMed] [Google Scholar]
- 26.Resende R, Ferreiro E, Pereira C, Oliveira CR. ER stress is involved in Aβ-induced GSK-3β activation and tau phosphorylation. J. Neurosci. Res. 2008;86:2091–2099. doi: 10.1002/jnr.21648. [DOI] [PubMed] [Google Scholar]
- 27.Huang WC, Lin YS, Chen CL, Wang CY, Chiu WH, Lin CF. Glycogen synthase kinase-3β mediates endoplasmic reticulum stress-induced lysosomal apoptosis in leukemia. J. Pharmacol. Exp. Ther. 2009;329:524–531. doi: 10.1124/jpet.108.148122. [DOI] [PubMed] [Google Scholar]
- 28.Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J. Biol. Chem. 2008;283:21934–21944. doi: 10.1074/jbc.M802481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eom TY, Roth KA, Jope RS. Neural precursor cells are protected from apoptosis induced by trophic factor withdrawal or genotoxic stress by inhibitors of glycogen synthase kinase 3. J. Biol. Chem. 2007;282:22856–22864. doi: 10.1074/jbc.M702973200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beurel E, Jope RS. Lipopolysaccharide-induced interleukin-6 production is controlled by glycogen synthase kinase-3 and STAT3 in the brain. J. Neuroinflammation. 2009;6:9–20. doi: 10.1186/1742-2094-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- 32.Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leost M, Schultz C, Link A, Wu YZ, Biernat J, Mandelkow EM, Bibb JA, Snyder GL, Greengard P, Zaharevitz DW, Gussio R, Senderowicz AM, Sausville EA, Kunick C, Meijer L. Paullones are potent inhibitors of glycogen synthase kinase-3β and cyclin-dependent kinase 5/p25. Eur. J. Biochem. 2000;267:5983–5994. doi: 10.1046/j.1432-1327.2000.01673.x. [DOI] [PubMed] [Google Scholar]
- 34.Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 35.Wagman AS, Johnson KW, Bussiere DE. Discovery and development of GSK3 inhibitors for the treatment of type 2 diabetes. Curr. Pharm. Des. 2004;10:1105–1137. doi: 10.2174/1381612043452668. [DOI] [PubMed] [Google Scholar]
- 36.White LA, Eaton MJ, Castro MC, Klose KJ, Globus MY, Shaw G, Whittemore SR. Distinct regulatory pathways control neurofilament expression and neurotransmitter synthesis in immortalized serotonergic neurons. J. Neurosci. 1994;14:6744–6753. doi: 10.1523/JNEUROSCI.14-11-06744.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- 38.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog. Neurobiol. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. Prog. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Somervaille TC, Linch DC, Khwaja A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- 42.Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Linseman DA, Butts BD, Precht TA, Phelps RA, Le SS, Laessig TA, Bouchard RJ, Florez-McClure ML, Heidenreich KA. Glycogen synthase kinase-3β phosphorylates Bax and promotes its mitochondrial localization during neuronal apoptosis. J Neurosci. 2004;24:9993–10002. doi: 10.1523/JNEUROSCI.2057-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 2005;65:10545–10554. doi: 10.1158/0008-5472.CAN-05-1925. [DOI] [PubMed] [Google Scholar]