

Abstract
Glioblastoma is the most malignant and prevalent brain tumor in humans. It is composed of heterogenic abnormal astroglial cells that avoid differentiation, maintain proliferation, and reluctant to apoptosis. N-(4-Hydroxyphenyl) retinamide (4-HPR) induced astrocytic differentiation and increased sensitivity to interferon-gamma (IFN-γ) for apoptosis in human glioblastoma A172, LN18, and SNB19 cells. Combination of 4-HPR and IFN-γ significantly inhibited human telomerase reverse transcriptase (hTERT), cyclin dependent kinase 2 (CDK2), and survivin to upregulate caspase-8, caspase-9, and caspase-3 for increasing apoptosis in all glioblastoma cell lines. Hence, combination of 4-HPR and IFN-γ should be considered for controlling growth of different human glioblastoma cells.
Keywords: Glioblastoma, 4-HPR, Differentiation, IFN-γ, Apoptosis
1. Introduction
Glioblastoma, which is originated from abnormal astroglial cells, is the most common primary brain tumor in humans. Although inadequate differentiation and aggressive proliferation make glioblastoma the most malignant brain tumor, it does harbor the molecular machinery for apoptosis [1]. Glioblastoma has extremely poor prognosis that distinguishes it from low grade astrocytomas [2]. Current treatment strategy for glioblastoma includes palliative therapy followed by surgery, radiotherapy, and chemotherapy. However, single chemotherapeutic agents for treating glioblastoma have been mostly ineffective necessitating the development of novel therapeutic strategies for effective management of glioblastoma [3]. One approach might be the synergistic combination chemotherapy to use a retinoid for inducing astrocytic differentiation and priming the glioblastoma cells to cytokine mediated apoptosis.
The intrinsic properties of glioblastoma make it avoid immunosurveillance and proliferate extensively with strikingly high rates of genomic instability [4]. This coupled with dysregulation of cell cycle results in the formation of multinucleate, polyploid tumor cells. Retinoids are known to influence several biological processes such as growth, development, and differentiation [5, 6]. The retinoid treatment is usually associated with induction of morphological changes and prolongation of doubling time [7]. This suppression of growth by retinoid treatment does not necessarily involve cell death but arrest the tumor cells in the G1 phase of the cell cycle [8].
N-(4-Hydroxyphenyl) retinamide, (4-HPR, also known as fenretinide), is a synthetic retinoid that has been effective in suppressing the growth of breast, ovarian, and small-cell-lung carcinomas [9–11]. Treatment with 4-HPR exerts restrictions on the cell cycle regulating cyclin dependent kinase (CDK) pathway [12] and phosphorylation of retinoblastoma protein (pRb), whose dephosphorylation results in cell cycle arrest. Inhibition of cell cycle check points would prolong the duration of cell cycle arrest, contributing to cytokine mediated activation of intrinsic apoptotic pathway.
Interferon-gamma (IFN-γ) is a homodimeric glycoprotein known to be involved in the immunosurveillance of solid tumors via a combination of lymphocyte mediated immune responses [13]. The direct mode of action of IFN-γ involves significant reduction of DNA synthesis along with induction of apoptosis [14]. The ability of IFN-γ to initiate the intrinsic and extrinsic apoptotic cascades through induction of pro-apoptotic genes along with regulation of members of the Bcl-2 family makes it a suitable candidate for combination with 4-HPR for treatment of glioblastoma.
It is now widely accepted that apoptotic signals are regulated by the members of the Bcl-2 and inhibitor-of-apoptosis protein (IAP) families. The pro-apoptotic and anti-apoptotic members of the Bcl-2 family homodimerize or heterodimerize at the mitochondrial membrane to tilt the balance between cell death and cell survival [15]. The members of the IAP family counteract the apoptotic stimuli by acting as endogenous inhibitors of caspases at a step down stream of the Bcl-2 family regulation [16]; however, an emerging line of evidence suggests that some of the IAP members such as survivin may have a putative role in regulation of cell cycle upstream to the Bcl-2 family [17] and down regulation of survivin in response to apoptotic stimuli may activate mitochondrial pathway of apoptosis [18, 19].
There is an urgent need to develop combination therapeutic regimens, which can prime the glioblastoma cells to down regulate the anti-apoptotic actions of the members of the Bcl-2 and IAP families and act synergistically for enhancement of apoptosis. This formed the rationale for our present study, where we sought to examine the synergistic action of 4-HPR and IFN-γ for induction of differentiation and activation of the molecular mechanisms for apoptosis in multiple glioblastoma cell lines of heterogeneous origin.
2. Materials and Methods
2.1. Cell cultures and treatments
Human glioblastoma A172 and LN18 cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in 1x Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (GIBCO/BRL, Grand Island, NY, USA). Another human glioblastoma SNB19 cell line was obtained from the National Cancer Institute (NCI, Bethesda, MD, USA) and maintained in DMEM/F12 medium (1:1 v/v) supplemented with 10% FBS and 1% penicillin and streptomycin (GIBCO/BRL). All cell lines were maintained in a humidified atmosphere containing 5% CO2 at 37°C. 4-HPR (Sigma Chemical, St. Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO) to make a stock solution and aliquots of stock solution were stored in the dark at 70°C. To avoid light sensitivity of 4-HPR, all treatments involving it were performed under subdued lighting. All experiments included control cultures, which contained the same volume of DMSO that was used in the 4-HPR treatment for a total of 5 days. The concentration of DMSO in each experiment was always less than or equal to 0.01%, which did not induce differentiation and cell death. Cells (not treated or treated with 0.5 μM 4-HPR for 3 days) were treated with 200 units/ml IFN-γ (Calbiochem, San Diego, CA, USA) for 48 h for induction of apoptosis. Following treatments, cells were used to examine morphological features of astrocytic differentiation, apoptosis, and expression of specific proteins.
2.2. Methylene blue staining to examine induction of astrocytic differentiation
Dose-response studies were carried out to find out the optimum concentration (0.5 μM) of 4-HPR for inducing astrocytic differentiation in all the glioblastoma cell lines employed in the present study. Cells were cultured in monolayer in 9-cm diameter plates in the absence and presence of 0.5 μM 4-HPR for 5 days. At the end of the treatment, the culture medium was aspirated and cells were washed twice with ice-cold phosphate-buffered saline (PBS), pH 7.4, in the culture plate. For in situ methylene blue staining of the cells, each plate was placed on ice and 5 ml of ice-cold 95% (v/v) ethanol was added to fix the cells for 5 min. After aspirating ethanol, 5 ml of ice-cold 0.2% (v/v) methylene blue solution (prepared in 50% ethanol) was added to each of the plates. Cells were stained with methylene blue for 30 sec and washed twice with ice-cold deionized distilled water. The plates were dried in air before being examined under the light microscope at 400× magnification.
2.3. Wright staining for detection of morphological features of apoptosis
After all treatments, both adherent and non-adherent cells were collected and washed twice with PBS, pH 7.4 before being fixed in 95% ethanol. The cells were allowed to dry before being stained. Finally the morphological features of the Wright-stained cells were observed under the light microscope. The morphological features of apoptotic cells included at least one of such characteristics as cell shrinkage, chromatin condensation, and membrane-bound apoptotic bodies. At least 600 cells were counted from each treatment and percentage of apoptosis was calculated.
2.4. Extraction of protein and Western blotting
We performed these procedures as described previously [20, 21]. Briefly, protein samples were extracted from glioblastoma A172, LN18, and SNB 19 cells, resolved by the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed by Western blotting using one of the primary IgG antibodies for β-actin, Bax, Bcl-2, Bid, caspase-3, caspase-8, caspase-9, cyclin dependent kinase 2 (CDK2), glial fibrillary acidic protein (GFAP), inhibitor of caspase-3-activated DNase (ICAD), phosphorylated (Ser 780)-retInoblastoma (pRb), survivin, and human telomerase reverse transcriptase (hTERT). With the exception of antibodies for β-actin (clone 15, Sigma Chemical), caspase-9 (BD Biosciences, San Jose, CA, USA), and GFAP (Chemicon International, Temecula, CA, USA), all other antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish peroxidase-conjugated goat anti-mouse or anti-rabbit secondary IgG antibody (ICN Biomedicals, Aurora, OH, USA) was used for detecting the primary antibody. Then, Western blots were incubated with the enhanced chemiluminescence (ECL) reagents (Amersham Pharmacia, Buckinghamshire, UK) and exposed to X-OMAT AR films (Eastman Kodak, Rochester, NY, USA). The ECL autoradiograms were scanned and optical density of each band was assessed using Quantity One software (Bio-Rad, Hercules, CA, USA).
2.5. Western blotting for determining mitochondrial release of cytochrome c into cytosol
We isolated mitochondrial and cytosolic fractions using the commercially available reagents (Pierce, Rockford, IL, USA) for determining any mitochondrial release of cytochrome c into cytosol after the treatments. The mitochondrial fractions were analyzed by Western blotting using primary IgG antibodies for cytochrome c oxidase 4 (COX4) (Molecular Probes, Eugene, OR, USA) and cytochrome c while the cytosolic fractions were analyzed using primary antibodies for β-actin and cytochrome c.
2.6. Statistical analysis
Results were analyzed using StatView software (Abacus Concepts, Berkeley, CA, USA) and compared using one-way analysis of variance (ANOVA) with Fisher’s post hoc test. Data were presented as mean ± standard error of mean (SEM) of separate experiments (n ≥ 3). Significant difference from control value was indicated by *P < 0.05 or **P < 0.005.
3. Results
3.1. Treatment with 4-HPR induced astrocytic differentiation
The induction of astrocytic differentiation in glioblastoma cells can be characterized by morphological and biochemical features. In situ methylene blue staining of cells after treatment with 4-HPR revealed morphological changes such as small and retracted cell bodies with thin elongated and branched processes indicating astrocytic differentiation while the control cells maintained wide cell body with short processes (Fig. 1). We performed Western blotting to examine the changes in biochemical features of astrocytic differentiation after the treatments (Fig. 2). Levels of β-actin were monitored to ensure equal loading of protein samples. Treatment with 4-HPR significantly increased (P<0.05) the levels of GFAP, a biochemical marker of astrocytic differentiation, in all three cell lines (Fig. 2A). Treatment with the combination of 4-HPR and IFN-γ significanty decreased hTERT (Fig. 2b), indicating induction of cell differentiation and restriction of cell division leading to cell death.
Fig. 1.
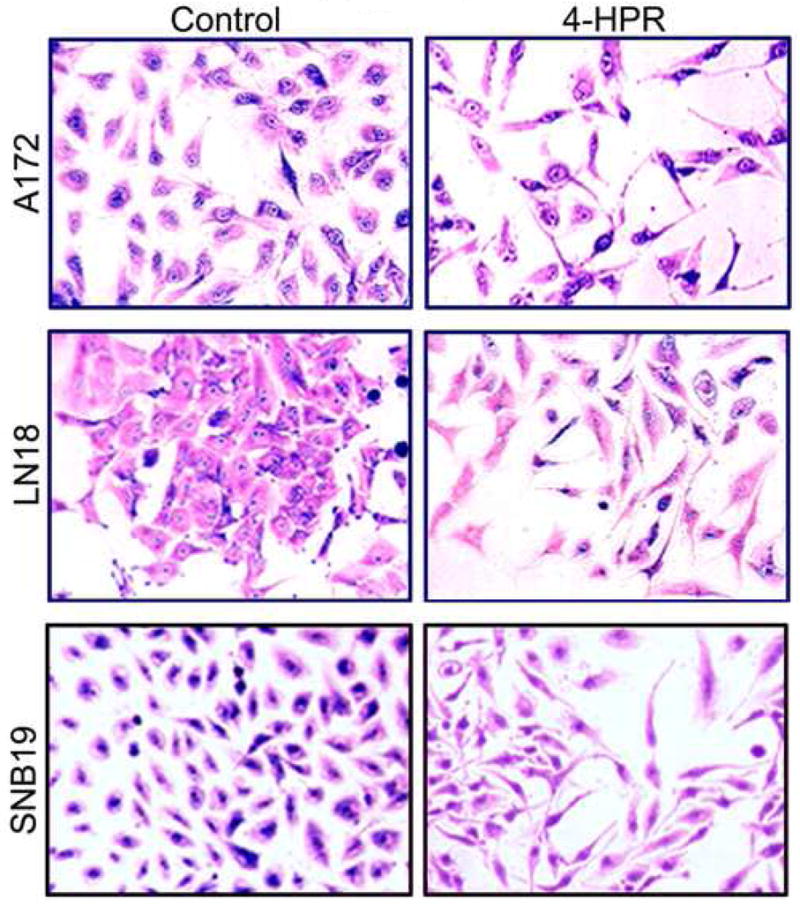
Morphological features of astrocytic differentiation in human glioblastoma A172, LN18, and SNB19 cells. Treatment of cells treated with 0.5 μM 4-HPR for 5 days induced morphological features such as small and retracted cell bodies with thin, elongated, and branched processes, indicating the prominence of differentiation process in these cells.
Fig. 2.
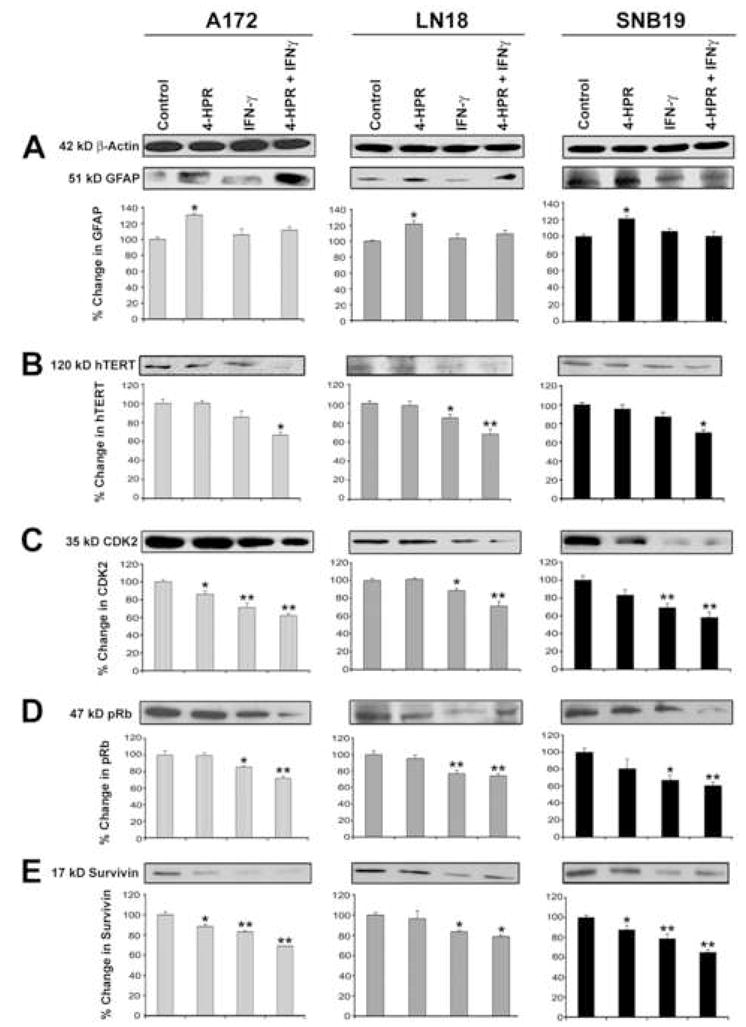
Western blotting for determination of changes in cytosolic levels of markers of astrocytic differentiation and cell cycle progression in glioblastoma A172, LN18, and SNB19 cells. Treatments: control, 0.5 μM 4-HPR (5 days), 200 units/ml IFN-γ (2 dads), and 0.5 μM 4-HPR (3 days) + 200 units/ml IFN-γ (2 days). Glioblastoma cells were grown to 70% confluency before being treated with 0.5 μM 4-HPR alone for 3 days and then in combination with 200 units/ml IFN-γ for 2 days. Representative Western blots (n ≥ 3) and bar graphs show levels of: (A) 51 kD GFAP, (B) 120 kD hTERT, (C) 35 kD CDK2, (D) 47 kD pRB, and (E) 17 kD survivin.
3.2. Combination of 4-HPR and IFN-γ down regulated markers of cell cycle progression
Next, we examined whether induction of astrocytic differentiation could be correlated with changes in levels of signaling molecules known to govern the G1 phase of cell cycle. Oncogenic/mitogenic signals stimulate G1 phase cyclins in presence of cyclin dependant kinases to phosphorylate Rb, a transcriptional repressor and critical component of the cell cycle machinery. We observed in all three cell lines that treatment with combination of 4-HPR and IFN-γ significantly decreased (P<0.005) the levels of CDK2 (Fig. 2C) with concomitant down regulation of pRb (Fig. 2D). Also, an interesting observation was the down regulation of survivin (Fig. 2E) in response to treatments with 4-HPR and IFN-γ alone and in combination, indicating the synergistic action of these agents. Results showed that treatments induced astrocytic differentiation, which in turn accentuated the process of cell cycle arrest in all three glioblastoma cell lines.
3.3. Morphological features of apoptotic cell death
The extent of apoptotic cell death was determined based on morphological features as revealed by in situ Wright staining (Fig. 3). The characteristic features of apoptotic cells such as shrinkage of cell volume, chromatin condensation, and membrane-bound apoptotic bodies were examined after the treatments of glioblastoma A172 (Fig. 3A), LN18 (Fig. 3B), and SNB19 (Fig. 3C) cells. Compared with control cells, treatment with 4-HPR caused non-significant change in percentage of apoptosis, IFN-γ alone showed significant increase (P<0.05) in apoptosis, and combination of 4-HPR and IFN-γ demonstrated the most significant increase (P<0.005) in apoptosis (Fig. 3).
Fig. 3.
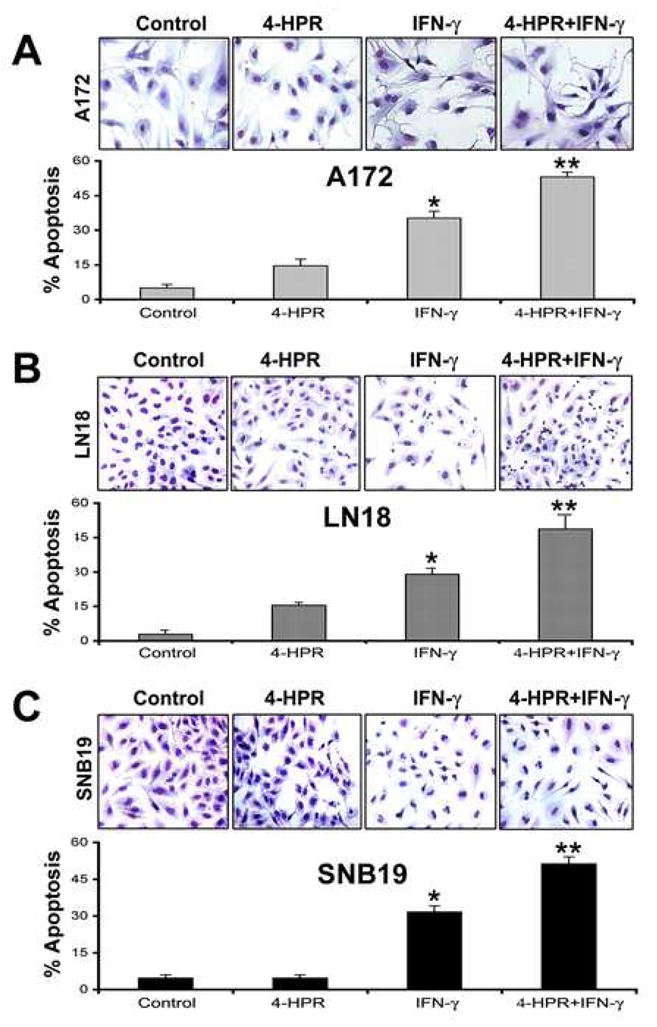
In situ Wright staining for morphological features of apoptosis in glioblastoma A172, LN18, and SNB19 cells. Treatments: control, 0.5 μM 4-HPR (5 days), 200 units/ml IFN-γ (2 dads), and 0.5 μM 4-HPR (3 days) + 200 units/ml IFN-γ (2 days). Based on the morphological features such as cell volume shrinkage along with chromatin condensation, and membrane-bound apoptotic bodies, changes in percent apoptosis were determined in glioblastoma (A) A172, (B) LN18, and (C) SNB19 cells.
3.4. Activation of caspase-8 and proteolytic cleavage of Bid
We performed Western blotting to examine the activation of extrinsic caspase cascade after the treatments (Fig. 4). β-Actin expression was monitored to ensure that equal amount of protein was loaded in each lane (Fig. 4A). Treatment with combination of 4-HPR and IFN-γ significantly increased active caspase-8 fragment in all glioblastoma cell lines (Fig. 4A). Activation of caspase-8 could cleave its substrate Bid to generate 15 kD truncated Bid (tBid), which could be translocated to mitochondria to induce the release of cytochrome c thereby activating mitochondrial pathway of apoptosis [22]. Moreover, activation of caspase cascade might be correlated with migration of cytosolic Bax to the mitochondria where it could assist tBid in releasing cytochrome c into the cytosol [23]. We found that treatment with 4-HPR and IFN-γ caused significant increases in proteolytic cleavage of Bid to tBid in all glioblastoma cell lines (Fig. 4B).
Fig. 4.
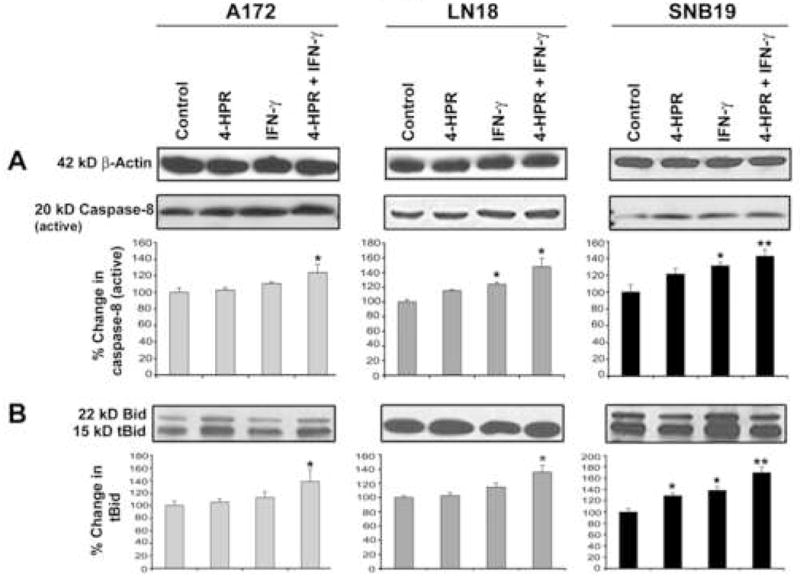
Western blotting for examination of activation of extrinsic pathway of apoptosis in glioblastoma A172, LN18, and SNB19 cells. Treatments: control, 0.5 μM 4-HPR (5 days), 200 units/ml IFN-γ (2 dads), and 0.5 μM 4-HPR (3 days) + 200 units/ml IFN-γ (2 days). Representative Western blots (n ≥ 3) and bar graphs show changes in: (A) 20 kD active caspase-8 and (B) 15 tBid.
3.5. Increase in Bax: Bcl-2 ratio and mitochondrial release of cytochrome c into cytosol
After the treatments, we examined increase in Bax:Bcl-2 to determine the commitment of cells to trigger mitochondrial release of cytochrome c into cytosol (Fig. 5). β-Actin expression was monitored to ensure that equal amounts of cytosolic protein samples were loaded in all lanes (Fig. 5A). We used a monoclonal antibody that could recognize both 21 kD Baxα and 24 kD Baxβ bands (Fig. 5A). We considered both bands in our estimation of total Bax expression. We used another monoclonal antibody to monitor the expression of 26 kD Bcl-2 protein (Fig. 5A). Treatments with IFN-γ alone and also combination of 4-HPR and IFN-γ changed the levels of expression of pro-apoptotic Bax (total) and anti-apoptotic Bcl-2 proteins resulting in significant increases in Bax:Bcl-2 ratio in all glioblastoma cell lines (Fig. 5A). Because increase in Bax:Bcl-2 ratio indicated mitochondrial release of cytochrome c into the cytosol, we extracted proteins from both cytosolic and mitochondrial fractions and examined alterations in the levels of cytochrome c by Western blotting. We monitored expression of COX4 as an internal control for the mitochondrial fractions in all cell lines (Fig. 5B). Treatment of cells with 4-HPR and IFN-γ promoted significantly the disappearance of 15 kD cytochrome c from the mitochondrial fraction (Fig. 5B) and its concomitant apprearance in the cytosolic fraction (Fig. 5C). Thus, the mechanism of cell death involved the mitochondrial release of cytochrome c, which could cause subsequential activation of caspase-9 and caspase-3.
Fig. 5.
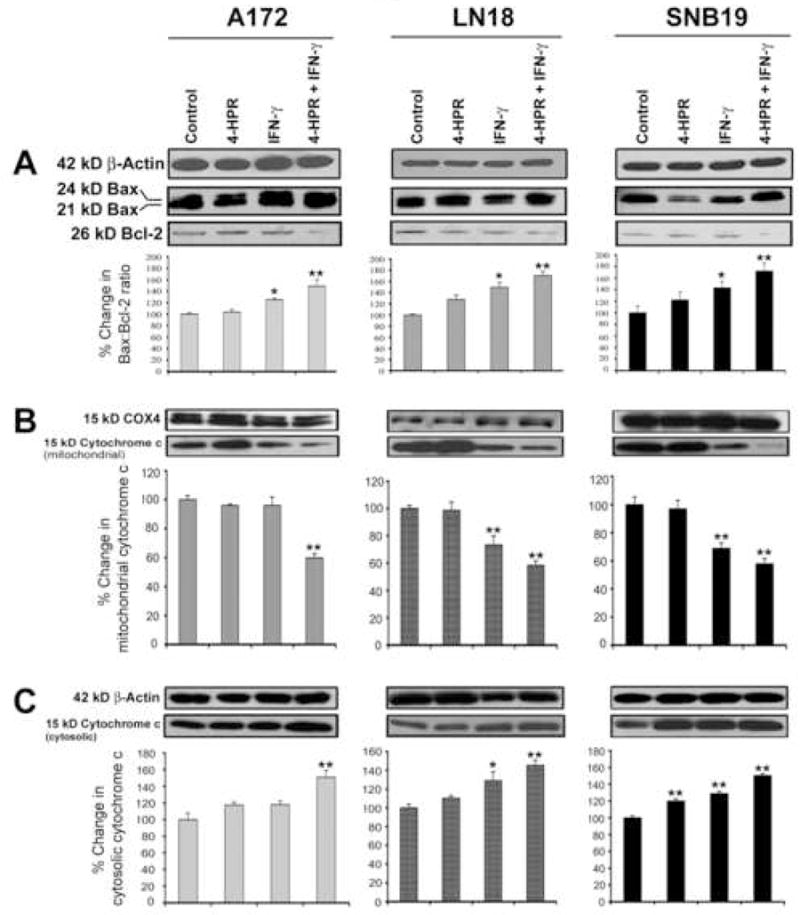
Western blotting for determination of increase in Bax:Bcl-2 ratio leading to mitochondrial release of cytochrome c into the cytosol in glioblastoma A172, LN18, and SNB19 cells. Treatments: control, 0.5 μM 4-HPR (5 days), 200 units/ml IFN-γ (2 dads), and 0.5 μM 4-HPR (3 days) + 200 units/ml IFN-γ (2 days). Representative Western blots (n ≥ 3) and bar graphs show changes in: (A) Bax:Bcl-2 ratio, (B) 15 KD cytochrome c in mitochondria, and (C) 15 kD cytochrome c in cytosol.
3.6. Activation of caspase-9 and caspase-3 leading to cleavage of ICAD
In mitochondria-dependent pathway of apoptosis, both cytosolic protein Apaf1 and cytochrome c participate in the activation of caspase-9 that in turn processes pro-caspase-3 to generate active caspases-3 [24, 25]. Out Western blotting showed the activation of mitochondria-dependent caspase cascade (Fig. 6). Treatment of cells with IFN-γ alone and combination of 4-HPR and IFN-γ significant increased the amounts of active caspase-9 (Fig. 6A) and active caspase-3 (Fig. 6B) fragments in all cell lines. Increase in aspase-3 activity caused significant increase in 40 kD ICAD fragment in all the cell lines in response to treatment with combination of 4-HPR and IFN-γ (Fig. 6C). Because of ICAD fragmentation, CAD could be set free to translocate to the nucleus for DNA fragmentation.
Fig. 6.
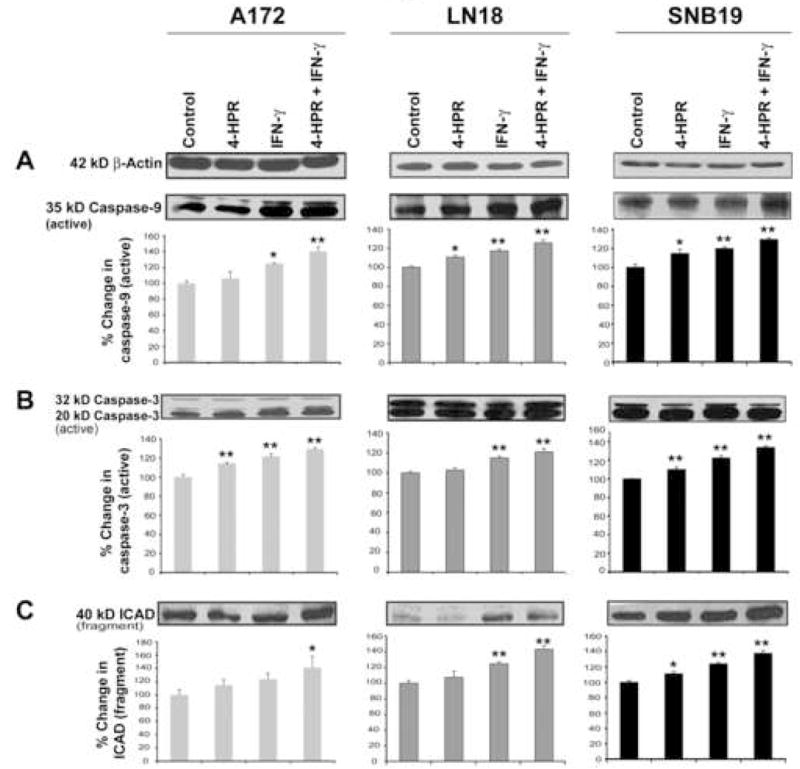
Western blotting for examination of activation of caspase-9 and caspase-3 and also formation of ICAD fragment in glioblastoma A172, LN18, and SNB19 cells. Treatments: control, 0.5 μM 4-HPR (5 days), 200 units/ml IFN-γ (2 dads), and 0.5 μM 4-HPR (3 days) + 200 units/ml IFN-γ (2 days). Representative Western blots (n ≥ 3) and bar graphs show changes in: (A) 35 kD active caspase-9, (B) 20 kD active caspase-3, and (C) 40 kD ICAD.
Based on the results of our investigation, we proposed a schematic model (Fig. 7) to show how 4-HPR and IFN-g changed different molecular events leading to differentiation and apoptosis in human glioblastoma cells.
Fig. 7.
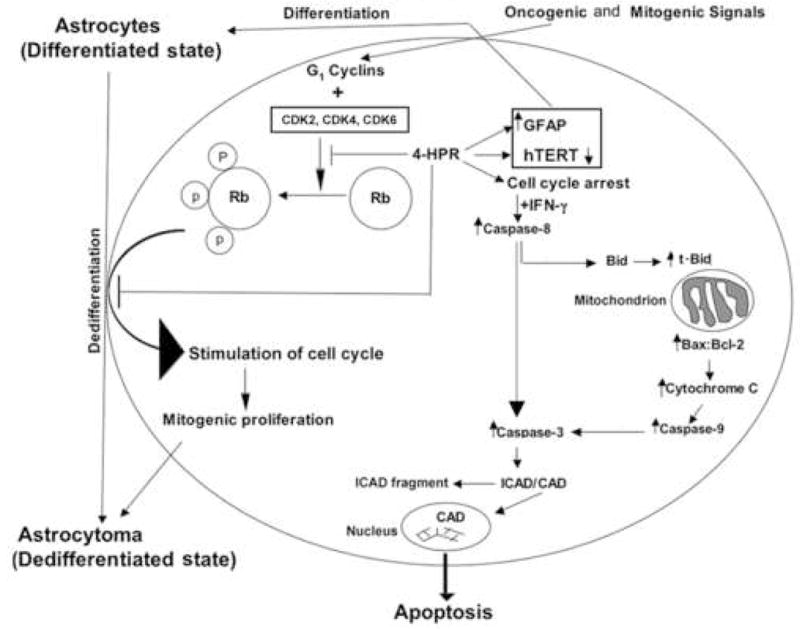
Schematic presentation of molecular events leading to astrocytic differentiation and apoptosis in glioblastoma cells after treatment with combination of 4-HPR and IFN-γ. Treatment with 4-HPR could down regulate hTERT, CDK2 and pRb, which would most likely result in cell cycle arrest. Prolongation of cell cycle arrest would enhance sensitivity of the cells to IFNγ for activation of caspase cascades for apoptosis.
4. Discussion
The results our present investigation strongly suggested that treatment of human heterogenic glioblastoma cells with 4-HPR induced astrocytic differentiation and down regulation of telomerase, CDK2, and survivin leading to increase in sensitivity to IFN-γ for activation of caspase cascades for apoptosis. Deletion of INK4a locus [26, 27] along with activation of Ras and Akt pathways can be responsible for oncogenic/mitogenic signals that lead to the dedifferentiation of the mature astroglial cells to astrocytoma [28]. Our results demonstrated that 4-HPR induced astrocytic differentiation with the down regulation of key signaling molecules to orchestrate prolongation of cell cycle arrest. Treatment of glioblastoma cells with 4-HPR triggered cell cycle arrest and thereby facilitated IFN-γ mediated activation of the apoptotic cascades.
The synthetic retinoid 4-HPR is widely used as an inducer of cellular differentiation in a wide variety of carcinomas [29–31]. One of the major advantages of using 4-HPR over other retinoids is its less toxicity profile [32] and its ability to display anti-proliferative activities at doses achievable pharmacologically [33]. Our observations suggested that 4-HPR even at a low concentration (0.5 μM) induced astrocytic differentiation in glioblastoma cells (Fig. 1). This phenomenon of astrocytic differentiation contributed to the down regulation of CDK2 and phosphorylation of its substrate Rb to pRb (Fig. 2). This event has been proposed to be accompanied by an early exit from the cell cycle [34].
The down regulation of CDK2 and pRb might be responsible for chromatin remodeling [35] that might contribute to sustained down regulation of survivin, the overexpression of which could enhance malignant potential of glioblastomas [36, 37]. One of the interesting observations made in the present study was the down regulation of survivin in glioblastoma cells after treatment with 4-HPR alone and also in combination with IFN-γ (Fig. 2). Previous studies attributed this down regulation of survivin to upregulation of caspase-3 mediated apoptotic cascade [38, 39]. Recent studies indicate a more refined role for survivin, which is postulated to be one of the three non-enzymatic subunits required for the enzymatic function of Aurora-B kinase [40]. Aurora-B kinase constitutes the enzymatic heart of the chromosomal passenger complex, which is responsible for chromosome alignment, segregation, and cytokinesis during mitosis. The down regulation of survivin would impede the activity of Aurora-B kinase, which would result in severe mitotic abnormality and result in non-homologous separation of the chromosomes. This aphorism was substantiated by transfecting mutated surviving to cells that were then found to progress normally from G1/S to G2/M phase, but majority of the cells made an early exit from the cell cycle and remained arrested with 4N DNA content [41].
Both hTERT and survivin genes control major steps in cancer development, ensuring indefinite cell growth and cell division [42]. Besides, hTERT and most of the genes capable of inhibiting apoptosis are located close to the telomeres in different species, suggesting that maintenance of telomeres might be connected with a correct balance of hTERT and survivin [43]. Down regulation of hTERT and survivin might contribute to prolongation of cell cycle arrest [44] and thereby contribute to activation of IFN-γ mediated apoptotic pathway.
The unique ability of 4-HPR to reduce cytokinesis at achievable pharmacological doses might contribute to the development of a new therapeutic regimen for treating glioblastoma. Further, treatment of glioblastoma cells with combination of 4-HPR and IFN-γ induced morphological features of apoptosis (Fig. 3), activated extrinsic caspase cascade to cause Bid cleavage to tBid (Fig. 4), and also triggered intrinsic caspase cascade due to increase in Bax:Bcl-2 ratio and mitochondrial release of cytochrome c into the cytosol (Fig. 5). The association of the cytosolic cytochrome c along with Apaf-1 and pro-caspase-9 contributes to the formation of apoptosome leading to activation of caspase-9 [45], which then activates effector caspases such as caspase-3 [46]. We found that apoptosis occurred in all three glioblastoma cell lines with activation of caspase-9 as well as of caspase-3 for apoptosis, which in turn caused ICAD fragmentation (Fig. 6) to release CAD for its translocation to the nucleus for degradation of genomic DNA.
5. Conclusion
Thus, our results showed that combination of 4-HPR and IFN-γ induced astrocytic differentiation with down regulation of markers of cell proliferation, contributing to sustained cell cycle arrest and increased apoptosis in human glioblastoma cells of heterogeneous origin. In order to harness the potential of such a combination of a chemotherapeutic agent and an immunotherapeutic agent, further studies need to be conducted in animal models of glioblastoma.
Footnotes
Grant support: This work was supported in part by the R01 grants (CA-91460 and NS-57811) from the National Institutes of Health (Bethesda, MD) to S.K.R.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ray SK, Patel SJ, Welsh CT, Wilford GG, Hogan EL, Banik NL. Molecular evidence of apoptotic death in malignant brain tumors including glioblastoma multiforme: upregulation of calpain and caspase-3. J Neurosci Res. 2002;69:197–206. doi: 10.1002/jnr.10265. [DOI] [PubMed] [Google Scholar]
- 2.Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 3.Parney IF, Chang SM. Current chemotherapy for glioblastoma. Cancer J. 2003;9:149–156. doi: 10.1097/00130404-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Parney IF, Hao C, Petruk KC. Glioblastoma immunology and immunotherapy. Neurosurgery. 2000;46:778–91. doi: 10.1097/00006123-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 5.Chambon PA. Decade of molecular biology of retinoic acid receptors. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 6.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 7.Soprano DR, Qin P, Soprano KJ. Retinoic acid receptors and cancers. Annu Rev Nutr. 2004;24:201–221. doi: 10.1146/annurev.nutr.24.012003.132407. [DOI] [PubMed] [Google Scholar]
- 8.Wu S, Zhang ZP, Zhang D, Soprano DR, Soprano KJ. Reduction of both RAR and RXR levels is required to maximally alter sensitivity of CA-OV3 ovarian tumor cells to growth suppression by all-trans retinoic acid. Exp Cell Res. 1997;237:118–126. doi: 10.1006/excr.1997.3769. [DOI] [PubMed] [Google Scholar]
- 9.Bhatnagar R, Abou-Issa H, Curley RW, Jr, Koolemans-Beynen A, Moeschberger ML, Webb TE. Growth suppression of human breast carcinoma cells in culture by N-(4-hydroxyphenyl) retinamide and its glucuronide and through synergism with glucarate. Biochem Pharmacol. 1991;41:1471–1477. doi: 10.1016/0006-2952(91)90563-k. [DOI] [PubMed] [Google Scholar]
- 10.Kalemkerian GP, Slusher R, Ramalingam S, Gadgeel S, Mabry M. Growth inhibition and induction of apoptosis by fenretinide in small-cell lung cancer cell lines. J Natl Cancer Inst. 1995;87:1674–1680. doi: 10.1093/jnci/87.22.1674. [DOI] [PubMed] [Google Scholar]
- 11.Sabichi AL, Hendricks DT, Bober MA, Birrer MJ. Retinoic acid receptor β expression and growth inhibition of gynecologic cancer cells by the synthetic retinoid N-(4-hydroxyphenyl) retinamide. J Natl Cancer Inst. 1998;90:597–605. doi: 10.1093/jnci/90.8.597. [DOI] [PubMed] [Google Scholar]
- 12.Panigone S, Debernardi S, Taya Y, Fontanella E, Airoldi R, Delia D. pRb and Cdk regulation by N-(4-hydroxyphenyl) retinamide. Oncogene. 2000;19:4035–4041. doi: 10.1038/sj.onc.1203743. [DOI] [PubMed] [Google Scholar]
- 13.Haque A, Das A, Hajiaghamohseni LM, Younger A, Banik NL, Ray SK. Induction of apoptosis and immune response by all-trans retinoic acid plus interferon-gamma in human malignant glioblastoma T98G and U87MG cells. Cancer Immunol Immunother. 2007;56:615–625. doi: 10.1007/s00262-006-0219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke F, East N, Upton C, Patel K, Balkwill FR. Interferon gamma induces cell cycle arrest and apoptosis in a model of ovarian cancer: enhancement of effect by batimastat. Eur J Cancer. 1997;33:1114–1121. doi: 10.1016/s0959-8049(97)88065-3. [DOI] [PubMed] [Google Scholar]
- 15.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 17.Altieri DC. Validating survivin as a cancer therapeutic target. Nat Rev Cancer. 2003;3:46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 18.Conway EM, Pollefeyt S, Steiner-Mosonyi M, Luo W, Devriese A, Lupu F, et al. Deficiency of survivin in transgenic mice exacerbates Fas-induced apoptosis via mitochondrial pathways. Gastroenterology. 2002;123:619–631. doi: 10.1053/gast.2002.34753. [DOI] [PubMed] [Google Scholar]
- 19.Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, et al. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karmakar S, Weinberg MS, Banik NL, Patel SJ, Ray SK. Activation of multiple molecular mechanisms for apoptosis in human malignant glioblastoma T98G and U87MG cells treated with sulforaphane. Neuroscience. 2006;141:1265–1280. doi: 10.1016/j.neuroscience.2006.04.075. [DOI] [PubMed] [Google Scholar]
- 21.Ray SK, Karmakar S, Nowak MW, Banik NL. Inhibition of calpain and caspase-3 prevented apoptosis and preserved electrophysiological properties of voltage-gated and ligand-gated ion channels in rat primary cortical neurons exposed to glutamate. Neuroscience. 2006;139:577–595. doi: 10.1016/j.neuroscience.2005.12.057. [DOI] [PubMed] [Google Scholar]
- 22.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 23.Murphy KM, Streips UN, Lock RB. Bax membrane insertion during Fas (CD95)-induced apoptosis precedes cytochrome c release and is inhibited by Bcl-2. Oncogene. 1999;18:5991–5999. doi: 10.1038/sj.onc.1203001. [DOI] [PubMed] [Google Scholar]
- 24.Cain K, Brown DG, Langlais C, Cohen GM. Caspase activation involves the formation of the aposome, a large (approximately 700 kD) caspase-activating complex. J Biol Chem. 1999;274:22686–22692. doi: 10.1074/jbc.274.32.22686. [DOI] [PubMed] [Google Scholar]
- 25.Saleh A, Srinivasula SM, Acharya S, Fishel R, Alnemri ES. Cytochrome c and dATP-mediated oligomerization of Apaf-1 is a prerequisite for procaspase-9 activation. J Biol Chem. 1999;274:17941–17945. doi: 10.1074/jbc.274.25.17941. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A. Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol. 1997;7:871–875. doi: 10.1111/j.1750-3639.1997.tb00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hegi ME, zur Hausen A, Ruedi D, Malin G, Kleihues P. Hemizygous or homozygous deletion of the chromosomal region containing the p16INK4a gene is associated with amplification of the EGF receptor gene in glioblastomas. Int J Cancer. 1997;73:57–63. doi: 10.1002/(sici)1097-0215(19970926)73:1<57::aid-ijc10>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Dai C, Holland EC. Astrocyte differentiation states and glioblastoma formation. Cancer J. 2003;9:72–81. doi: 10.1097/00130404-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 29.Dowlatshahi K, Mehta RG, Thomas CF, Dinger NM, Moon RC. Therapeutic effect of N-(4-hydroxyphenyl) retinamide on N-methyl-N-nitrosourea-induced rat mammary cancer. Cancer Lett. 1989;47:187–192. doi: 10.1016/0304-3835(89)90089-x. [DOI] [PubMed] [Google Scholar]
- 30.Igawa M, Tanabe T, Chodak GW, Rukstalis DB. N-(4-hydroxyphenyl) retinamide induces cell cycle specific growth inhibition in PC3 cells. Prostate. 1994;24:299–305. doi: 10.1002/pros.2990240605. [DOI] [PubMed] [Google Scholar]
- 31.Pienta KJ. The epidemiology of prostate cancer: clues for chemoprevention. In Vivo. 1994;8:419–422. [PubMed] [Google Scholar]
- 32.Veronesi U, Mariani L, Decensi A, Formelli F, Camerini T, Miceli R, et al. Fifteen-year results of a randomized phase III trial of fenretinide to prevent second breast cancer. Ann Oncol. 2006;17:1065–1071. doi: 10.1093/annonc/mdl047. [DOI] [PubMed] [Google Scholar]
- 33.Naik HR, Kalemkerian G, Pienta KJ. 4-Hydroxyphenylretinamide in the chemoprevention of cancer. Adv Pharmacol. 1995;33:315–347. doi: 10.1016/s1054-3589(08)60673-0. [DOI] [PubMed] [Google Scholar]
- 34.Galderisi U, Jori FP, Giordano A. Cell cycle regulation and neural differentiation. Oncogene. 2003;22:5208–5219. doi: 10.1038/sj.onc.1206558. [DOI] [PubMed] [Google Scholar]
- 35.Macaluso M, Montanari M, Giordano A. Rb family proteins as modulators of gene expression and new aspects regarding the interaction with chromatin remodeling enzymes. Oncogene. 2006;25:5263–5267. doi: 10.1038/sj.onc.1209680. [DOI] [PubMed] [Google Scholar]
- 36.Chakravarti A, Zhai GG, Zhang M, Malhotra R, Latham DE, Delaney MA, et al. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene. 2004;23:7494–7506. doi: 10.1038/sj.onc.1208049. [DOI] [PubMed] [Google Scholar]
- 37.Katoh M, Wilmotte R, Belkouch MC, de Tribolet N, Pizzolato G, Dietrich PY. Survivin in brain tumors: an attractive target for immunotherapy. J Neurooncol. 2003;64:71–76. doi: 10.1007/BF02700022. [DOI] [PubMed] [Google Scholar]
- 38.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi K, Hatano M, Otaki M, Ogasawara T, Tokuhisa T. Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation. Proc Natl Acad Sci USA. 1999;96:1457–1462. doi: 10.1073/pnas.96.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vader G, Medema RH, Lens SM. The chromosomal passenger complex: guiding Aurora-B through mitosis. J Cell Biol. 2006;173:833–837. doi: 10.1083/jcb.200604032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lens SM, Medema RH. The survivin/Aurora B complex: its role in coordinating tension and attachment. Cell Cycle. 2003;2:507–510. doi: 10.4161/cc.2.6.559. [DOI] [PubMed] [Google Scholar]
- 42.Park HR, Min SK, Cho HD, Kim KH, Shin HS, Park YE. Expression profiles of p63, p53, survivin, and hTERT in skin tumors. J Cutan Pathol. 2004;31:544–549. doi: 10.1111/j.0303-6987.2004.00228.x. [DOI] [PubMed] [Google Scholar]
- 43.Wurl P, Kappler M, Meye A, Bartel F, Kohler T, Lautenschlager C, et al. Co-expression of survivin and TERT and risk of tumour-related death in patients with soft-tissue sarcoma. Lancet. 2002;359:943–945. doi: 10.1016/S0140-6736(02)07990-4. [DOI] [PubMed] [Google Scholar]
- 44.Vogel C, Hager C, Bastians H. Mechanisms of mitotic cell death induced by chemotherapy-mediated G2 checkpoint abrogation. Cancer Res. 2007;67:339–345. doi: 10.1158/0008-5472.CAN-06-2548. [DOI] [PubMed] [Google Scholar]
- 45.Zou H, Yang R, Hao J, Wang J, Sun C, Fesik SW, et al. Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J Biol Chem. 2003;278:8091–8098. doi: 10.1074/jbc.M204783200. [DOI] [PubMed] [Google Scholar]
- 46.Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES. Autoactivation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell. 1998;1:949–957. doi: 10.1016/s1097-2765(00)80095-7. [DOI] [PubMed] [Google Scholar]