

Abstract
Background
Transfusion is a cornerstone of the management of sickle cell disease but carries a high risk of hemolytic transfusion reaction, probably because of differences in erythrocyte antigens between blood donors of European descent and patients of African descent. Patients may experience hemolytic transfusion reactions that are delayed by from a few days to two weeks and manifest as acute hemolysis (hemoglobinuria, jaundice, and pallor), symptoms suggesting severe vaso-occlusive crisis (pain, fever, and acute chest syndrome), and profound anemia, often with reticulocytopenia. This case-series study aims to describe the main characteristics of this syndrome, to discuss its pathophysiology, and to propose a management strategy.
Design and Methods
We identified 8 pediatric cases of delayed hemolytic transfusion reactions between 2006 and 2009 in the database of the Necker Hospital, France. All patients had received cross-matched red cell units compatible in the ABO, RH, and KEL systems. We reviewed the medical charts in the computerized blood transfusion databases. All patients were admitted to the intensive care unit. We progressively adopted the following strategy: intravenous immunoglobulins, and darbopoietin alpha when the reticulocyte count was below 150×109/L, without further blood transfusion during the acute episode unless absolutely necessary.
Results
The median time between the transfusion and the diagnosis of delayed hemolytic transfusion reaction was six days. All patients had severe bone pain; all but one had a high-grade fever. Five patients had hemoglobin levels less than than 4g/dL and 3 had reticulocytopenia. In 5 patients, no new antibody was found; one patient had weakly reactive antibodies. Only 2 patients had new allo-antibodies possibly responsible for the delayed hemolytic reaction.
Conclusions
The initial symptoms of delayed hemolytic transfusion reaction were complex and mimicked other complications of sickle cell disease. In most of our cases, no new antibody was identified, which underlines the complexity of the pathophysiology of this syndrome.
Keywords: sickle cell disease, transfusion, allo-immunization, intravenous immunoglobulin, rituximab
Introduction
Blood transfusion is a cornerstone of the management of sickle cell disease (SCD).1,2 The goals of blood transfusion are to increase oxygen distribution to the tissues and/or to replace the rigid sickle-shaped red blood cells (RBCs) by deformable RBCs. However, blood transfusion in patients affected with SCD is associated with a high rate of hemolytic transfusion reactions. The differences in RBC antigens between blood donors of European descent and patients with SCD of Afro-Caribbean descent often result in allo-immunization. Antigenic differences are found within common variants and within variant antigens.3,4 Some partial antigens are found exclusively in the Afro-Caribbean population, and carriers of these variant antigens can produce allo-antibodies when exposed to the complete antigen. These antibodies can cause severe delayed hemolytic transfusion reactions (DHTRs), which present specific features in SCD patients.5–9 DHTR has been reported to occur in 4–11% of SCD patients given blood transfusions.6,10–12 DHTRs occur from a few days to two weeks after a blood transfusion and manifest as clinical features of acute hemolysis (hemoglobinuria, jaundice, and pallor) combined with symptoms suggesting severe vaso-occlusive crisis (pain, fever, and sometimes acute chest syndrome). The destruction of both the donor’s and the recipient’s RBCs leads to an abrupt drop in the hemoglobin level, to a value lower than the pre-transfusion value. Reticulocytopenia is common. Screening tests may show allo-antibody reactivation, sometimes with clinically relevant auto-antibodies.13 However, cases with no detectable antibody are not infrequent. Corticosteroid and/or intravenous immunoglobulin (IVIg) therapy has been reported to ensure recovery from DHTR.5–9,13,14 More recently, two studies reported the use of rituximab for preventing DHTR.13,15 The pathophysiology of this syndrome remains unclear, especially when there is no detectable antibody. A recent study suggests accelerated apoptosis of donor RBCs.12
This article describes a series of patients with SCD and DHTR in order to underline several important points. First, our cases illustrate the complexity of the initial clinical picture in DHTR, which mimics other complications of SCD, leading to a risk of misdiagnosis. Further blood transfusions given to treat a mistakenly diagnosed SCD complication can cause a life-threatening exacerbation of the hemolysis. Second, many of our patients had no detectable antibody. Finally, we describe the treatments used for our patients and we suggest a management strategy.
Design and Methods
Patients
We identified 8 pediatric patients (5 girls and 3 boys) with SCD and DHTR in the Necker Hospital database. DHTR was defined as the abrupt onset, at least three days after an RBC transfusion, of signs indicating accelerated hemolysis: hemoglobinuria, jaundice, pallor, hemoglobin (Hb) level drop, lactic dehydrogenase (LDH) elevation, and bilirubin elevation above the baseline value. In patients with more than one DHTR, the index episode was the most recent one. All 8 patients had homozygous SCD (SS disease); 7 were from Africa and one from the Caribbean. Median age was 10.5 years (range 2–15 years). Of these 8 patients, 2 were receiving follow up for SCD at the Necker Hospital before the DHTR, 4 were transferred to the intensive care unit of the Necker Hospital for management of the DHTR, and 2 were managed at their usual reference center (Poissy-Saint Germain en Laye Hospital and Limoges Hospital, respectively) according to the protocol suggested by the Necker Hospital. All DHTRs occurred between 2006 and 2010.
All 8 patients received cross-matched RBC units compatible in the ABO, RH, and KEL systems. Patients with positive screening tests received units extensively matched for the other immunogenic blood groups (JK, FY, and MNS). When DHTR was suspected, a new screening test, a direct antiglobulin test (DAT), and an elution test were performed. Finally, so far, molecular analysis has been performed in two settings: patients with Rh antibodies against expressed Rh antigens to detect Rh variants prone to allo-immunization, thereby distinguishing between allo- and auto-antibodies; and in patients with depressed expression of Rh antigens.
Methods
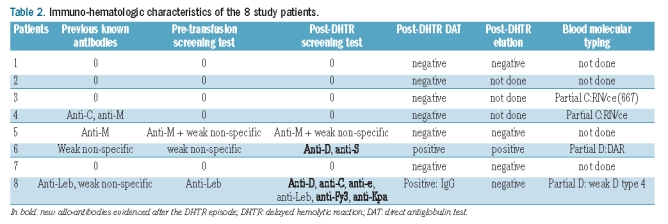
We retrospectively reviewed the medical charts of the 8 patients. Further information on their transfusion history was obtained from the computerized blood transfusion databanks for the Paris area (where the Necker, Ambroise Paré, Trousseau, Le Chesnay, and Poissy-St Germain en Laye Hospitals are located) and for the Aquitaine Limousin area (where the Limoges Hospital is located). We recorded the number of previous transfusions, any past history of erythrocyte allo-immunization and/or DHTR, and reasons for the transfusion responsible for the current DHTR (Table 1 and Figure 1). We may have underestimated the number of previous transfusions, as some patients may have received blood transfusions outside the Paris or Aquitaine Limousin areas that were not recorded in their medical charts, although this seems highly unlikely. The patients are numbered according to the chronological order of DHTR diagnosis. We performed molecular analysis in patients 4, 6, and 8 who exhibited previous (patient 4) or new (patients 6 and 8) allo-antibodies; and in patient 3 who had decreased expression of the RH antigen. Results are shown in Table 2.
Table 1.
Clinical and laboratory characteristics of the delayed hemolytic transfusion reactions
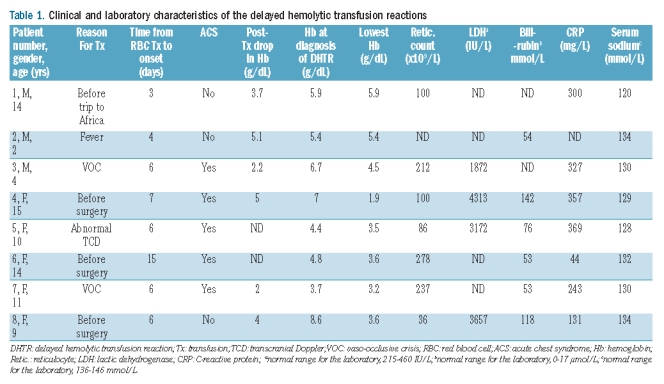
Figure 1.

Transfusion and immuno-hematologic findings.
Table 2.
Immuno-hematologic characteristics of the 8 study patients.
We had no standardized protocol for managing DHTR from 2006 to the beginning of 2009 (patients 1, 2 and 3). We progressively adopted the following strategy: the child is transferred to the intensive care unit and given oxygen, fluids, and analgesics; 1g/kg intravenous immunoglobulins are infused, and if needed, immunoglobulin infusion is repeated on the following day; if the reticulocyte count is less than 150×109/L, darbopoietin alpha 5 μg/kg is administered. Most importantly, further blood transfusions are avoided during the DHTR. After the DHTR is resolved, hydroxyurea therapy is considered to minimize transfusion requirements.
Results
Clinical presentation
The main clinical and laboratory findings (except the immuno-hematologic data) are reported in Table 1. The main past immuno-hematologic data are reported in Table 2 and Figure 1. The median time from the last transfusion to the diagnosis of DHTR was six days (range 3–15). All children had severe bone pain and all but one had a high-grade fever. The 7 febrile children had blood cultures performed, with negative results. In 3 patients, severe bone pain with a fever and C-reactive protein elevation to more than 300 mg/L led to an initial diagnosis of osteomyelitis (patients 1, 4 and 5); of these 3 patients, one underwent a bone aspiration which showed no organisms. In 5 patients (patients 3, 4, 5, 6 and 7), the combination of a fever, chest pain, hypoxemia, and a new pulmonary infiltrate led to a diagnosis of acute chest syndrome. In 2 patients (patients 1 and 5), renal infarction was suspected then ruled out by ultrasonography of the kidneys. The median Hb level at diagnosis was 5.65 g/dL (range 3.7–8.6 g/dL). The median fall in Hb from the post-transfusion value was 3.85 g/dL (range 2–5.1 g/dL). Reticulocyte counts were decreased (≤100×109/L) in 4 patients, unchanged in 3 patients, and not determined in one patient. In all patients who had LDH and bilirubin assays, the values were increased compared to baseline. Serum sodium was low in all patients (mean 129±4.4 mmol/L; normal range for the laboratory 136–146 mmol/L). In the 3 patients who had their urinary sodium measured, the values were excessively high given the low serum sodium levels. Renal function was temporarily impaired in one patient (#6) who had an increase in creatinine level to 155 μmol/L (normal range for the laboratory 20–75 μmol/L) with a concomitant β2-microglobulin value of 10 mg/mL (normal range for the laboratory 0–0.35 mg/mL). This renal function impairment with tubulopathy resolved within five days.
History of delayed hemolytic transfusion reaction
Patients 1, 2, 3, and 7 had no prior history of DHTR and their pre-transfusion screening tests were negative. Their transfusions were cross-match compatible. The screening tests and DATs remained negative during the DHTR. No antibodies against RBCs were detected at a distance from the DHTR episode (data not shown).
Patient 4 had had two previous DHTRs. The first episode occurred in 2003 and was due to the production of anti-M antibody. A blood transfusion in 2004 was uneventful. In 2007, she again received a blood transfusion after which she experienced a DHTR due to anti-C antibody. Tests performed before the 2009 transfusion that caused the last DHTR were negative for anti-M and antiC antibodies. She was given cross-match-compatible blood units negative for C, E, K, Fya, M, and S. Screening tests performed after the DHTR were negative. She had an RN variant allele, producing a partial C antigen. Blood group evaluation in the RH system showed no decrease in the expression of this variant antigen. Patient 5 had a serum anti-M antibody before the transfusion and received M-negative cross-matched units. After onset of the DHTR, only weak reactions were evidenced, with no detectable specificity. The DAT was negative. Patient 6 had no prior history of DHTR but had a weakly reactive screening test with no detectable specificity. The RH phenotype was normal. The post-DHTR screening test showed anti-D and anti-S antibodies, and the DAT was IgG-positive. This patient had received D+, S+ RBC units. In this case, anti-S and anti-D antibody production could be the cause of the DHTR. Molecular analysis showed a partial D antigen (DAR variant). Finally, patient 8 had had a DHTR in 2008. Her phenotype was D+C-E-c+e+, K-, Fya-Fyb-, S-s+, Lea-Leb-. The DHTR in 2008 occurred after a transfusion of 5 units, one of which was Leb+ and another S+. All units were D-, E-, C-, and Fya-. The only antibody detected after the DHTR was anti-Leb. In 2010, before a tonsillectomy, she received 3 units including two Fya+, Fyb+ units and one D+ unit. She developed new antibodies (anti-D, anti-C, anti-e, anti-Fy3, and anti-Kpa) during this second DHTR episode. Molecular analysis showed weak D type 4, which could be considered a partial D antigen.
Treatments and outcomes (Table 3)
Table 3.
Treatments and outcomes.
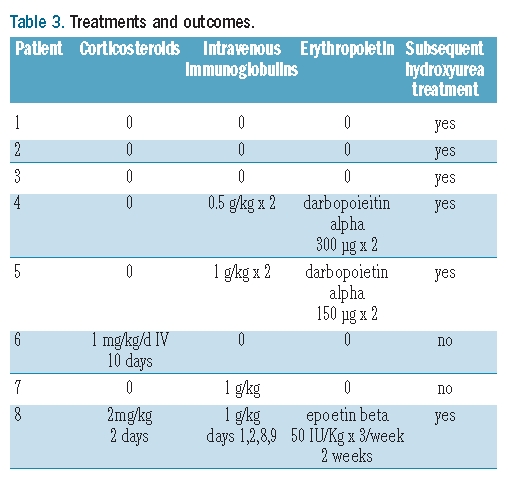
The first 3 patients experienced DHTRs in 2006, when we had no standardized protocol for managing DHTRs in SCD patients. Given that the minimal Hb levels were not very low (4.5 to 5.9 g/dL), no specific treatment was given. A spontaneous favourable outcome was seen in all 3 patients. The next 5 cases occurred in 2009 and 2010. Diagnosis was rapid, and the Hb levels at diagnosis ranged from 3.7 to 8.6 g/dL. Treatment for DHTR was started immediately and consisted of corticosteroids in one patient (patient 6), IVIg in 3 patients (patients 4, 5 and 7), and both in one patient (patient 8). Three patients (patients 4, 5 and 8) had reticulocytopenia (100×109/L, 86×109/L, and 36×109/L) and received erythropoietin. Hb levels continued to decrease in 6 patients in the days following diagnosis and reached a median minimum value of 3.60 g/dL (1.9–5.9) after a median of two days (range 1–5). In patient 4, the Hb level plateaued at about 4 g/dL before dropping abruptly to 1.9 g/dL. Usually, the drop in Hb level stopped one day after the end of the clinical hemoglobinuria. A further transfusion was considered indispensable in patients 4 and 5, whose Hb levels just before the repeat transfusion were 1.9 g/dL and 3.5 g/dL, respectively. Both patients received IVIg before the new transfusion which was well tolerated with no further hemolytic reaction.
The patients who had a fever received intravenous cefo-taxime (n=6) or ceftriaxone (n=1). The antibiotics were always started after the onset of hemoglobinuria. The 3 patients with suspected osteomyelitis received intravenous rifampin combined with a 3rd generation cephalosporin, started after the diagnosis of DHTR. Patients with suspected ACS were managed with macrolides and non-invasive ventilation.
Hydroxyurea was started just after the DHTR in 4 of the 8 patients (patients 3, 4, 5 and 8) in the hope of decreasing transfusion requirements. In 2 additional patients (patients 1 and 2), hydroxyurea was started later because of recurrent painful crises. The appropriateness of hydroxyurea therapy in the remaining 2 patients is still under debate. Of the 8 patients, only one (patient 2) has received further blood transfusions since the index DHTR episode, in 2006. The transfusions were given for pulmonary complications and followed by hydroxyurea therapy: these were well tolerated. The patient died suddenly in 2010 during hospitalization for a moderate painful crisis treated with intravenous hydration and paracetamol, with no transfusion. No autopsy was performed and the cause of death was not determined.
Discussion
We studied 8 children with SCD who experienced DHTRs, including 5 with Hb levels less than 4 g/dL. These cases illustrate the need for routinely considering DHTR in SCD patients whose clinical and laboratory signs of hemolysis suddenly worsen within two weeks of a blood transfusion. Hemoglobinuria is strongly suggestive of DHTR in SCD. DHTR can occur even after transfusions that were matched at least in the ABO, RH, and KEL systems and that produced a negative serological cross-match test. Negative immunological tests do not exclude a DHTR. The increase in HbS percentages indicating destruction of the transfused RBCs confirms the diagnosis of DHTR. In most of our patients, DHTR was not suspected initially: the diagnosis initially considered most likely was osteomyelitis in 3 patients and renal infarction in 2 patients. Maintaining a high index of suspicion for DHTR is crucial as mistakenly diagnosing another complication (e.g. osteomyelitis or ACS) may lead to a further transfusion which may exacerbate the hemolysis. Tubulopathy related to intravascular hemolysis is extremely common in DHTR, as shown by the presence of hyponatremia in all our patients, with transient renal failure in one patient.
A limitation of our study is that we cannot assess the occurrence rate of DHTR in the pediatric population given the retrospective study design and our status as a reference center that serves not only our area but also patients referred to us from other areas. To determine the occurrence rate of DHTR, and to gather additional information on the spectrum of clinical and laboratory presentations, we are planning a one year prospective study in the Paris area which will be conducted both by the reference centers for SCD and by the French blood transfusion service.
The pathophysiology of DHTR in SCD patients is unclear. Classically, DHTR is ascribed to a reaction between anti-RBC antibodies produced by the recipient and antigens expressed by the donor RBCs. However, anti-RBC antibodies are often undetectable in SCD patients with DHTR.5 Of our 8 patients, 5 (patients 1, 2, 3, 4 and 7) had no detectable new antibodies. Patient 5 had weakly reactive antibodies. Only in 2 patients (patients 6 and 8) were we able to ascribe the DHTR to transfusion-induced reactivation of allo-antibody production. Thus, in three-quarters of our patients the routine immuno-hematologic tests failed to identify the cause of the DHTR. Of the 5 patients with no detectable antibodies, all but patients 4 and 8 had no prior history of DHTR despite having had several previous transfusions. In this situation, the clinical and laboratory status at the time of transfusion may influence the outcome of the transfusion, independently of the allo-immunization process. According to one hypothesis, stored donor RBCs may experience accelerated eryptosis in the bloodstream of SCD patients, with externalization of the phosphatidylserine membrane at the outer surface of the cell.12,16 Eryptosis may be the first step in a cascade of events which together cause accelerated RBC destruction and hemolysis. RBCs may be rapidly destroyed by macrophages, which are probably activated. Another possibility is that inflammatory enzymes, such as phospholipase A2, hydrolyze the senescent phosphatidylserine-exposing RBCs.17 DHTR may result from a combination of several factors18 and the role of concomitant inflammation is under debate. SCD has been postulated to be a chronic inflammatory disease.19 In our patient 1, the transfusion was planned before a trip to Africa in the absence of any identifiable inflammatory risk factors. In patients 4 and 6, however, the transfusion was given before a surgical procedure. Conceivably, cytokine production during surgery may have led to an inflammatory reaction in these patients.20 Patient 4 received several transfusions between 2003 and 2009, and 2 of the 3 transfusions followed by DHTRs were given before a surgical procedure (tonsillectomy in 2003 and cholecystectomy in 2009). In patient 5, the pre-transfusion screening test showed only weak non-specific reactivity. One possibility is that a non-clinically significant antibody may become significant when the donor RBCs undergo accelerated senescence in the bloodstream. Patients 6 and 8 had allo-antibodies that can be considered responsible for the DHTR. In patient 6, a common anti-S antibody and an uncommon anti-D antibody were detected. The development of anti-S antibody could not be prevented because of the shortage in France of donors of African descent having the same extended phenotype as SCD patients. The anti-D antibody was probably present before the transfusion but not detected because of its weak reactivity. This anti-D antibody was related to the presence of a partial D antigen of the DAR type in this patient. Similarly, no strictly antigen-identical units were available for patient 8, in whom the production of anti-Fy3 could not be prevented. Although the units were C-negative, anti-C antibodies developed, via a mechanism that remains unclear. This patient also developed anti-e antibodies, although her phenotype was e+. Molecular typing of the RHCE found no variant, making an auto-anti-e the most probable hypothesis. On the contrary, the presence of anti-D antibody can be ascribed to the partial D antigen identified by molecular typing. However, this anti-D antibody was produced after a single D-positive transfusion.
Finally, the risk factors for DHTR in patients without detectable antibodies are unknown. Consequently, patients at risk for a first or recurrent episode of DHTR cannot be identified.
The uncertainties about the pathophysiology of DHTR in SCD patients complicate the identification of the best treatment approach. Previously described patients usually received corticosteroid therapy,6,7,14 IVIg,8 or both.5,9,21 The protocol currently used at our institution involves IVIg infusion. However, no randomized controlled trials have been carried out to assess treatments for SCD patients with DHTR. All previous publications insist on the importance of avoiding further RBC transfusions. We minimize the use of corticosteroids, which can induce severe side effects in children with SCD.22 Reversible posterior leukoencephalopathy syndrome was recently reported after corticosteroid therapy in a child with SCD and DHTR.14 Given the observational nature of our study, no conclusions can be drawn about the efficacy of IVIg therapy. Nevertheless, the 4 patients who received IVIg therapy (0.5–1 g/kg, 1–4 injections) experienced resolution of the hemolytic process and tolerated the infusions well. The effectiveness of IVIg therapy in patients 4 and 7, who had no pre-existing antibodies or new antibodies detected, does not rule out accelerated RBC senescence. Although competitive mononuclear phagocytic system blockade is among the most widely accepted mechanisms for explaining the therapeutic effects of IVIg therapy in patients with detectable antibodies, other mechanisms may be involved in patients without detectable antibodies. Thus, IVIg therapy affects the cellular immune response: for instance, IVIg therapy alters the apoptosis of lymphocytes and monocytes via a Fas (CD95)-dependent effect that is probably due to naturally occurring anti-Fas antibodies contained in IVIg and capable of blocking Fas-FasL binding.23 Thus, IVIg therapy may act by diminishing the destruction by macrophages of donor RBCs undergoing accelerated senescence. Another possible effect of IVIg therapy is cytokine modulation. IVIg therapy induces the production of interleukin-1 receptor antagonist, which can inhibit inflammation and macrophage phagocytic function.24,25
Erythropoietin-stimulating agents were given to the 3 patients with reticulocytopenia because we could not predict the duration of this manifestation; its mechanism is unknown.
Finally, the indications for rituximab therapy in SCD patients with DHTR remain unclear. Rituximab was used preventively in a poly-immunized 33-year old SCD patient who had had a history of DHTR two years earlier with a positive DAT and who required another transfusion in preparation for hip surgery. Rituximab was given before and after this transfusion, which was well tolerated.13 Rituximab combined with corticosteroid therapy and darbopoietin alpha was used in a 30-year old SCD patient with DHTR, previously known anti-MNS antibodies, and newly detected anti-Leb antibodies.15 New antibodies to RBCs were detectable in both these patients. Of our 8 patients, only 2 had detectable anti-RBC antibodies. They may be candidates for rituximab therapy should further blood transfusions prove to be unavoidable in the future.
Whether molecular analysis should be performed routinely in SCD patients merits discussion. Of our 8 patients, 4 underwent molecular analysis, which consistently showed a variant RH antigen. Patient 6 had a partial D, type DAR, and an anti-D antibody was considered responsible for the DHTR. In patient 4, a partial C variant had caused a prior DHTR, although the associated anti-C antibody was not involved in the index DHTR. A previous study showed that the C antigen should always be evaluated for a partial variant in SCD patients.11 In patient 3, a complex combination of rare alleles was found but was not involved in the index DHTR. Finally, patient 8, who had a weak D type 4, developed anti-D antibodies after receiving a single D+ unit. All previous transfusions in this patient were D-negative. It is important to point out that, in patients with variants, serological blood-group determination showed no evidence of depressed expression. This finding suggests that molecular analysis may be warranted in all patients and not only in those with altered serological phenotypes. This issue is currently under review at our institution.
In conclusion, we believe that DHTR may be underestimated in SCD patients as the manifestations mimic other complications of SCD and post-transfusion screening tests are usually negative. Patients in whom the diagnosis of DHTR is missed may receive repeat transfusions, which may contribute to the mortality associated with SCD. The high proportion of DHTR cases without identifiable antibodies suggests that multiple factors are involved in the pathophysiology of this life-threatening complication. Among these factors, inflammation may play a substantial role.
Supplementary Material
Acknowledgments
We are grateful to Dr Descazeaud, EFS Aquitaine Limousin, Limoges, and to Dr Wolfe for technical help.
Footnotes
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Ohene-Frempong K. Indications for red cell transfusion in sickle cell disease. Semin Hematol. 2001;38(1 Suppl 1):5–13. doi: 10.1053/shem.2001.20139. [DOI] [PubMed] [Google Scholar]
- 2.Vichinsky EP, Earles A, Johnson RA, Hoag MS, Williams A, Lubin B. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. N Engl J Med. 1990;322(23):1617–21. doi: 10.1056/NEJM199006073222301. [DOI] [PubMed] [Google Scholar]
- 3.Noizat-Pirenne F, Lee K, Le Pennec PY, Simon P, Kazup P, Bachir D, et al. Rare RHCE phenotypes in black individuals of Afro-Caribbean origin: identification and transfusion safety. Blood. 2002;100(12):4223–31. doi: 10.1182/blood-2002-01-0229. [DOI] [PubMed] [Google Scholar]
- 4.Noizat-Pirenne F. Immunohematologic characteristics in the Afro-caribbean population. Consequences for transfusion safety. Transfus Clin Biol. 2003;10(3):185–91. doi: 10.1016/s1246-7820(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 5.Scheunemann LP, Ataga KI. Delayed haemolytic transfusion reaction in sickle cell disease. Am J Med Sci. 2010;339(3):266–9. doi: 10.1097/MAJ.0b013e3181c70e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Talano JA, Hillery CA, Gottschall JL, Baylerian DM, Scott JP. Delayed haemolytic transfusion reaction/hyperhemolysis syndrome in children with sickle cell disease. Pediatrics. 2003;111(6 Pt 1):e661–5. doi: 10.1542/peds.111.6.e661. [DOI] [PubMed] [Google Scholar]
- 7.Petz LD, Calhoun L, Shulman IA, Johnson C, Herron RM. The sickle cell hemolytic transfusion reaction syndrome. Transfusion. 1997;37(4):382–92. doi: 10.1046/j.1537-2995.1997.37497265338.x. [DOI] [PubMed] [Google Scholar]
- 8.Cullis JO, Win N, Dudley JM, Kaye T. Post-transfusion hyperhaemolysis in a patient with sickle cell disease: use of steroids and intravenous immunoglobulin to prevent further red cell destruction. Vox Sang. 1995;69(4):355–7. doi: 10.1111/j.1423-0410.1995.tb00373.x. [DOI] [PubMed] [Google Scholar]
- 9.Win N, Doughty H, Telfer P, Wild BJ, Pearson TC. Hyperhemolytic transfusion reaction in sickle cell disease. Transfusion. 2001;41(3):323–8. doi: 10.1046/j.1537-2995.2001.41030323.x. [DOI] [PubMed] [Google Scholar]
- 10.Cox JV, Steane E, Cunningham G, Frenkel EP. Risk of alloimmunization and delayed haemolytic transfusions reactions in patients with sickle cell disease. Arch Intern Med. 1988;148(11):2485–9. [PubMed] [Google Scholar]
- 11.Tournamille C, Meunier-Costes N, Costes B, Martret J, Barrault A, Gauthier P, et al. Partial C antigen in sickle cell disease patients: clinical relevance and prevention of allommunization. Transfusion. 2010;50(1):13–9. doi: 10.1111/j.1537-2995.2009.02382.x. [DOI] [PubMed] [Google Scholar]
- 12.Chadebech P, Habibi A, Nzouakou R, Bachir D, Meunier-Costes N, Bonin P, et al. Delayed hemolytic transfusion reaction in sickle cell disease patients: evidence of an emerging syndrome with suicidal red blood cell death. Transfusion. 2009;49(9):1785–92. doi: 10.1111/j.1537-2995.2009.02199.x. [DOI] [PubMed] [Google Scholar]
- 13.Noizat-Pirenne F, Bachir D, Chadebech P, Michel M, Plonquet A, Lecron JC, et al. Rituximab for prevention of delayed haemolytic transfusion reaction in sickle cell disease. Haematologica. 2007;92(12):e132–e135. doi: 10.3324/haematol.12074. [DOI] [PubMed] [Google Scholar]
- 14.Elenga N, Mialou V, Kebaili K, Galambrun C, Bertrand Y, Pondarre C. Severe neurologic complication after delayed haemolytic transfusion reaction in 2 children with sickle cell anemia. J Pediatr Hematol Oncol. 2008;30(12):928–30. doi: 10.1097/MPH.0b013e31818c9172. [DOI] [PubMed] [Google Scholar]
- 15.Bachmeyer C, Maury J, Parrot A, Bachir D, Stankovic K, Girot R, et al. Rituximab as an effective treatment of hyperhemolysis syndrome in sickle cell anemia. Am J Hematol. 2010;85(1):91–2. doi: 10.1002/ajh.21578. [DOI] [PubMed] [Google Scholar]
- 16.Zimring JC, Spitalnik SL. To RBC or not to RBC: the role of suicidal death in hemolytic transfusion reactions. Transfusion. 2009;49(9):1776–8. doi: 10.1111/j.1537-2995.2009.02339.x. [DOI] [PubMed] [Google Scholar]
- 17.Neidlinger NA, Larkin SK, Bhagat A, Victorino GP, Kuypers FA. Hydrolysis of phosphatidylserine-exposing red blood cells by secretory phospholipase A2 generates lysophosphatidic acid and results in vascular dysfunction. J Biol Chem. 2006;281(2):775–81. doi: 10.1074/jbc.M505790200. [DOI] [PubMed] [Google Scholar]
- 18.Hod EA, Sokol SA, Zimring JC, Spitalnik SL. Hypothesis: hemolytic transfusion reactions represent an alternative type of anaphylaxis. Int J Clin Exp Pathol. 2009;2(1):71–82. [PMC free article] [PubMed] [Google Scholar]
- 19.Platt OS. Sickle cell disease as an inflammatory disease. J Clin Invest. 2000;106(3):337–8. doi: 10.1172/JCI10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni Choileain N, Redmond HP. Cell response to surgery. Arch Surg. 2006;141(11):1132–40. doi: 10.1001/archsurg.141.11.1132. [DOI] [PubMed] [Google Scholar]
- 21.Win N, Sinha S, Lee E, Mills W. Treatment with intravenous immunoglobuline and steroids may correct severe anemia in hyper-hemolytic transfusion reactions: case report and literature review. Transfus Med Rev. 2010;24(1):64–7. doi: 10.1016/j.tmrv.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Couillard S, Benkerrou M, Girot R, Brousse V, Ferster A, Bader-Meunier B. Steroid treatment in children with sickle cell disease. Haematologica. 2007;92(3):425–6. doi: 10.3324/haematol.10800. [DOI] [PubMed] [Google Scholar]
- 23.Crow AR, Lazarus AH. The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: what do we really know? Transfus Med Rev. 2008;22(2):103–16. doi: 10.1016/j.tmrv.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Crow AR, Song S, Semple JW, Freedman J, Lazarus AH. A role for IL-1 receptor antagonist or other cytokines in the acute therapeutic effects of IVIG? Blood. 2007;109(1):155–8. doi: 10.1182/blood-2006-05-023796. [DOI] [PubMed] [Google Scholar]
- 25.Kurosawa S, Kato M. Anesthetics, immune cells, and immune responses. J Anesth. 2008;22(3):263–77. doi: 10.1007/s00540-008-0626-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.