

Abstract
Observations of crystallization experiments are classified as specific outcomes and integrated through a phase diagram to visualize solubility and thereby direct subsequent experiments. Specific examples are taken from our high-throughput crystallization laboratory which provided a broad scope of data from 20 million crystallization experiments on 12,500 different biological macromolecules. The methods and rationale are broadly and generally applicable in any crystallization laboratory. Through a combination of incomplete factorial sampling of crystallization cocktails, standard outcome classifications, visualization of outcomes as they relate chemically and application of a simple phase diagram approach we demonstrate how to logically design subsequent crystallization experiments.
1. Introduction
Of the over 60,000 macromolecular structures currently deposited in the PDB1 approximately 86% were determined by single crystal X-ray diffraction methods. The importance of X-ray crystallography is reflected in the twenty-five Nobel prizes associated with the technique, including the determination of the structure of DNA,2 the structure of vitamin B12,3 the structure of the photosynthetic reaction center,4 the enzymatic mechanism underlying the synthesis of adenosine triphosphate,5 the structure of potassium channels,6 the molecular basis of eukaryotic transcription7 and most recently the ribosome.8 However, most proteins do not readily produce diffraction-quality crystals. In the case of the systematic work of structural genomics consortia during the past 10 years, failure as well as success has been tracked.9 Only 34% of expressed and purified targets provide a crystal and out of that only 12% result in a structure deposited in the PDB. Crystallographic structures require crystals, while crystallization remains fundamentally a hit-or-miss proposition.
Currently it is not possible to predict crystallization conditions of biological macromolecules from primary sequence alone. Crystallization is typically approached in an empirical manner by combining the protein (used in the generic for biological macromolecules) with scores of biochemical screens that have a history of success with other proteins. Commercial screening kits have been produced to speed the process, but these kits by necessity use either coarse-sampling of the available chemical space, or focus on a small area of chemical space with fine-sampling. In either case, potential crystallization conditions are easily missed. The approach for high-throughput crystallization screening largely adopted by structural factories such as those funded by the NIH Protein Structure Initiative (PSI) has been to increase both the biochemical conditions for crystallization and the target variants. This approach is denominator-dependent, that is it achieves a high structural output where structural output is dependent upon the number of proteins entering the pipeline. In its initial inception this approach was thought adequate to build up a library of fold space. The PSI emphasis now targets important biological problems. The failure of the crystallization step is even more significant. Fortunately, the resources we have developed supported by the PSI initiative are well placed to explore this area.
In the high-throughput screening laboratory at the Hauptman-Woodward Institute (HTSlab) we run a crystallization screening service used by over 1,000 laboratories worldwide and readily available as a resource to the general biomedical community. To date we have built up an archive of data from over 12,500 samples submitted to the laboratory. We screen these samples with three groups of chemical conditions looking at the sample’s response to salts, PEGS and making use of commercial crystallization screens. Crystallization experiments are set up in 1536-well experiment plates (Greiner BioOne, Frickenhausen, Germany) using the microbatch-under-oil method.10 Full details are described elsewhere.11 An automated imaging system records the experiments’ outcomes one day after the addition of the protein solution, and weekly thereafter for six weeks. Images are archived and the resulting quantity of information produced can be considerable. In this paper we summarize some of the observations from our 10 years of experience and nearly 20 million experiments set up and recorded to date. We cover options for initial characterization, examples of typical outcomes and what they mean in terms of solubility phase space, and methods to verify crystals are protein. We start from the individual experiment, expand to show the effect of considering neighboring results and finally show how time can be a useful tool in understanding the process. The goal of this paper is to provide practical and generally applicable methods to interrogate and interpret crystallization results, based upon our own observations and backed by literature supporting and explaining those observations. Our aim is to demonstrate that careful consideration of “what’s in the drop” can point the way toward successful crystallization when coupled with a phase diagram approach.
2. The phase diagram
A crystal typically results when protein molecules come out of solution in an ordered manner. The isoelectric point of a protein, the solution pH at which the polar amino acid residues have a net charge of zero, is often cited as a chemical condition where the protein solubility is near a minimum. These studies date back to the work of Arda Alden Green who considered pH the most important solution property to affect solubility12. While generally true, this has some exceptions. In conditions where the protein is salting-out (protein solubility decreases as salt is added to the solution and binds water required to hydrate the protein molecules), electrostatic interactions are significantly screened and protein net charge does not determine protein interactions. In many cases pH will be an important variable but in others it will have less influence. Multiple variables influence crystallization. We can represent the interaction of multiple variables and resulting solubility with respect to crystallization outcomes using a phase diagram, Figure 1. In this case we show an idealized schematic of two dimensional space; the diagram is a representation of the phase of a protein with respect to the concentrations of protein and crystallizing agent. In reality the multiple variables and degree of influence complicate this simplified vision and the figure represents a slice through a multidimensional space but illustrates the key regions important to the interpretation of empirical results.
Figure 1.
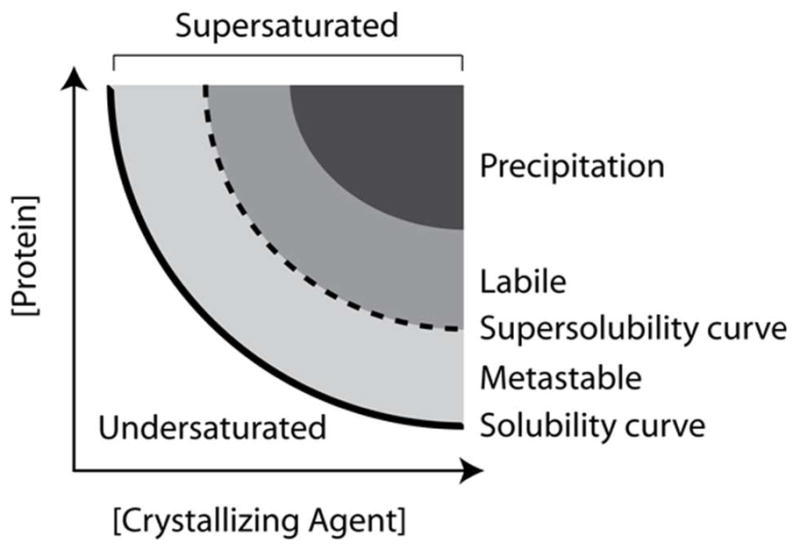
Schematic illustration of a typical crystallization phase diagram using protein concentration and crystallizing agent concentration as axes. This is an idealized two-dimensional projection of multidimensional space. Depending on how this space is sampled the different regions can have variable shapes and sizes. Not shown is the liquid phase separation which falls in the metastable zone at high protein concentrations.
Within the phase diagram there are distinct, thermodynamically defined zones where the physical-chemical conditions of the solution determine the phase of the protein. These zones are defined as undersaturated, saturated, metastable, labile, and precipitation. Undersaturated solutions will remain a single liquid phase. Saturation is a solution state where a crystal, if present, would remain in equilibrium with the surrounding solution. It is defined by the solubility curve on the phase diagram. If the same crystal were placed in slightly undersaturated conditions it would dissolve until the solution reached saturation. If the crystal was in a clear metastable solution (supersaturated), the protein would add to the surface of the growing crystal until the solution reached saturation. The two zones most relevant to crystallization are the metastable and labile zones. In the ideal case, solution parameters (commonly a precipitating agent, pH or temperature) drive a protein solution to the labile zone that is highly supersaturated, where the protein undergoes spontaneous, homogeneous nucleation. Once the nuclei form, the level of supersaturation can decrease to the metastable zone that will not produce additional crystal nuclei, but will support crystal growth. While a metastable state is thermodynamically supersaturated, kinetically there is an infinite lag time for crystallization. Finally, we have the precipitation zone, which is too supersaturated to support ordered aggregation and results in amorphous precipitate. Not shown on the typical phase diagram is the point of liquid-liquid phase separation. This occurs in the metastable zone at high protein concentrations. Each of these has different weightings on their influence. The phase diagram is typically presented as a two-dimensional plot with the major factors influencing outcome as the axes. In reality it is a multidimensional sampling of phase space and the two-dimensional representation is a slice through this space. Consequently, there can be considerable variability influenced by apparently small changes. Empirical approaches currently offer the best methods of probing the phase diagram.
What does the phase diagram tell us about crystallization? Crystals form under solution conditions that fall between those producing clear drops (solution conditions where protein-solvent interactions are stronger than protein-protein interactions) and those producing precipitate (solution conditions where protein-protein interactions are stronger than protein-solvent interactions). The phase diagram offers a perspective to help interpret the crystallization outcomes by providing a vector between these two outcomes. There is a region where protein-protein interactions are weakly attractive, termed the crystallization slot, that was proposed and defined by light-scattering studies.13 The osmotic second virial coefficient (B22), as measured by static light scattering, is a thermodynamic parameter that reflects the magnitude and direction of two-body, dilute solution interactions. The ‘crystallization slot’ defines a range of B22 values that are slightly attractive and indicate an increase in the probability of successful crystallization. Crystals can appear outside of the crystallization slot, although it is less probable. An approximation of this crystallization slot, though far less rigorous than measured B22 values, can be implied through an empirically derived phase diagram. In a phase diagram, the region between those conditions that produce clear results (positive repulsive B22) and those that result in precipitation (negative attractive B22) have the highest probability of falling within a crystallization slot. As noted by Dumetz, et al., “Even the most economical methods … cannot match simple crystallization screens” in their ability to probe this crystallization slot.14 The graphical representation offered by the phase diagram sets boundaries for chemically defined regions with a higher probability to produce crystals. Results are easier to interpret in a non-subjective manner when crystallization outcomes are considered in terms of the second virial coefficient and represented graphically.
Phase diagrams in the context of understanding the influence of variables and identifying conditions close to the ‘crystallization slot’ provide a useful framework to rationalize and direct crystallization experiments. Fine-sampling of a protein’s solubility and precision concentrations of protein remaining in solution are not needed to produce a useable phase diagram. A few well-designed screening experiments are sufficient to make this simple sketch of a protein’s solubility as it relates to crystallization. We can make use of the phase diagram with observation of individual experiments, characterization of them over time and comparison with results in other chemical conditions, to drive a rational approach to successful crystallization. Microfluidic approaches have been ingeniously and successfully adapted to derive phase knowledge.15 We will focus our discussions on characterizing typical crystallization results, their relative location in an ideal phase diagram and using that knowledge to aid further crystallization experiments.
3. Characterizing the drop
The first step in the analysis of crystallization results is to classify the outcomes. While there are now many automated imaging systems a simple, stereomicroscope is second only to a pipette in its value to a crystallization laboratory. There is significant flexibility in regards to magnification, plane of focus, lighting and polarization; all extremely useful when trying to observe crystals. As an example, in Figure 2, two drops are shown imaged at different focal depths (a) and (b) and with different polarization settings (c) and (d). Focusing to image different depths revealed two sets of crystals, one set is sedimented on the bottom of the well, 2(a) and the other nearer the surface, 2(b). In 2(c) and 2(d) different polarization settings accent the crystals. Automated systems have their place when the number of experiments increases, but one has to remember that manual analysis can reveal results that may be missed in more automated systems. Whether using a microscope or automated imaging system, note taking and record keeping is critical. Electronic notes, even a simple Word document or Excel spreadsheet, make it possible to readily search for and analyze data.16 Similarly, a laboratory standard template sheet with reference images can help to consistently describe results especially with multiple projects. This can be important in cases where one sample may produce many potential leads and another only a few. Consistency in recording results allows one to identify multiple chemically distinct conditions in the former, while being realistic in the effort that is required to optimize the latter.
Figure 2.

Examples of initial imaging where changing focus within the drop shows crystals (a) at the bottom of the well in the lower part of the image and (b) near the surface in the upper part of the well as imaged. In (c) and (d) different polarization settings have been used for identical results clearly indicating crystalline nature.
There are a limited set of outcomes that can be clearly identified by eye from images of crystallization experiments. From our studies we have identified a set that includes clear, phase separation, precipitate, skin formation, crystals, and junk (lint, or other debris that inadvertently fall into experiments). To focus on the most common outcomes we defined the ten highest frequency single and combination outcomes for a group of 147,456 (96 proteins × 1536 cocktails) crystallization experiments; type 1 being clear, type 2 showing phase separation, type 3 with phase separation and precipitate, type 4 with phase separation and skin, type 5 with phase separation and crystals, type 6 with precipitate, type 7 with precipitate and skin, type 8 with precipitate and crystal, type 9 with a crystal and type 10 for anything that is undefined or possible contamination 17, 18, Figure 3. Although crystals, type 9, did not fall within the most frequent outcomes, they were included as an outcome definition as they are the eventual goal of crystallization screening. We have not subdivided or classified these further but there is certainly scope to do so. Below, we detail the interpretation of these outcomes in relation to the phase diagram.
Figure 3.

A representative example of typical crystallization screening outcomes with a description of those outcomes given in the text.
3.1 Clear drops
A clear drop is a single liquid phase that indicates the sample is completely in solution, within the limits of the imaging system, Figure 3, type 1. The solution could simply be undersaturated (away from crystallization conditions), but if chemical conditions are suitable for crystallization, then the system can be in a metastable supersaturated state. Although based upon outward appearances a clear drop in the undersaturated zone looks identical to a clear drop in the metastable zone, the two are decidedly different thermodynamic states. The former provides a boundary for crystallization optimization while the latter a central point for optimization. Clear drops in isolation provide limited information; undersaturated solutions have to be distinguished from metastable solutions. The simplest way of doing this is based upon their chemical proximity to conditions that crystallized (see section 4.1). Those conditions that are undersaturated will largely show clear drops in experiments that are closely related chemically. Those experiments that are at or near metastable conditions will often contain, precipitate, a liquid-liquid phase separation or even crystals in adjacent, closely related chemical conditions.
The metastable zone can be defined by its borders.19 The lower border is a loci of points called the solubility curve, painstakingly measured for a limited number of proteins 20, 21. In the metastable case the solution conditions are at equilibrium with a single morphology of a crystalline phase. The curve is thermodynamic and notwithstanding any chemical or physical changes the solution will remain stable with zero probability of nucleation and an infinite induction time for nucleation. The upper boundary of the metastable zone is set by a limit where the probability of a nucleation event is certain with zero nucleation lag time. Crossing the upper boundary you would experimentally observe instantaneous, spontaneous, homogeneous nucleation. This is defined as the supersolubility curve. If a crystallization experiment appears clear, and the chemical conditions are very similar to conditions in the labile zone, then it is likely that the drop is at or near metastable supersaturation. Seeding methods such as those described by Bergfors can be applied very effectively to crystallization experiments falling in this metastable zone.22
3.2 Liquid-liquid phase separation
Liquid-liquid phase separation is often seen as drops within drops, cloud-like patterns of liquid within the drop, or a similar appearance to a shaken bottle of oil and vinegar, the drop can even have a dimpled appearance similar to the surface of a golf ball. An example is shown in Figure 3, type 2. There are protein-rich and protein-poor liquid phases. Protein concentrations of 400mg/mL have been measured in the protein-rich phase, a concentration comparable to that found in crystals.23 Experimental and theoretical studies demonstrate the formation of immiscible liquid-liquid (L-L) phase separation in the metastable region of the phase diagram. This L-L phase separation occurs only where there are short range, and/or highly anisotropic interactions between protein molecules; further experimental evidence demonstrates this region is connected with conditions for growing crystals.24
When the temperature of crystallization is near or below the formation temperature of a metastable, immiscible L-L phase separation, at high levels of supersaturation, experimental data and numerical simulations support a two-step, non-classical nucleation process.24 In this mechanism a protein-rich liquid phase first forms. Nucleation takes place from this phase followed by initial growth of the nuclei sometimes into the protein-rich and other times into the protein-poor environment. Haas and Drenth24 suggest that this growth mechanism can lead to fewer crystal defects and more rapid crystal growth. In this case, molecules in the concentrated liquid protein phase surrounding the crystal are not driven to the surface of the crystal by diffusion; therefore, misaligned molecules can be more readily exchanged. Literature also supports that it is not the higher protein concentration within the coacervate droplets or the molecular fluidity that may initiate nucleation, but rather an interface effect between the dense liquid of high-protein concentration in the droplet and the immiscible surrounding liquid of low-protein concentration.25
When a L-L phase separation is observed, e.g. Figure 4(a), if one phase is protein-rich and the other protein-poor, then the system is very close to conditions that have the potential to produce crystals. If the protein contains tryptophan residues, then the presence of a protein-rich phase can be verified using UV fluorescence, Figure 4(b). Crystals will sometimes form from the dense liquid phase without intervention; Figure 4(c). As is the case with metastable conditions, this protein-rich immiscible liquid phase can be used for seeding.26 The other useful and effective option to induce crystal formation is to drive the system towards a higher level of supersaturation, the labile state, using temperature. The rationale for this approach is to increase the attraction between protein molecules by decreasing the temperature.27 However, this process will be dependent upon the solubility properties of the protein/solvent. Protein solubility is dictated by the combination of the protein and its chemical environment. The same protein can have increased solubility at higher temperatures in one chemical environment, and lower temperatures in a different chemical environment. If the protein/solvent is more soluble at higher temperatures and L-L phase separation is seen in the drop, then decreasing the temperature will drive the system towards a higher level of supersaturation. The opposite applies in cases where the protein/solvent exhibits retro-solubility, i.e. the protein is more soluble at lower temperatures. In this case the experiments would be moved to a higher temperature environment, or set up at a higher temperature in a replicate experiment.
Figure 4.

An illustration of liquid-liquid phase separation in (a) visible light and (b) under UV illumination and examples (c and d) where crystals are growing at the interface of the phase separation.
3.3 Precipitation
Precipitation is identified by a graininess appearing in the crystallization plate, Figure 3, types 4–8. This result indicates that the biochemical conditions have driven the protein sample into a state of supersaturation many times beyond the level required for growth of single crystals. Precipitation can be of two forms, bad (typically amorphous) or good (microcrystalline). Amorphous precipitate can be brownish in color, frequently has a skin on the drop, will not re-dissolve, will not absorb dye and will not act as a successful seed. This type of precipitation occurs due to non-native protein aggregation and is a subject of much interest in the food industry and of significant biomedical importance.28 Crystalline precipitation is protein aggregation where the chemical environment permits the native conformation to remain intact i.e. those likely to lead to a crystal with minimal optimization effort. Crystalline precipitates are patterned, can have a sandy appearance, typically but not always show bi-refringence (depending on the orientation and symmetry of the crystals), will re-dissolve, will absorb dye and can successfully act as seeds. In Figure 5, a number of examples are shown, (a and b) amorphous precipitate, (c) a good patterned precipitate (optimized to produce crystals), (d) microcrystalline precipitate with a sandy appearance, (e) a crystalline precipitate with the dye being taken up and (f) a mixture of amorphous precipitate and a protein crystal identified through its uptake of Chromotrope 2B(red) dye. In these examples, the advantage of color imaging is highlighted by the ability to clearly distinguish the red (Chromotrope 2B) and blue (methylene blue) color dyes that can be used to check that crystals are indeed protein.
Figure 5.

Examples of bad precipitates (a) and (b) - despite looking good, good precipitates (c–e), and (f) the presence of crystals growing in the presence of bad precipitate.
3.4 Skin formation
Skin formation is a thin layer of denatured protein that under the microscope can look similar to a layer of wrinkled plastic wrap on the surface of the drop. This skin can be removed from the drop using microtools, such as an acupuncture needle, and often has a consistency similar to a spider’s web which can be difficult to remove from fragile crystals. The skin is a form of interfacial adsorption of the protein onto the interface whether it is solution/oil or solution/air, or solution/surface such as the plastic and glass materials that typically support the protein drop; examples are shown in Figure 6. There is an induction period or lag-time involved in this process due to diffusive and convective transport as the protein begins to concentrate at the interface.29 For protein solutions with concentrations greater than 50 to 100 μg/mL, two orders of magnitude less than typical crystallization experiments, the lag-time is not resolved, having a time-scale faster than the experimental technique used for observation.30 Layers of protein molecules undergo conformational changes, proteins aggregate, form branches, and can produce a viscoelastic, gel-like network structure (skin) that is often an irreversible process permanently denaturing the protein. Both Wei31 and Beverung30 have proposed that hydrophobic residues on the surface or interior of the protein, and conformational stability mediate this process. As the protein molecules continue to unfold at the interface they seek energetically favorable conformations for hydrophobic and hydrophilic side chains. These changes in protein conformation exposed to the aqueous phase lead to interactions with proteins in the solution. A hypothesis by Beverung et al. 29 based on their studies of bovine serum albumin, β-casein, ovalbumin, wheat germ lipase, Pyrococcus furiosus glutamate dehydrogenase, and lysozyme, proposes that a protein’s calculated surface hydrophobicity and the instability index32 are directly related to strong, irreversible adsorption at a water/oil interface and the onset of gelation.30 Earlier research hypothesized that the presence of intramolecular disulfide bridges within the protein molecule (increasing the stability) may slow the formation of the viscoelastic film.32 This is consistent with the correlation between the stability of the protein and formation of skin. There are also anecdotal reports that suggest higher concentrations of reducing agents, such as dithiothreitol, may prevent the formation of intermolecular disulfide bonds between the solvent accessible free cysteines of neighboring proteins to help alleviate skin formation.
Figure 6.

Examples of skin formation within crystallization screening experiments. Typically skin formation is seen to start from the interface of the drop with a surface, e.g. the edge closest to the support from which the drop is sitting or hanging.
Oil has been used to reduce the formation of skin on the surface of protein crystallization drops for Pseudomonas aeruginosa AmiC.33, 34 Studies specific to the dynamics of adsorption at an oil-water interface make it clear that there is a difference between air-water and air-oil interfacial phenomena.35 These differences offer an orthogonal explanation beyond the kinetics of equilibration, for differences observed between microbatch-under-oil and vapor-diffusion crystallization experiments and provides a rationale based on interfacial phenomena for adding a thin layer of oil to support the drop, or surround the drop, to change the interfacial properties to support crystallization. Silicon grease36 and Fluorinert37 have been deposited onto a surface to float the crystallization drop to prevent crystals from adhering. While intended to ease removal of the fragile crystals that form on the substrate’s surface, these protocols also change critical interfacial properties which can affect crystallization. The Venn diagram of successful microbatch-under-oil versus successful vapor-diffusion crystallization experiments for the same set of proteins does not completely overlap38 (observed in our own studies). There are occasions, fortunately rare, when it has been impossible to reproduce crystallization leads from our microbatch-under-oil experiments using vapor diffusion methods. A remedy that has been successful for many of these cases has been to add a volume of the same oil used in the microbatch experiments to the surface of the protein drop in the vapor diffusion experiment. While this retards evaporation and equilibration of the drop we have proven that the effect is oil and not equilibration dependent by setting up identical batch experiments, where the vapor pressure of water over the reservoir and drop at the onset of equilibration were essentially identical. Crystals would only form when the oil was present.
3.5 Combination results
We have described outcomes in singular cases, but our top ten categories described at the beginning of this section include combinations of those outcomes. In our study to determine these outcomes we looked at results from two and four weeks after setting up the initial experiment. Had we chosen a few days, most would be clear and conversely, had we chosen several months, most would be precipitate. Crystallization is a dynamic process and conditions change. This is obvious in the case of vapor diffusion where the concentration of the crystallization drop is influenced by vapor loss or gain due to that of the reservoir solution. Changes in concentration, important to the phase diagram, also occur when protein comes out of solution either as a precipitate (amorphous or crystalline) or crystal. This is an effective reduction in protein concentration in solution and shifts the location on the phase diagram. The ten categories occurring most frequently can easily be explained by an appropriate trajectory on the phase diagram and the addition of time as a variable in the analysis (described later).
3.6 What to after observing a crystal hit
The best case scenario is to see a large and visually perfect crystal directly from screening experiments. This is rare, but it does happen. Unfortunately, visually beautiful crystals do not infallibly diffract X-rays, or diffract sufficiently for structural solution which can be a great source of frustration. The cause of this malady is a lack of short range order within the crystal. The solution to this problem can be as simple as more careful control of variables such as temperature and pH during crystallization. Short range order occurs over very small length scales and small changes in these variables can have a big influence. Another point to note is that when crystals fail to diffract X-rays under cryogenic conditions one must always consider if the cryoprotectant or cryocooling procedure is at fault. Crystals should always be studied at the temperature they were grown at before proceeding to cryocooling techniques. This is easy to say, but the ease of looping and cooling versus other methods of ambient mounting often makes cryocooling the first rather than last step of examining the resulting crystal. Ambient temperature diffraction experiments clearly identify cases where the cryopreservation process is sub-optimal and the root cause of loss of diffracting power. X-ray analysis of crystals before cryocooling will focus subsequent experimental efforts on improving cryopreservation protocols rather than crystallization conditions and save significant time, sample and aggravation.
Sometimes, despite onerous efforts, crystals that form under a set of chemical conditions are recalcitrant to optimization. When this is the case, it is often beneficial to move on to optimize crystals produced from a different set of chemical conditions if available. We have witnessed numerous cases where a researcher reports spending months, or even years trying to optimize a particular crystallization condition and fails to improve diffraction significantly, or is unable to produce crystals that diffract X-rays. Shifting efforts to a different crystallization condition or conditions is a rapid process and can help produce diffraction suitable for structural solution. Alternatively, if no alternative conditions producing hits are observed (an uncommon situation from our observations), molecular biology approaches or orthologs should be considered.
Studies suggest proteins that crystallize by ‘salting-out’ (typically the main precipitating agent being a salt at high concentration) will do so independent of their isoelectric point.39 In these cases fine-sampling pH around the pKa values of charged residues that promote the formation of salt bridges and polar crystal contacts is suggested as an effective strategy.40 An experimental approach by McPherson reported that the fine-screening of pH in 0.05 pH unit increments, for five different buffers at 277 and 295K was a successful strategy to optimize crystallization conditions for a number of different proteins.41
When crystals are of insufficient size, or appear poorly formed a simple grid screen, varying the pH and precipitant concentration for example, surrounding the original hit, can be an effective means to produce diffraction-quality crystals, Figure 7.42 Even a small change in the pH, or precipitant concentration can produce significant changes in the outcome of the experiments, changing the outcomes from a L-L phase separation to crystals, or precipitate.
Figure 7.

Even a slight change in the pH, or concentration of precipitating agent can produce a significant change in the experiments outcome. In the case of protein P6305, all of the drops contain 18%(w/v) PEG 400 with slight changes in the pH shifting the conditions from the precipitation zone (left), to the labile (center) and finally L-L phase separation of the metastable zone(right) of the phase diagram.
In the high-throughput scenario we use a process we have termed Drop Volume Ratio and Temperature or DVR/T to optimize an initial lead condition. 43 We scan a range of inverse volume ratios between the protein and crystallization cocktail solutions, gradually increasing the volume of protein in the drop while gradually decreasing the volume of cocktail, similar to the method used by Rayment et al. 44 By adding temperature we slice through the phase diagram over the most interesting region of the chemical space. Using this approach even if an optimized crystal does not result, it is possible to define the phase diagram with far greater fidelity distinguishing all, or most of the zones and enabling a finer iteration of the process, Figure 8.
Figure 8.

Results from a DVR/T optimization experiment. The numbers (1–16) indicate decreasing volumes of protein with increasing volumes of cocktail in the experiment drop. Each experiment is set up in replicate and incubated at five different temperatures as indicated in the figure. The central point, an equal volume ratio of protein to cocktail (ratio 8 at 23°C) replicates the screening experiment conditions that produced the initial crystals. The results of this experiment are a projection through the phase diagram with clear drops indicating undersaturated, saturated, or metastable conditions (red outline), with those conditions most likely to be at or near metastable bordering the labile zone (blue outline) where spontaneous homogeneous nucleation takes place and the precipitation zone (black outline). This empirical phase diagram is easily produced and can help to identify conditions for seeding and further optimization.
4. Results from multiple conditions
4.1 Chemical space mapping
Aristotle stated “The whole is more than the sum of its parts”, this is especially relevant for crystallization. By screening multiple chemical conditions and comparing the results between them we can begin to map the phase space by putting the results in a sensible chemical landscape. When we do this we can visualize how to drive the crystallization to optimal conditions. This is described in detail elsewhere45, 46 and is a specific design principle of the crystallization screening performed at the HTSlab. Rather than a single crystal hit, we strive to produce phase information enabling more detailed screening for recalcitrant samples and optimization strategies for those that give a hit. We utilize a 1536 cocktail screen with 916 of those conditions fall into two groups. Group 1, Salt/buffers and group 2, PEG/Salt (at low concentrations)/buffers) are both constructed using an incomplete factorial design47. We sample pH with a salt and PEG group buffered with 100mM concentrations of CAPS (10.0), TAPS (9.0), Tris (8.0), HEPES (7.5), Bis-Tris Propane (7.0), MES (6.0), sodium acetate (5.0), and sodium citrate (4.0). The 229 salt cocktails contain 33 different, highly soluble salts at ~ 30, 60, and 90% saturation. The 687 PEG cocktails include 5 different molecular weights of PEG, 20 KDa, 8 KDa, 4 KDa, 1 KDa and 400 Da combined with 36 salts at 100mM concentration. By scoring outcomes and mapping that data onto the incomplete factorial design, arranged in a chemically sensible layout, we can readily visualize the effects of different chemistries on the outcome and construct a rudimentary phase diagram, Figure 9. We gain a more complete understanding of crystallization through parallel implementation of incomplete-factorial based in-house screens and well-designed, more chemically diverse commercial screens. The 620 conditions from Hampton Research (Aliso Viejo, CA) include Silver Bullets™, Silver Bullets Bio™, PEG/Ion HT™, Crystal Screen HT™, Index™, Crystal Screen Cryo™ and Grid Screen Salt HT™. The combination of the incomplete factorial sampling of chemical space used in our in-house cocktails, coupled with a commonly used set of chemically diverse commercial screens, enables us to correlate our crystallization results to those reported by the general community.
Figure 9.

Chemical space mapping (bottom right) of crystallization outcomes can be used to construct a rudimentary phase diagram (left). The phase diagram displays thermodynamic relationships that exist between the outcomes and points toward significant variables, in this case pH, to direct subsequent crystallization trials.
5. Time as a crystallization variable
5.1 Time as it relates to equilibration, crystal nucleation and crystal growth
Crystallization is a process that takes time, and is dependent on the biochemical environment; this can either be an incredibly slow, or surprisingly rapid event. In our HTSlab we typically image weekly over a period of 6 weeks, representing a compromise between duration, sampling and storage capacity. There are distinct advantages to both capturing static images and slower, dynamic microscopic observation. In the fastest cases, crystals can appear minutes after the protein and cocktail are combined - rapid nucleation and growth occurs. As an example, canavalin crystals can have growth rates that reach 5000Å per minute, with crystals reaching full size in only 3 hours.48 In our own laboratory we have observed similar growth rates with a truncated tRNA synthetase. While the full-length tRNA synthetase took weeks to crystallize, a truncated form of the enzyme crystallized in minutes. A compromise has to be made between imaging intervals, instrumentation able to perform the imaging and the number of samples in the queue. In a situation with few samples, a more appropriate approach would be initial shorter periods of observation followed by observation at increasing time intervals as the experiment progresses matching the observation to the kinetics. An ideal approach would take into account the equilibration time of the individual crystallization cocktails but this proves difficult to accomplish in practice.
Our own experiments make use of the micro-batch under oil crystallization technique where the conditions are immediately at equilibrium. Vapor-diffusion experiments, the most common crystallization technique, are designed to dehydrate and increase the relative concentrations of non-volatile solutes in the experiment drop. The time required to equilibrate the drop can be on the scale of 24 hours for drops and reservoirs having concentrated salts and on the scale of weeks for drops and reservoirs with a low ionic strength. Slow equilibration times often result when polyethylene glycol is used as a precipitant with mM concentrations of salts.49, 50 Knowledge of the kinetics of equilibration can be used to devise more efficient observation schedules. These kinetics can be exploited by use of diffusion based optimization techniques that increase or decrease the rate of equilibration.51
The time required for the first observable crystals to form is important and relevant data for optimization. If crystals appear immediately after adding protein to cocktail then the system is at labile supersaturation from the onset. If crystals are observed shortly after the initial set up, equilibration was not required to drive the system to a state of labile supersaturation. Regardless of the crystallization method, the experiment is essentially a batch experiment. What does this mean in terms of optimization? If the crystals are reproducible and of sufficient diffraction quality to answer the biochemical question at hand, then there is no need to change any parameters or think further about the crystallization. If however, there is difficulty reproducing the crystals, experiments set up under seemingly identical conditions produce outcomes that are often microcrystalline, or otherwise unsuitable for structural solution, then it is beneficial to consider the rapid onset of supersaturation. Decreasing the starting concentration of the precipitating agent, or the protein may prove beneficial, especially for vapor-diffusion experiments, where this would provide a starting point for the experiment that is not sufficiently supersaturated for crystallization at the onset, but rather approaches supersaturation during equilibration with the reservoir solution as the experiment drop dehydrates.
5.2 Delayed crystallization
When a crystallization event takes several months, long after equilibration takes place, it can be caused by chemical or physical changes to the protein.52 Recognized causes of delayed onset crystallization are limited in situ proteolysis, chemical changes such as the loss of bound metal ion or cofactor, or a change in the protein’s conformation. Second, the experiment drop can simply become more concentrated with respect to non-volatile components, the drop volume decreases as volatile components evaporate while the solutes that remain in the smaller volume increase in relative concentration. This will, under the appropriate biochemical conditions, lead to supersaturation, nucleation and crystal growth. Most plastics, especially polystyrene, the material from which many crystallization plates are fabricated, are water permeable.53 No matter how good a seal is used on the plate, unless stored in a humid environment, water will slowly evaporate through the plastic plate, or plastic seal, to simultaneously concentrate the drop and reservoir solutions. The relative change in concentration per unit volume of water loss will be more significant at lower volumes. This should not be considered detrimental, but it is something that one should be aware of when trying to reproduce crystal hits from plates that have been incubated at room temperature for extended periods of time. The evaporative loss is mitigated at lower temperatures, e.g. 4°C, where the vapor pressure of the solution will be lower, and by storing the plates in a controlled environment having high-relative humidity. In an extreme case, depending on the local environmental control, it may make a difference whether an experiment is set up during the winter or summer months.
Reproducing a crystal hit that appears shortly after adding protein to the cocktail can require a different tact than the approach used to reproduce a crystal that is not observed until six weeks after adding the protein to the cocktail. In the latter case, consideration of the factors described above can be critical for successful replication of the crystals. The availability of kits for in-situ proteolysis, such as the Proti-Ace (Hampton Research, Aliso Viejo, CA) can shorten the time required to reproduce crystal hits that will appear only after extended incubation and may require the consideration of extenuating circumstance (variables) to effectively reproduce the hit.
5.3 Ostwald’s rule of stages and Ostwald ripening
In some cases amorphous precipitate may be observed, followed over time by a small crystal which slowly grows while the precipitate recedes. This is described by Ostwald’s rule of stages which simply states, “When leaving a given state and in transforming to another state, the state which is sought out is not the thermodynamically stable one, but the state nearest in stability to the original state.” 54 In terms of crystallization, this means that the least soluble solid state will be the first to come out of solution. The next form to appear is not necessarily the most thermodynamically stable, but rather the form that is closest in energy to the first material to phase separate from the solution. This process continues, with a series of intermediate metastable forms, whose appearance is dependent on kinetics and not solely thermodynamics, until the formation of the most thermodynamically stable state, the form with the lowest Gibb’s free energy. Eloquently stated by Threfall, “The very existence of different forms at a given temperature is proof of the triumph of kinetics over thermodynamics”.55 Another common example is when different morphologies of a crystalline protein are observed in a single drop where they can co-exist for some time. However over time, one crystal form, the most thermodynamically stable form, will increase in size at the expense of the other. Ostwald’s rule of stages is not the same as Ostwald ripening. Ostwald’s rule of stages transitions between different states to decrease the free energy of the system, while Ostwald ripening will decrease the surface free energy of a system of single small crystals through mass-controlled transport to larger crystals of the same form.
Evidence for Ostwald ripening or coarsening can also be seen through periodic recording of experimental outcomes. First order phase transitions involve a latent heat; crystallization is such a transition. These transitions have a kinetic component and take place over three stages. The nucleation stage typically produces a narrow size distribution of a large number of particles. This is followed by the second phase, “dispersed cluster growth by monomer deposition”,56 where the nuclei will increase rapidly in size; however, the system has not yet reached equilibrium. The third stage, Ostwald ripening, minimizes the surface free energy.57 Smaller clusters are less stable than their larger counterparts. They will dissolve and through a diffusional mass transfer process become part of the larger clusters, Figure 10. Ostwald ripening can take place in a solution having two liquid phases, or in this example a solution having a single crystalline phase where the smaller crystals are depleted to feed the energetically favored larger crystals. Evidence to support Ostwald ripening will include the initial formation of small crystals or microcrystalline precipitates (which can often be verified by examination with cross-polarized filters to observe birefringence), a circular depletion zone of the microcrystals which forms concentrically around the single, larger crystal and conditions.58 To verify Ostwald ripening over effects described by Ostwald’s rule of stages, one would have to determine that the microcrystals differ only in size from the macrocrystals.
Figure 10.

An example of either Ostwald ripening, or Ostwald’s rule of stages. We can’t readily verify ripening without evidence that the precipitate is microcrystalline and of the same morphology as the larger crystals. Experimental images shown at 1 hour, 2.5 hours and 48 hours left to right. The precipitate which was not verified to be microcrystalline, has re-dissolved and been incorporated into the larger crystals, demonstrating either Ostwald ripening, or Ostwald’s rule of stages.
6. Temperature
In a study of 30 randomly selected, commercially available proteins 86% showed a temperature-dependent solubility.59 When crystallization plates are stored or left on an illuminated microscope stage without proper temperature control, crystals can dissolve and crystallization outcomes can become irreproducible; temperature variations of even a few degrees Celsius can cause crystals to appear and then disappear from the drop.60 Protein solubility is most sensitive to temperature under conditions of low ionic strength.61
Temperature can affect crystallization in ways that can go unnoticed by the observer. An atomic force microscopy study of growing tRNAPHE crystals demonstrated that in a 5°C range of temperature there were three different growth mechanisms.62 The way molecules attach themselves to the surface of a growing crystal can affect the quality of the crystal.63 When crystals are poorly diffracting, it can be beneficial to set up the experiments over a range of temperatures and to keep in mind that visual observations, the size or number of crystals, does not necessarily relate to diffraction quality.
7. Differences in crystallization methods
The crystallization method affects the experiment’s outcome. The method can dictate both thermodynamic and kinetic components of the experiment including, the starting and equilibrium concentrations of the solutes, the rate of equilibration and the path taken through the phase diagram. As mentioned previously the experiments set up in the HTSlab and therefore the majority of our experience comes from the microbatch-under-oil technique. Unlike vapor diffusion where the initial crystallization conditions fall somewhere along the equilibration path, microbatch experiments sample a single point; this allows us to accurately understand the biochemical conditions leading to a result. The majority of crystallization lead conditions generated by our laboratory are translated from batch to vapor diffusion experiments by our users. One way to understand how to translate between crystallization methods is to compare the different paths taken by each method through an idealized phase diagram as demonstrated in Figure 11.
Figure 11.

An example of idealized phase diagrams comparing the paths taken by comparable batch and vapor diffusion experiments. The starting concentration of the protein is 10mg/ml and the starting concentration of the crystallizing agent is 2M salt. We will use equal volumes of protein and crystallizing agent to form the experiment drop. For the batch experiment the protein and cocktail concentration are one half the starting concentrations of the stock solutions after initial set up due to co-dilution, 5mg/ml for the protein and 1M for the salt. The batch container is sealed to prevent evaporation. Therefore, the chemical concentrations in the drop will remain constant until there is a change of phase. The experiment has to be set up in a supersaturated state that ideally would fall in the labile zone where spontaneous, homogeneous nucleation can occur. As protein phase separation takes place in the form of a solid (crystal in this idealized example) the solution conditions will reach equilibrium when the solution conditions fall on the solubility curve. Compare this to a vapor diffusion experiment. The experiment drop will initially match that of the batch experiment due to co-dilution of the protein and salt solutions during initial set up of the experiment. In the case of vapor diffusion experiments, the drop will dehydrate as it equilibrates through the vapor phase with a reservoir solution. This increases the relative concentrations of solutes in the experiment drop. Following the idealized path the experiment will supersaturate and cross the supersolubility curve as it enters the labile zone. Spontaneous, homogeneous nucleation takes place. The level of supersaturation decreases as protein comes out of solution during crystal growth. Equilibration is reached where the solution conditions are on the solubility curve.38, 64
8. Salt or protein
Protein crystals are frequently produced from complex chemical environments. There can be interactions between the chemicals required to stabilize the protein and the chemicals in the cocktails. The formation of inorganic crystals does not preclude the formation of protein crystals in same drop. The complex chemical environments, covering a broad range of pH, temperature, chemical additives are very likely to produce false positives, crystals that are not proteins. There number of methods available to verify crystals are protein. These methods are summarized in table Oftentimes these methods do not provide absolute confirmation as they all have caveats and at best a case towards the crystal’s composition. It is important to be aware of the strengths and weaknesses each method. In the case of dyes, protein crystals will not always absorb dye. When dye absorption take place it can happen quickly or take several hours. Dye crystals can appear in the drop. Adding can initiate crystallization of the protein. When using SDS-PAGE analysis it is possible to carry-protein precipitate, or liquid phase on the salt crystals which can lead to false positives. UV will be ineffective unless the sequence contains tryptophan residues. Again, protein liquid phase concentrate on inorganic crystals and lead to difficulty interpreting the results. Very small crystals difficult to mechanically crush and very small inorganic crystals can be fragile. Birefringence, the refraction of light, is a property of anisotropic crystals. Birefringence is not a property of isotropic such as glass, nor is it a property of isotropic crystals which have equivalent crystallographic Birefringence under crossed-polarization is therefore dependent upon both the symmetry and of the crystals. It can be difficult to see evidence of birefringence for small protein crystals, most plates and sealing films will rotate polarized light, making interpretation difficult unless crystals removed to a glass, or low-birefringence plastic. When verifying the composition of crystals, destructive methods should generally be attempted before destructive methods, especially when are in limited supply. The best method to distinguish protein from salt crystals is by X-ray diffraction. This the only way to quantify the diffraction properties of the resulting crystals and to be certain the are of the intended target. If you only have one crystal, try the non-destructive methods first, and then X-ray diffraction.
9. Discussion and conclusions
In this paper we have tried to demonstrate that the outcomes of crystallization experiments should not be thought of individually, nor as a binary result i.e. crystal or no crystal. Information derived from crystallization screening outcomes can provide a guide to the protein’s solubility that compares favorably to B22 measurements provided by static light scattering experiments. There is significantly more to be learned from crystallization screening outcomes than which condition or conditions to optimize. When these outcomes are considered in the context of a phase diagram they increase the chances of crystallization success and direct the optimization of initial hits into crystals suitable for diffraction.
The phase diagram is unique for each protein. The examples presented in this paper are simplified, two-dimensional schematics. The actual response of the protein to a multi-parametric biochemical environment is multi-dimensional. Almost two decades ago it was estimated that there were at least 23 parameters that can affect crystallization65, some factors having more influence than others. The coverage of these parameters is limited experimentally. From a practical perspective, we can only explore and comprehend a finite number of variables. We can visualize complex experimental results by examining subsets of variables. When we consider the outcomes of the crystallization experiments globally, in terms of how outcomes are positioned in biochemical and biophysical space, we develop an empirical picture of the phase diagram. From crystallization theory we can then predict the regions where crystallization is more probable and derive appropriate experiments to probe these regions. We can also use the phase diagram to understand the relative placement of outcomes with respect to both chemical conditions and other outcomes and with this knowledge alter the environment to conditions that are more favorable for crystallization. A seemingly insignificant shift in chemical conditions or temperature can transform a metastable clear drop into a crystal. The problem is one of scale. There are often many clear drops produced by screening experiments. Individually, we can’t see the difference between an undersaturated clear drop and a metastable clear drop. Simultaneous, global consideration of the experimental outcomes helps to distinguish undersaturated from metastable conditions which efficiently and rationally directs subsequent crystallization trials.
Our initial data is based on visual observation. This is adequate to obtain a crystallization lead, a condition that can be optimized to produce diffraction ready crystals. It is impossible through visual inspection to know how well crystals will diffract X-rays, or if the crystals will diffract at all. An empirical knowledge of the protein’s phase diagram provides data that can determine the sensitivity of the protein’s solubility to specific biochemical conditions. As an example, if a protein is very sensitive to pH, a small change in pH causes precipitation, fine-sampling of pH may be beneficial. Chemically distinct crystallization conditions can be exploited to optimize and obtain structural information of the different crystal forms. Crystals of the same protein produced from different biochemical conditions will vary in their physical properties such as solvent content, fragility, ease of cryopreservation, and X-ray diffraction. In cases where poor diffraction is observed, diffraction can often be improved with alternative crystallization conditions, which are frequently less labor intensive than molecular biology approaches but which can be equally effective. We use 1536 different chemical conditions with the majority derived from an incomplete factorial sampling. In addition to identifying a promising lead, this sampling provides additional information on the response of the protein to a wide range of chemical space. We require 5mg of protein to set up 1536 crystallization screening experiments. We observe an initial crystallization lead for ~50% of samples; oftentimes there are multiple chemically distinct conditions that produce leads. With a smaller sampling of chemical space, either in breadth or in fidelity, these leads may not be observed. A tradeoff exists between coarse and fine sampling of chemical space. We want to sample finely enough, over a wide range of chemical space to both get a lead, and to provide information we can use to map a phase diagram. If we sample inadequately, we will lose this information. Crystallization leads can be obtained with far fewer than 1536 biochemical conditions. A study by Page et al.66 on 1,539 different soluble purified proteins from the T. maritima proteome gave crystals for 465 of these proteins using a 480 condition screen. For those that crystallized a subset of only 67 of these conditions would have produced crystals in 86% of these cases. Clearly crystallization can be accomplished with a smaller sampling of chemical space albeit at the expense of information on the protein phase diagram. For a limited number of samples an appropriate approach would be pre-screening the sample to get an idea of the precipitate/clear boundary region. A finer sampling of conditions near this border region could identify hits which would be followed by optimization. In a high-throughput laboratory this is possible but would benefit from the development of accurate image analysis, the use of formulation robotics and comprehensive feedback within the pipeline. This is a current area of research within our group.
Time is an important and useful variable in the context of crystallization. The kinetics of equilibration for vapor diffusion experiments and their affect on crystallization outcomes have been well documented.67 Most successful crystallization experiments start with a purified, soluble sample and after a period of time end with a crystal. Crystallization is a time-dependent process. Changes that take place over time as the experiment equilibrates add a kinetic component to our empirical understanding of the phase diagram. This contributes to our knowledge and increases the probability of successful optimization. We noted that image analysis is a requirement to perform these studies in high-throughput. Using data from our own crystallization efforts, automated classification has been developed that correctly identifies 98% of all clear drops, and 89% of all precipitate only images.68 It is also correct in identifying 80% of crystal cases. While this accuracy may seem to have an unacceptably high failure rate, missing too many crystals, it is actually somewhat better than the human case where 78% of images are correctly and consistently identified69. However, the bad news is that this image analysis study required the computational resources of the World Wide Computing Grid - processing time was intensive. The power of computational capability is increasing daily and this is a task well suited to multi-processer computing. In the near future we see the addition of the time component into the analysis to improve the accuracy and identification of crystal cases by focusing more computational resources on those images that show significant changes over time, versus those that remain unchanged, i.e. those conditions that are well into the undersaturated region or precipitation region of the phase diagram. Notation of the time when a change is first observed, and a description of the outcome gives a temporal location in the phase diagram and provides kinetic information for subsequent experiments. Time is also important in those samples that may produce crystals through Ostwald-related processed (section 5.5) and proteins that change during the course of the experiment through in situ proteolysis, oxidation or reduction.
Crystallization is a stochastic process and there will be variability in outcomes in apparently identical experiments. Perhaps the best example of this was an investigation of redundancy in the vapor diffusion crystallization of lysozyme by Newman et al.70 Two studies were conducted, one replicating 4 crystallization conditions 96 times with two different lysozyme concentrations and the other using the Hampton Research HT crystallization screen replicating it 28 times with two different lysozyme drop volumes (and screening at two different temperatures). The results were illuminating and showed a significant difference in crystallization results between plates. Within our own crystallization facility we sample a large number of conditions in a comprehensive manner. We can relate results to each other and identify trends for further investigation that may have been missed without requiring oversampling or a coarser screen. Given the stochastic nature of crystallization, if a condition is identified that is close to a metastable region (by interpretation of conditions around it) then experiments should be repeated around this condition.
Unfortunately, a salt or PEG crystal is a potential outcome in any crystallization experiment. A variety of methods are available to distinguish protein crystals from artifacts that form in the experiment drop, but when tryptophan is present in the amino acid sequence the most promising method appears to be UV florescence.71 Another emerging technology, i.e. second-order non-linear imaging of chiral crystals (SONICC), shows tremendous promise.72 We believe that we are close, if not already at the point where non-protein crystals can be identified in the initial screening, well before optimization, and X-ray analysis is attempted. These systems can be expensive for individual laboratories and a variety of other less costly, but highly effective techniques are available, table 1.
Table 1.
Methods for identifying salt crystals
| Properties | Protein crystals | Inorganic crystals | Notes |
|---|---|---|---|
| If identical crystals are in the reservoir | No | Yes | Good control observation |
| If identical crystals appear in a control experiment that does not contain the protein | No | Yes | A good control experiment to setup |
| If crystals dehydrate upon exposure to air | Likely | Unlikely | Protein crystals are typically ~30–70% water |
| If crystals can be readily crushed by mechanical stress | Likely | Unlikely | Protein crystals are held together with weak interactions |
| If the crystals absorb dye | Likely | Unlikely | Dye can diffuse into protein crystal’s solvent channels |
| If washed crystals give an SDS-PAGE band at the expected MW | Likely | Unlikely | Useful check to run if enough sample is available |
| If washed crystals give an SDS-PAGE band at a different MW | Likely | Unlikely | Possible contamination or proteolysis, proceed with caution |
| If there is weak birefringence under crossed-polarization | Likely | Unlikely | Cubic systems will not show this Inorganic crystals extinguish rapidly |
| If there is fluorescence at 280nm | Likely | Unlikely | If tryptophan is present, some inorganic chemicals can fluoresce |
| If there is no X-ray diffraction | Likely | Unlikely | Some inorganic crystals may not diffract X-rays |
| If X-ray diffraction shows a few well spaced reflections | Unlikely | Likely | Characteristic of salt diffraction |
A potentially exciting problem for crystallizers is the introduction of microfocus beamlines and further down the way, the routine use of 4th generation X-ray sources for crystallography. On existing synchrotron beamlines data can be collected from crystals on the order of 5 μm in size. It is technically feasible to collect a complete X-ray data set from a crystal on the order of 1–2 μm in size.73 At 4th generation sources, e.g. the LCLS, X-ray data is likely to be collected on crystals fractions of this size. The problem for the crystallizer becomes knowing that crystallization has succeeded. Observation of these crystals is difficult and technically challenging. The standard microscope in a laboratory is more suited to imaging the entire crystallization drop rather than focusing in on a single 10 micron crystal. Imaging these miniscule crystals poses a technical challenge that we have to face. However, it is likely that the reduction in required crystal volume will result in samples that are physically more perfect and easier to cryoprotect.74 Less and perhaps even no optimization may be needed beyond the initial crystal hit as beamline technology development makes routine the handling of micron-sized crystals.
In summary, it is possible to obtain an empirical picture of the crystallization phase diagram by relating crystallization outcomes and biochemical environments together to provide a global perspective of the protein’s solubility. The chemical, physical and theoretical explanations for each type of outcome are available and can rationally guide the crystallization process. Time is a critical element of the crystallization experiment. To make full use of the information provided by these experiments requires careful note taking and the representation of results in a manner that allows simple human interpretation. As a final word we would point out that many unintended variables are easily and often invisibly added to crystallization experiments; these are sometimes difficult to determine because they go unnoticed and play a lead role in the ‘art’ misnomer that seems to be inextricably tied to protein crystallization75; when crystallizing proteins, everything matters.
Acknowledgments
JRL and EHS would like to acknowledge NIH for funding support through U54 GM074899 and R01 GM088396. Dr. George DeTitta and members of the crystallization screening laboratory are acknowledged for their tireless effort in maintaining an efficient laboratory and generating may of the images examined in putting this paper together. The many users of the screening laboratory are also thanked in particular Guy Montelione from the NESG for access to detailed information about all of the samples run through the screening laboratory.
Footnotes
Suggested referees:
Abraham Lenoff, lenhoff@udel.edu
Terese Bergfors, terese@xray.bmc.uu.se
Alexander McPherson, amcphers@uci.edu
Neer Asherie, asherie@yu.edu
Allan D’Arcy, allan.darcy@novartis.com
References
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171(4356):737–8. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 3.Hodgkin DC, Kamper J, MacKay M, Pickworth JJHR, Shoemaker CB, White JG, Prosen RJ, Trueblood KN. The Structure of Vitamin B12 I. An Outline of the Crystallographic Investigation of Vitamin B12. Proc R Soc Lond A. 1957;242(1229):228–263. [Google Scholar]
- 4.Deisenhofer J, Epp O, Miki K, Huber R, Michel H. X-ray structure analysis of a membrane protein complex. Electron density map at 3 A resolution and a model of the chromophores of the photosynthetic reaction center from Rhodopseudomonas viridis. J Mol Biol. 1984;180(2):385–98. doi: 10.1016/s0022-2836(84)80011-x. [DOI] [PubMed] [Google Scholar]
- 5.Abrahams JP, Leslie AG, Lutter R, Walker JE. Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370(6491):621–8. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 6.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 7.Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292(5523):1863–76. doi: 10.1126/science.1059493. [DOI] [PubMed] [Google Scholar]
- 8.Schlunzen F, Hansen HA, Thygesen J, Bennett WS, Volkmann N, Levin I, Harms J, Bartels H, Zaytzev-Bashan A, Berkovitch-Yellin Z, et al. A milestone in ribosomal crystallography: the construction of preliminary electron density maps at intermediate resolution. Biochem Cell Biol. 1995;73(11–12):739–49. doi: 10.1139/o95-082. [DOI] [PubMed] [Google Scholar]
- 9.Chen L, Oughtred R, Berman HM, Westbrook J. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20(16):2860–2. doi: 10.1093/bioinformatics/bth300. [DOI] [PubMed] [Google Scholar]
- 10.Chayen NE, Stewart PDS, Maeder DL, Blow DM. An Automated-System for Microbatch Protein Crystallization and Screening. Journal of Applied Crystallography. 1990;23:297–302. [Google Scholar]
- 11.Luft JR, Collins RJ, Fehrman NA, Lauricella AM, Veatch CK, DeTitta GT. A deliberate approach to screening for initial crystallization conditions of biological macromolecules. Journal of Structural Biology. 2003;142(1):170–179. doi: 10.1016/s1047-8477(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 12.Green AA. Studies in the physical chemistry of the proteins. IX. The effect of electrolytes on the solubility of hemoglobin in solutions of varying hydrogen ion activity with a note on the comnparable behavior of casein. Journal of BIological Chemistry. 1931;93:517–542. [Google Scholar]
- 13.Wilson WW. Light scattering as a diagnostic for protein crystal growth - A practical approach. Journal of Structural Biology. 2003;142(1):56–65. doi: 10.1016/s1047-8477(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 14.Dumetz AC, Snellinger-O’Brien AM, Kaler EW, Lenhoff AM. Patterns of protein - protein interactions in salt solutions and implications for protein crystallization. Protein Science. 2007;16(9):1867–1877. doi: 10.1110/ps.072957907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson MJ, Hansen CL, Quake SR. Phase knowledge enables rational screens for protein crystallization. Proc Natl Acad Sci U S A. 2006;103(45):16746–51. doi: 10.1073/pnas.0605293103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hannick LI, Perozzo MA, Schultz LW, Ward KB. A Pc-Based Spreadsheet for Tracking Results of Crystallization Experiments. Journal of Crystal Growth. 1992;122(1–4):303–305. [Google Scholar]
- 17.Snell EH, Luft JR, Potter SA, Lauricella AM, Gulde SM, Malkowski MG, Rosenblum M, Said MI, Smith JL, Veatch CK, Wolfley JL, Collins RJ, Franks G, Thayer M, Cumbaa C, Jurisica I, DeTitta GT. Establishing a training set through the visual analysis of crystallization trials part I: ~150,000 images. Acta Cryst D. 2008;64:1123–1130. doi: 10.1107/S0907444908028047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snell EH, Lauricella AM, Potter SA, Luft JR, Gulde SM, Collins RJ, Franks G, Malkowski MG, Cumbaa C, Jurisica I, DeTitta GT. Establishing a training set through the visual analysis of crystallization trials. Part II: crystal examples. Acta Crystallogr D. 2008;64:1131–1137. doi: 10.1107/S0907444908028059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Ruiz JM. Nucleation of protein crystals. Journal of Structural Biology. 2003;142(1):22–31. doi: 10.1016/s1047-8477(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 20.Forsythe EL, Judge RA, Pusey ML. Tetragonal chicken egg white lysozyme solubility in sodium chloride solutions. Journal of Chemical and Engineering Data. 1999;44(3):637–640. [Google Scholar]
- 21.Rosenberger F, Howard SB, Sowers JW, Nyce TA. Temperature-Dependence of Protein Solubility - Determination and Application to Crystallization in X-Ray Capillaries. Journal of Crystal Growth. 1993;129(1–2):1–12. [Google Scholar]
- 22.Bergfors T. Seeds to crystals. Journal of Structural Biology. 2003;142(1):66–76. doi: 10.1016/s1047-8477(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 23.Kuznetsov YG, Malkin AJ, McPherson A. The liquid protein phase in crystallization: a case study - intact immunoglobulins. Journal of Crystal Growth. 2001;232(1–4):30–39. [Google Scholar]
- 24.Haas C, Drenth J. The interface between a protein crystal and anaqueous solution and its effects on nucleation and crystal growth. Journal of Physical Chemistry B. 2000;104(2):368–377. [Google Scholar]
- 25.Vekilov PG. Dense liquid precursor for the nucleation of ordered solid phases from solution. Crystal Growth & Design. 2004;4(4):671–685. [Google Scholar]
- 26.Bergfors T. Seeds to crystals. J Struct Biol. 2003;142(1):66–76. doi: 10.1016/s1047-8477(03)00039-x. [DOI] [PubMed] [Google Scholar]
- 27.Dumetz AC, Chockla AM, Kaler EW, Lenhoff AM. Protein phase behavior in aqueous solutions: Crystallization, liquid-liquid phase separation, gels, and aggregates. Biophys J. 2008;94(2):570–583. doi: 10.1529/biophysj.107.116152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krebs MRH, Devlin GL, Donald AM. Protein particulates: Another generic form of protein aggregation? Biophysical Journal. 2007;92(4):1336–1342. doi: 10.1529/biophysj.106.094342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ybert C, di Meglio JM. Study of protein adsorption by dynamic surface tension measurements: Diffusive regime. Langmuir. 1998;14(2):471–475. [Google Scholar]
- 30.Beverung CJ, Radke CJ, Blanch HW. Protein adsorption at the oil/water interface: characterization of adsorption kinetics by dynamic interfacial tension measurements. Biophys Chem. 1999;81(1):59–80. doi: 10.1016/s0301-4622(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 31.Wei A-P, Herron JN, Andrade JD. The role of protein structure in surface tension kinetics. In: Crommelin DJA, Schellekens H, editors. From Clone to Clinic. Kluwer Academic Publishers; The Netherlands: 1990. pp. 305–313. [Google Scholar]
- 32.Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4(2):155–61. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- 33.Wilson SA, Chayen NE, Hemmings AM, Drew RE, Pearl LH. Crystallization of and preliminary X-ray data for the negative regulator (AmiC) of the amidase operon of Pseudomonas aeruginosa. J Mol Biol. 1991;222(4):869–71. doi: 10.1016/0022-2836(91)90579-u. [DOI] [PubMed] [Google Scholar]
- 34.Pearl L, O’Hara B, Drew R, Wilson S. Crystal structure of AmiC: the controller of transcription antitermination in the amidase operon of Pseudomonas aeruginosa. EMBO J. 1994;13(24):5810–7. doi: 10.1002/j.1460-2075.1994.tb06924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maldonado-Valderrama J, Fainerman VB, Aksenenko E, Galvez-Ruiz MJ, Cabrerizo-Vilchez MA, Miller R. Dynamics of protein adsorption at the oil-water interface: comparison with a theoretical model. Colloids and Surfaces a-Physicochemical and Engineering Aspects. 2005;261(1–3):85–92. [Google Scholar]
- 36.Ray WJ, Puvathingal JM. The Use of Silicones in Protein Crystallizations Involving Single or Multiple Growth Cycles. Journal of Applied Crystallography. 1984 Oct;17:370–371. [Google Scholar]
- 37.Adachi H, Takano K, Morikawa M, Kanaya S, Yoshimura M, Mori Y, Sasaki T. Application of a two-liquid system to sitting-drop vapour-diffusion protein crystallization. Acta Crystallogr D. 2003;59:194–196. doi: 10.1107/s0907444902019741. [DOI] [PubMed] [Google Scholar]
- 38.Chayen NE. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallogr D. 1998;54:8–15. doi: 10.1107/s0907444997005374. [DOI] [PubMed] [Google Scholar]
- 39.Trevino SR, Scholtz JM, Pace CN. Measuring and increasing protein solubility. Journal of Pharmaceutical Sciences. 2008;97(10):4155–4166. doi: 10.1002/jps.21327. [DOI] [PubMed] [Google Scholar]
- 40.Dumetz AC, Chockla AM, Kaler EW, Lenhoff AM. Effects of pH on protein-protein interactions and implications for protein phase behavior. Biochimica Et Biophysica Acta-Proteins and Proteomics. 2008;1784(4):600–610. doi: 10.1016/j.bbapap.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 41.McPherson A. Increasing the Size of Microcrystals by Fine Sampling of Ph Limits. Journal of Applied Crystallography. 1995;28:362–365. [Google Scholar]
- 42.Cox MJ, Weber PC. An Investigation of Protein Crystallization Parameters Using Successive Automated Grid Searches (Sags) Journal of Crystal Growth. 1988;90(1–3):318–324. [Google Scholar]
- 43.Luft JR, Wolfley JR, Said MI, Nagel RM, Lauricella AM, Smith JL, Thayer MH, Veatch CK, Snell EH, Malkowski MG, Detitta GT. Efficient optimization of crystallization conditions by manipulation of drop volume ratio and temperature. Protein Science. 2007;16(4):715–722. doi: 10.1110/ps.062699707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rayment I. Small-scale batch crystallization of proteins revisited: an underutilized way to grow large protein crystals. Structure. 2002;10(2):147–51. doi: 10.1016/s0969-2126(02)00711-6. [DOI] [PubMed] [Google Scholar]
- 45.Nagel RM, Luft JR, Snell EH. AutoSherlock: a program for effective crystallization data analysis. J Appl Crystallogr. 2008;41(Pt 6):1173–1176. doi: 10.1107/S0021889808028938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Snell EH, Nagel RM, Wojtaszcyk A, O’Neill H, Wolfley JL, Luft JR. The application and use of chemical space mapping to interpret crystallization screening results. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 12):1240–9. doi: 10.1107/S0907444908032411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Audic S, Lopez F, Claverie JM, Poirot O, Abergel C. SAmBA: An interactive software for optimizing the design of biological macromolecules crystallization experiments. Proteins-Structure Function and Genetics. 1997;29(2):252–257. doi: 10.1002/(sici)1097-0134(199710)29:2<252::aid-prot12>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 48.Koszelak S, Mcpherson A. Time-Lapse Microphotography of Protein Crystal-Growth Using a Color Vcr. Journal of Crystal Growth. 1988;90(1–3):340–343. [Google Scholar]
- 49.Arakali SV, Luft JR, DeTitta GT. Non-ideality of aqueous solutions of polyethylene glycol: consequences for its use as a macromolecular crystallizing agent in vapor-diffusion experiments. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 5):772–9. doi: 10.1107/S0907444995000436. [DOI] [PubMed] [Google Scholar]
- 50.Luft JR, DeTitta GT. Chaperone salts, polyethylene glycol and rates of equilibration in vapor- diffusion crystallization. Acta Crystallogr D Biol Crystallogr. 1995;51(Pt 5):780–5. doi: 10.1107/S0907444995002277. [DOI] [PubMed] [Google Scholar]
- 51.Luft JR, DeTitta GT. Kinetic aspects of macromolecular crystallization. Methods Enzymol. 1997;276:110–31. [PubMed] [Google Scholar]
- 52.McPherson A. Current Approaches to Macromolecular Crystallization. European Journal of Biochemistry. 1990;189(1):1–23. doi: 10.1111/j.1432-1033.1990.tb15454.x. [DOI] [PubMed] [Google Scholar]
- 53.Massey LK. Permeability Properties of Plastics and Elastomers - A Guide to Packaging and Barrier Materials. 2. William Andrew Publishing/Plastics Design Library; 2003. p. 610. [Google Scholar]
- 54.Ostwald W. Studies upon the forming and changing solid bodies. Zeitschrift fur Physikalische Chemie. 1897;22:289–330. [Google Scholar]
- 55.Threlfall T. Structural and thermodynamic explanations of Ostwald’s rule. Organic Process Research & Development. 2003;7(6):1017–1027. [Google Scholar]
- 56.Madras G, McCoy BJ. Ostwald ripening with size-dependent rates: Similarity and power-law solutions. Journal of Chemical Physics. 2002;117(17):8042–8049. [Google Scholar]
- 57.Marqusee JA, Ross J. Kinetics of Phase-Transitions - Theory of Ostwald Ripening. Journal of Chemical Physics. 1983;79(1):373–378. [Google Scholar]
- 58.Ng JD, Lorber B, Witz J, TheobaldDietrich A, Kern D, Giege R. The crystallization of biological macromolecules from precipitates: Evidence for Ostwald ripening. Journal of Crystal Growth. 1996;168(1–4):50–62. [Google Scholar]
- 59.Christopher GK, Phipps AG, Gray RJ. Temperature-dependent solubility of selected proteins. Journal of Crystal Growth. 1998;191(4):820–826. [Google Scholar]
- 60.Cudney B. Protein Crystallization and Dumb Luck. The Rigaku Journal. 1999;16(1):1–7. [Google Scholar]
- 61.Jones WF, Wiencek JM, Darcy PA. Improvements in lysozyme crystal quality via temperature-controlled growth at low ionic strength. Journal of Crystal Growth. 2001;232(1–4):221–228. [Google Scholar]
- 62.Ng JD, Kuznetsov YG, Malkin AJ, Keith G, Giege R, McPherson A. Visualization of RNA crystal growth by atomic force microscopy. Nucleic Acids Research. 1997;25(13):2582–2588. doi: 10.1093/nar/25.13.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McPherson A. Crystallization of biological macromolecules. Cold Spring Harbor Laboratory Press; New York: 1999. pp. 218–219. [Google Scholar]
- 64.Luft JR, DeTitta GT. Rational Selection of Crystallization Techniques. In: Bergfors T, editor. Protein Crystallization. 2. International University Line; LaJolla: 2009. pp. 11–45. [Google Scholar]
- 65.Giege R, Ducruix A. An introduction to the crystallogenesis of biological macromolecules. In: Ducruix A, Giege R, editors. Crystallization of Nucleic Acids and Proteins: A Practical Approach. Oxford University Press; New York: 1992. pp. 1–18. [Google Scholar]
- 66.Page R, Grzechnik SK, Canaves JM, Spraggon G, Kreusch A, Kuhn P, Stevens RC, Lesley SA. Shotgun crystallization strategy for structural genomics: an optimized two-tiered crystallization screen against the Thermotoga maritima proteome. Acta Crystallogr D. 2003;59:1028–1037. doi: 10.1107/s0907444903007790. [DOI] [PubMed] [Google Scholar]
- 67.Luft JR, DeTitta G. Kinetic aspects of macromolecular crystallization. Macromolecular Crystallography, Pt A. 1997;276:110–131. [PubMed] [Google Scholar]
- 68.Cumbaa CA, Jurisica I. Protein crystallization analysis on the World Community Grid. J Struct Funct Genomics. 11(1):61–9. doi: 10.1007/s10969-009-9076-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Snell EH, Luft JR, Potter SA, Lauricella AM, Gulde SM, Malkowski MG, Koszelak-Rosenblum M, Said MI, Smith JL, Veatch CK, Collins RJ, Franks G, Thayer M, Cumbaa C, Jurisica I, DeTitta GT. Establishing a training set through the visual analysis of crystallization trials. Part I: approximately 150,000 images. Acta Crystallogr D Biol Crystallogr. 2008;64(Pt 11):1123–30. doi: 10.1107/S0907444908028047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newman J, Xu J, Willis MC. Initial evaluations of the reproducibility of vapor-diffusion crystallization. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 7):826–32. doi: 10.1107/S0907444907025784. [DOI] [PubMed] [Google Scholar]
- 71.Judge RA, Swift K, Gonzalez C. An ultraviolet fluorescence-based method for identifying and distinguishing protein crystals. Acta Crystallogr D. 2005;61:60–66. doi: 10.1107/S0907444904026538. [DOI] [PubMed] [Google Scholar]
- 72.Kissick DJ, Gualtieri EJ, Simpson GJ, Cherezov V. Nonlinear Optical Imaging of Integral Membrane Protein Crystals in Lipidic Mesophases. Analytical Chemistry. 2010;82(2):491–497. doi: 10.1021/ac902139w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holton JM, Frankel KA. The minimum crystal size needed for a complete diffraction data set. Acta Crystallogr D Biol Crystallogr. 66(Pt 4):393–408. doi: 10.1107/S0907444910007262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chayen NE, Helliwell JR, Snell EH. Macromolecular Crystallization and Crystal Perfection. Oxford University Press; Oxford: 2010. [Google Scholar]
- 75.Chayen NE. Turning protein crystallisation from an art into a science. Current Opinion in Structural Biology. 2004;14(5):577–583. doi: 10.1016/j.sbi.2004.08.002. [DOI] [PubMed] [Google Scholar]