

Abstract
Subcellular fractionation of proteins is a preferred method of choice for detection and identification of proteins from complex mixtures such as bacterial cells. To characterize the membrane proteins of the Antarctic bacterium Pseudomonas syringae Lz4W, the membrane fractions were prepared using three different methods, namely Triton X-100 solubilization, sucrose density gradient, and carbonate extraction methods. The proteins were separated on one-dimensional polyacrylamide gels and analyzed using a combination of liquid chromatography-coupled electrospray ionization-MS. The membrane proteins that were prepared by carbonate extraction were separated on two-dimensional PAGE in different pI ranges using the detergent 2% amidosulfobetaine (ASB). The proteins were then subjected to matrix-assisted laser desorption ionization-time-of-flight/time-of-flight for analysis and identification. Because the genome sequence of P. syringae Lz4W is not known, the proteins were identified by using the relevant sequence databases of the Pseudomonas sp available at National Centre for Biotechnology Information (NCBI). The sequence identification of some tryptic peptides were validated by de novo sequencing and others by chemical modification and mass spectrometry. The peptide sequences of P. syringae Lz4W were then matched with the sequences of the peptides from different Pseudomonas sp. by similarity search of the proteins from different species using clustal W2 program. Thus by using a combination of the methods, we have been able to identify large number of proteins of this bacterial strain, which include most of the outer membrane proteins.
Subcellular fractionation of proteins improves the detection of low abundant proteins and helps in the identification using mass spectrometric methods. As the cytoplasmic proteins of an organism can be identified with the routinely used procedures in the laboratories, most studies have recently focused at identification of membrane proteins (1–4). The hydrophobic character of these latter molecules makes them poorly soluble in aqueous solvents and hence their separation by two-dimensional PAGE is very complicated (5–10). Membrane proteins are invariably found to be associated with the cytoplasmic proteins even after using stringent preparation procedures (3, 8, 9). Therefore, preparation and fractionation of membrane proteins remains a problem (11).
For sequence determination and identification, these proteins are subjected to liquid chromatography-electrospray ionization (LC-ESI)1 tandem MS (MS/MS) studies. MS/MS followed by database search tools such as MASCOT, SEQUEST, and Sonar are generally used for peptide sequence determination and the identification of proteins (12–15). Because proteins are identified based on matches to their sequences resulting from comparison of observed peptide MS/MS spectra to the theoretical spectra, the theoretical spectra are generated from the sequence database of different organisms. Peptides are considered identified if they pass certain preset scoring thresholds based on the native scores of the database search algorithms (15–18). Because of the large number of spectral data produced per typical experiment, even a very low error rate results in false positive identifications (19, 20). Therefore, a general criteria has been recommended requiring more than two unique peptides to be identified within a single protein for positive identification (21, 22). Recent studies have developed new models for identifying proteins by single peptide match also, which proposed to include these single peptide hits in the list of identified proteins (23, 24).
Peptide sequences can also be validated by using a combination of chemical and mass spectrometric methods (25–27). Identification of proteins is heavily dependent on the accuracy of the sequence of the tryptic peptides, particularly when the genome sequence of the organism under study is not available.
De novo sequencing methods and sequence validation methods are very useful in the sequence determination of the tryptic peptides for correct identification of the proteins. De novo sequencing programs such as Lutefisk, AUDENS are also used for cross species protein identifications (28, 29). The peptide fragmentation fingerprinting approach also generates partial sequence information in order to filter the candidate peptide before identification (30). Research studies have also shown that N- terminal tagging with some chemical agents aids in the de novo sequencing of peptides using ESI-MS/MS and matrix-assisted laser desorption ionization (MALDI) MS/MS (31).
Several researchers including our laboratory are investigating the mechanism of cold adaptation using Antarctic bacteria as model system (32–36). In the present study we focused our attention to the membrane proteins. For this, the membrane proteins of the Antarctic bacterium Pseudomonas syringae Lz4W were prepared by three different methods to identify as many proteins as possible. We also, compared the membrane protein preparation by Triton X-100 solubilization method and sucrose density gradient method. The data obtained from proteins prepared by carbonate extraction were also used to validate the peptide sequences and compared with the identifications obtained from the other methods. Because the genome sequence of this Antarctic bacterium is not known, the proteins were identified using the relevant data bases of other Pseudomonas sp. available at NCBI. The proteins of P. syringae Lz4W were fractionated by subcellular fractionation methods, separated on one-dimensional and two-dimensional gels, identified by using a combination of different ionization methods (MALDI and ESI), database searches (MASCOT and SEQUEST), and bioinformatics procedures. Thus, using a combination of different methods, about 1290 proteins of this bacterium, which included most of the outer membrane proteins have been identified in this study.
EXPERIMENTAL PROCEDURES
Growth Conditions of the Bacterium
The Antarctic psychotropic bacterium Pseudomonas syringae Lz4W has the ability to grow between 0 °C and 30 °C with an optimum growth at ∼22 °C. The bacterium was grown routinely at 22 °C in Antarctic bacterial medium, consisting of bactopeptone 0.5% (w/v) and yeast extract 0.25 (w/v) as described earlier (37), to an optical density to 1.0 at 600 nm and harvested. About 500 mg of the wet cells were taken for analysis.
Isolation and Preparation of Membrane Proteins
The bacterial cell pellet was suspended in 6 ml of 50 mm Tris.HCl, pH 7.3, and after adding lysozyme (60 μg/ml), 0.7 mg of DNase and RNase it was sonicated for 2 min in a Branson sonifier. The cell debris and the remaining intact cells were removed by centrifugation at 8000 rpm for 10 min. The cellular fractionation of the proteins of P. syringae Lz4W was done by either Triton X-100 solubilization as described earlier (38).The membrane proteins using carbonate extraction were prepared essentially as described earlier (39), with minor modifications. The supernatant was diluted with 0.1 m sodium carbonate (pH 11) and stirred for 1 h on an ice bath. The carbonate- treated membranes were collected by centrifugation at 115,000 × g for 1 h at 4 °C. The membrane pellet was washed with 2 ml of 50 mm Tris.HCl, pH 7.3 and centrifuged again at 115,000 × g for 20 min at 4 °C. The pellet containing membrane proteins were solubilized for two-dimensional electrophoresis with 1 ml of isoelectric focusing (IEF) solution as described below and subjected to two-dimensional SDS gel after IEF.
The membrane proteins were also prepared by sucrose density gradient method as described earlier with minor modifications (40). After removing the cell debris as described above, the supernatant was loaded on a two step gradient of sucrose (60 and 70% sucrose layers) and ultracentrifuged at 38,500 rpm for 18 h at 4 °C. The two layers were mixed and centrifuged again at 55,000 rpm for 1 h at 4 °C. The pellet was solubilized in 2% Triton X-100 and mixed with the supernatant. The proteins in the combined fractions were precipitated with 10% trichloroacetic acid (TCA) and left for 12 h at −20 °C. The contents were centrifuged at 14,000 rpm for 10 min at 4 °C and the pellet washed with acetone twice and resuspended in the loading buffer and subjected to SDS gel electrophoresis.
One-dimensional SDS-PAGE
The membrane proteins of P. syringae Lz4W obtained from triton X-100 solubilization or sucrose-density gradient were separated on a 12% SDS-PAGE gel according to the Laemmli protocol (41). About 150 μg of the protein reduced and alkylated, dissolved in SDS-PAGE loading buffer, boiled for 5 min and loaded in different wells in the 1 mm thick gel.
Two-dimensional Gel Electrophoresis After Carbonate Extraction
IEF was performed with BIORAD 11 cm immobilized pH gradient strips with 3–10, 4–7, and 5–8. The IPG strips were rehydrated over night (12–16 h) in 30 mm Tris.HCl pH 8, 7 m urea and 2 m thiourea containing 20 mm dithiotreitol (DTT) and 0.4% carrier ampholyte and 2% ASB-14 as described before for membrane proteins (42). The IEF run was set from 0–300 V in 1 min, 300 V for 3 h, from 300–3500 for 1 h and set at 3500 V for 20 h. After the IEF run the IPG strips were incubated at room temperature for 10 min in 6 m urea, 30% (v/v) glycerol, 2% (w/v) SDS, 15 mm DTT, 0.125 m Tris, and 0.1 m HCl. The second equilibration step was carried out for 5 min in the same solution except that DTT was replaced with iodoacetamide. The second dimension (SDS-PAGE) was carried out on a 12% SDS gel without a stacking gel. After keeping the IPG strip on the SDS gel, the strip was sealed with a layer of agarose before the electrophoresis run. All the gels were stained with Coomassie blue.
In-gel Digestion of Proteins with Trypsin
The prominent bands of the one-dimensional gel were cut and subjected to in-gel digestion as described earlier (43). Briefly, the gel pieces were further cut to smallest size, and washed with water followed by washing with 50% of 25 mm ammonium bicarbonate and acetonitrile (ACN) till the stain is removed. Subsequently the gel pieces were treated with ACN and dried. Depending on the size of the band, 10–20 μl of trypsin (10 μg/ml) was added to the gel pieces and incubated at 37 °C for 16–18 h. The resultant peptides were extracted twice with 100 μl of 5% trifluoroacetic acid (TFA) in 50% ACN pooled together and dried in a speed vac concentrator. The samples were redissolved in 5% ACN/H20 before loading for LC-MS/MS. The protein bands of two-dimensional gels were also digested similarly and spotted on a MALDI plate for analysis.
MALDI TOF/TOF MS
The mass spectra of the trypsin-digested proteins from two-dimensional gels were analyzed on a 4800 MALDI TOF/TOF mass analyzer obtained from Applied Biosystems (Foster city, CA). The mass spectra were recorded in reflector mode using the matrix α-cyano-4-hydroxycinnamic acid (5 mg/ml in 50% ACN). The individual peptides for MS/MS were selected by timed ion selection method, and the collision induced dissociation mass spectra recorded using air as collision gas with 1KeV energy. The top 5 peptides were selected for MS/MS based on the intensity of the precursor ion. Proteins were identified using the GPS explorer software (Ver.3.5) supplied by the manufacturer. This program uses MASCOT search tools for the identification of proteins. Because the genome sequence of the bacterium is not known, the proteins were identified using the relevant database of the Pseudomonas species available at the NCBI database. The database includes 99128 protein sequences (updated up to July 2008). Using MS/MS spectra, the top scoring peptides were selected and several proteins could be identified. The precursor ion mass tolerance and the fragment ion mass tolerance were set at 0.2 and 0.25 respectively. The signal to noise ratio was set at 10 and 50 peaks per 200 Da were taken for analysis with maximum number of peaks set to 65. Methionine oxidation was set as the variable modification and carboxyamidomethylation as the fixed modification for cysteine. Enzyme trypsin was selected with one missed cleavage. The peptide summary report groups the peptide matches into protein hits and the protein score was derived by combining the ion scores for ranking the proteins. The highest scoring proteins that contained one or more peptides were selected for identification. The MASCOT acceptance score for the peptide and for protein were 18 and 34 respectively. As a default MASCOT uses a threshold of 5% probability that protein identification is incorrect. The de novo sequencing of the peptides was determined by submitting the MS/MS spectra to the MASCOT program setting the relevant parameters as described above.
LC-MS/MS of Trypsin Digested Proteins
The mass spectra (LC-ESI MS/MS) of the tryptic digested gel bands from one-dimensional SDS gels were analyzed by a Finnigan linear ion trap mass analyzer (Thermo Electron corporation, San Jose, CA) equipped with a surveyor MS pump plus. The MS data acquired in the mass range m/z 400–2000 and the MS/MS of the most abundant ions was acquired in the LTQ using collision induced dissociation. Briefly, the tryptic peptides obtained from each band were manually loaded on a C-18 precolumn (Applied Biosystems) connected to a Biobasic C-18 (100 mmX0.18) RP column, with a pore size of 300 Å and particle size of 5 μm. The flow rate was set at 3 μl/min. The mobile phases A and B were 0.2% formic acid in water and 0.2% formic acid in 95% ACN respectively. The gradient was started at 10min and increased to 60% B in 40 min and to 100% B in 55 min and retained at 100% B till 65 min. The column was equilibrated for 10 min before the next injection. MS and MS/MS spectra were obtained at a heated capillary temperature of 200 °C and the ESI voltage was set at 4 KV. The peptides were fragmented using normalized collision energy of 35%. The MS/MS spectrum of the top seven peptides with a signal threshold of 500 counts was acquired with 30 msec activation time and a repeat duration of 30 s. The LC-MS/MS data was deposited at PRIDE (www.ebi.ac.uk/pride) for validation (44). Accession numbers of 12279 to 12291 for the triton X-100 solubilized membrane protein preparation and 13524 for the sucrose density gradient membrane protein preparation were obtained.
Data Analysis
LC ESI MS/MS spectra were analyzed using the Bioworks browser (Ver 3.2) supplied by the manufacturer. All the MS/MS spectra were analyzed using SEQUEST (Thermo fisher scientific) selecting the enzyme trypsin and applying the search parameters of precursor tolerance of 1.0 Da and a fragment tolerance of 1.4 Da, and b-ion and y-ion series for sequence determination; oxidation of methionine (15.99 Da) and carboxyamidomethylation (57.02 Da) of cysteine were considered as variable and fixed modification respectively. As the genome sequence of the Pseudomonas syringae Lz4W is not available all the data were searched using the relevant database of Pseudomonas sp. available at NCBI as described earlier (38). Same database used for MASCOT (for MALDI) was used for SEQUEST (linear ion trap MS/MS) searches also. The MS/MS spectra of the multiply charged peptides were searched against the database of Pseudomonas sp. The cross correlation scores (X corr) of singly-, doubly-, and triply-charged peptides were greater than 1.8, 2.5, and 3.5 respectively, were fixed for protein identification. The program listed the peptides corresponding to the proteins. A list of peptide sequences that had highest X corr values was identified (13, 45). Other parameters of Δ Cn > 0.1 are selected for anticipated results in addition to Xcorr score. The proteins were identified either by sufficient number of peptides or identified at least by one peptide that is redundant enough to be considered reliable with acceptable scores. Because several other criteria were used to identify the sequence of peptides, after identifying the proteins with the set threshold values, even the peptides below the defined thresholds were also retained in the supplemental Table S1 to increase the sequence coverage.
The values of ΔCn obtained for a large number of peptides are very much higher than the selected criteria, suggesting the sequences obtained are exact matches. In addition, a filter for estimation for false positives was adapted. The false detection rates (FDR) can be minimized by fixing X corr values and manually increase Δ Cn to get the peptide identification with specific FDR (46, 47), or use fixed Δ Cn value and manually increase X corr values (48, 49).The algorithm calculated the probability scores. P (pro) value can be defined as an extrapolation protein probability. A minimum value of P (pep) was set at 0.001, which allows a cut off of less than 0.1% false positives. The procedure adapted in this method uses the highest P (pep) within each protein to be equivalent to P (pro). The peptides generated from the psychotropic Antarctic bacterium P. syringae Lz4W picked up maximum homology hits with the proteins from different mesophilic Pseudomonas sp. The peptides corresponding to each protein obtained from different Pseudomonas sp. were listed together after checking the sequence similarity by Clustal W. The peptide identification results were integrated by DTA select to display the list of proteins identified from all the fractions and peptide lists belonging to each protein (50). The final list of proteins was prepared by combining all the proteins obtained from different LC-MS/MS runs after manual verification. The redundancy of peptides within the list of peptides from each protein was removed by verifying both the m/z value and the corresponding sequence. The redundant proteins were also removed.
Clustal W2 for Sequence Alignment of Proteins
The basic principle of Clustal W2 is the progressive alignment of the given sequences. It is a data exploration tool rather than a definitive analysis method. It has an improved sensitivity for multiple sequence alignments (51). The program starts with pair-wise comparison between two sequences initially and proceeds toward the multiple alignments and aligns sequences globally. The default parameters were used for sequence alignment. Thus, inferring from the results of clustal W2, the peptides obtained from proteins with the same name, but matched with different Pseudomonas sp were listed into a single entry as belonging to P. syringae Lz4W.
Subcellular Localization of the Identified Proteins
The subcellular localization of the identified proteins was carried out using a program PSORT b Ver 3.0 (http://www.psort.org) by choosing Gram-negative strains and normal output format that displays results with the final predictions and associated scores as reported earlier (52, 53). SCL scores of greater than 7.5 in a particular localization are considered as a confirmed localization. Cytosolic proteins are identified clearly with good scores (8–9.5 on a scale of 10) and some outer membrane proteins with a score of 10. This program cannot detect lipoprotein motifs. In general, this method was useful to compartmentalize the identified proteins obtained from P. syringae Lz4W. The GRAVY scores of the peptides derived from outer membrane proteins were determined using the web site at (http://www.gravy-calculator.de) to identify the hydrophobic nature of these peptides.
RESULTS
Preparation, Separation, and Analysis of Membrane Proteins
The main objective of this study was to identify the membrane proteins of P. syringae Lz4W. Toward this, different protocols were employed for the isolation of membrane proteins that were then separated on one-dimensional and two-dimensional gels. The three protocols used for the preparation of membrane proteins are shown in Fig. 1. Fig. 2 and 3 shows the typical separations of the membrane proteins by one-dimensional SDS-PAGE and two-dimensional respectively. The proteins separated on one-dimensional gels were analyzed on a LC coupled ESI MS/MS followed by trypsin digestion. The protein separated on two-dimensional gels was analyzed on MALDI TOF/TOF as described under materials and methods. The results of protein identification were provided in supplemental Tables S1, S2 and S3.
Fig. 1.

The methods used for the preparation of membrane proteins. A, shows the sucrose density gradient method, B, shows the Triton X-100 solubilization method and panel C exhibits the carbonate extraction procedure. The procedure used in panels A and B generally used to compare the efficiency of these methods to prepare as many membrane proteins as possible from P. syringae Lz4W. The procedure used in panel C was used for identification of more proteins and validate the protein/peptide identifications.
Fig. 2.
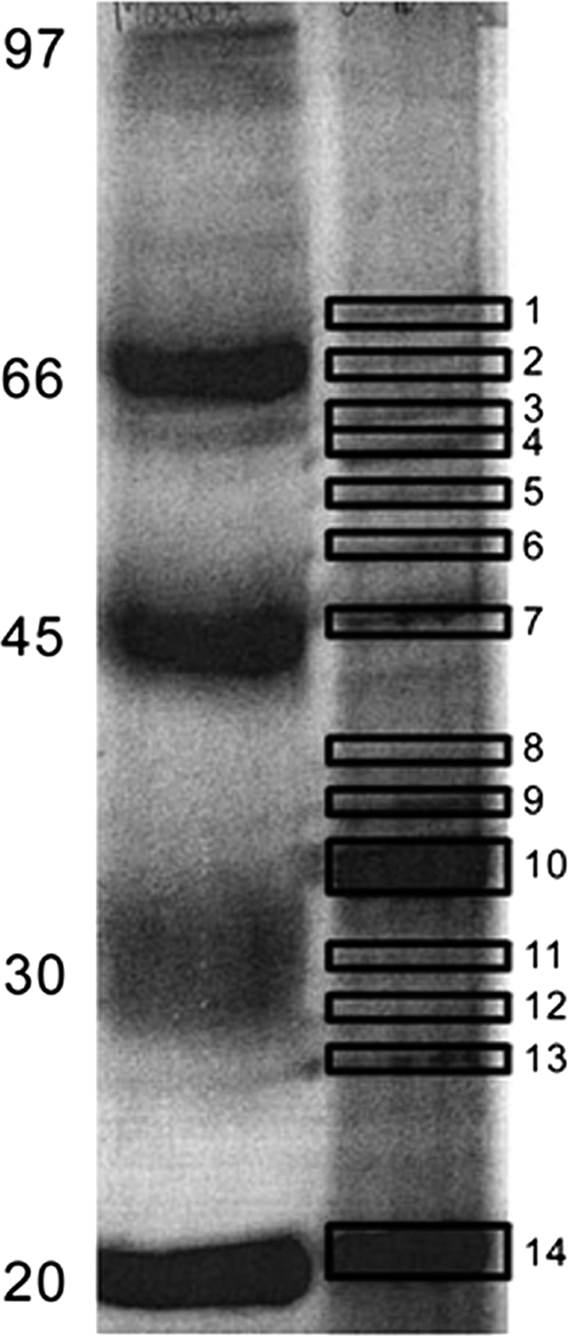
A representative gel picture of one-dimensional gel with 1 mm thickness, 12% SDS-PAGE of the membrane proteins of the Antarctic bacterium P. syringae prepared by sucrose density gradient method. 150 μg of the protein was loaded on the gel and stained the gel with coomassie blue.
Fig. 3.

Two-dimensional gel pictures of the membrane proteins of the Antarctic bacterium P. syringae prepared by carbonate extraction procedure separated on a 12% SDS gel. Proteins separated in the pI ranges 3–10 (panel A), 4–7 (panel B) and 5–8 (panel C) are shown. The gel was stained with Coomassie blue.
Identification of Proteins
Proteins Separated on One-dimensional Gel
The scheme adapted for the identification of proteins is shown in the Fig. 4. A total of 1151 proteins were identified from the LC coupled ESI MS/MS spectral data using SEQUEST program. From these proteins 642 were identified with hits of a minimum of two or more than two peptides, and 509 proteins were identified with single peptide hits. It is also noteworthy that about 170 hypothetical proteins were identified (supplemental Table S1). Some of the proteins were represented by many peptides. For example, translational initiation factor IF2 was identified with as many as 30 peptides, Polyribonucleotide nucleotidyl transferase with 29 peptides and elongation factor G with 34 peptides. On the other hand, the outer membrane porin (OMP) was identified with 6 peptides. All these peptides of OMP matched highly with the protein of Pseudomonas fluorescence pf-5. Another protein, organic solvent tolerance protein was identified with 9 peptides which matched to the protein from different Pseudomonas sp: 6 peptides were from P. fluorescence Pf-O1, two peptides from P.syringae pv. syringae B728 and one peptide from P. mendocina ymp (see supplementary tables). The proteins identified with peptides from different Pseudomonas sp were aligned using Clustal W2 to examine their sequence similarity, which suggest that the protein is highly conserved among them. The sequence alignment of the protein ATP synthetase subunit beta of different Pseudomonas sp. is shown in the supplemental Fig. S1 highlighting the peptides obtained in the analysis.
Fig. 4.

The work flow adapted for the identification of proteins of P. syringae Lz4W.
Proteins Separated on Two-dimensional Gel After Carbonate Extraction
Identification of proteins and validation of peptides sequences was also carried out using MALDI TOF/TOF. In all, 206 proteins were identified from this mass spectral data using MASCOT search. Out of them 104 proteins were identified with a minimum of two or more than two peptide hits, whereas, 102 proteins were identified with single peptide hits using the database search algorithms. The MS/MS spectra were also used to identify the sequence of the peptide by de novo sequence program of MASCOT. These results showed that the de novo sequence of 61 peptides matched exactly with the database searches, 281 peptides showed scrambled sequence or partial matches. The de novo sequence could not be obtained for 62 peptides. Scrambled sequences and partially matched sequences are also indicators for the correct identification of peptide sequences (54). Accordingly, some algorithms have been developed to correlate de novo sequences with database searches (54). However, in the present study, manual verification method has been used, as the data size is small and easy to handle. Thus, the protein identifications were verified both by de novo sequences and database search.
In addition, we observed that five proteins with single peptide hits using ESI MS/MS could be identified with multiple peptides in MALDI results. Similarly, 19 proteins with single peptide hits from MALDI results were found to match with multiple peptides in the ESI results. Thus, a combination of these ionization methods appears to be helpful in identifying proteins confidently. It is to be noted that we identified 64 proteins commonly from both these ionization methods.
Validation of Peptide Sequences Using Chemical/MS Methods
For validating the peptide sequence results, chemical markers like methionine oxidation can also be used. Because methionine oxidation occurs commonly during the sample preparation, enzymatic digestion, and other processes, it can be used for validating the peptides as described earlier (55). More than 100 proteins were verified for the peptides containing methionine/oxidized methionine and phenylalanine. The MALDI mass spectrum was manually verified to confirm these sequences (see supplemental Table S2). Wherever significant amounts of protein were available, the tryptic peptides were acetylated and an increase was observed in the b ion intensity in the collision induced dissociation mass spectra (data not shown). This observation was helpful to validate the sequence of the peptide as described earlier (27). Using a combination of N-terminal analysis and proteomics approaches some of these proteins were identified earlier from this bacterium (38, 56). Altogether, using a combination of methods described above we have identified 1293 proteins in this study including 112 outer membrane proteins from the Antarctic bacteria P. syringae Lz 4W, whose genome sequence is not known.
Prediction of Subcellular Localization
We have also examined the subcellular localization of the identified proteins using the program PSORT b V.3 as shown in Fig. 5. These results reveal that 112 are outer membrane proteins, 135 cytoplsamic membrane proteins, and 38 periplasmic proteins. Although, total membrane proteins identified were 364 many of them were found to have multiple localization sites, and many were predicted to be cytoplasmic proteins but with multiple localization sites (See Fig. 5). In addition, there are 88 proteins whose localization could not be predicted using this program. The 112 outer membrane proteins shown in this table were identified from 376 tryptic peptides generated from them. Out of these tryptic peptides 297 peptides exhibited negative GRAVY score and 79 peptides positive score. This indicates that the peptides generated from outside the trans membrane domains of these proteins which was as expected (supplemental Table S3).
Fig. 5.
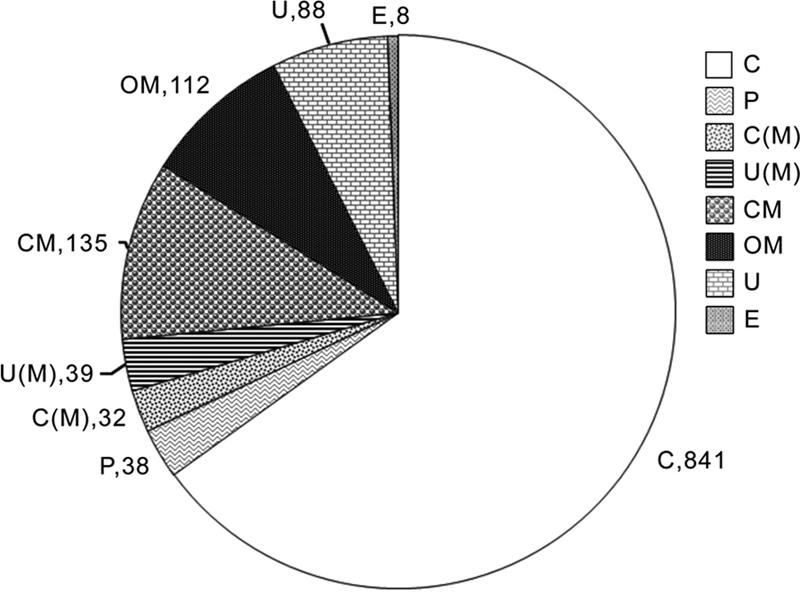
The prediction of sub cellular localization of different proteins identified using PSORT b V3 from P. syringae Lz 4W. The representation of different labels are as follows: C, cytoplasmic; OM, outer membrane; CM, cytoplasmic membrane; C(M), cytoplamic with multiple localization sites; U, unknown; U(M), unknown with multiple localization sites; P, periplasmic; E, extracellular proteins.
Comparison of Membrane Protein Preparation Methods
Fig. 6 and supplemental Tables S2 and S3 shows the comparison of different membrane proteins prepared by both the methods. We compared the two membrane protein preparation methods (Triton X-100 solubilization and sucrose gradient) for their relative moieties (hydrophobic peptides) in identifying the membrane proteins as both of them were identified by identical procedures (one-dimensional-LC-ESI MS/MS). We observed that sucrose density gradient method resulted in the identification of more outer membrane proteins. The yield of membrane proteins was higher/better with sucrose density gradient method when compared with Triton X-100 solubilization method.
Fig. 6.

Comparison of different membrane proteins obtained from sucrose density gradient method (light bars) and triton X-100 solubilization methods (dark bars) from the Antarctic bacterium P. syringae. Same abbreviations are used in Fig. 5 and Fig. 6.
DISCUSSION
Even with the development of different methods/protocols for identification of membrane proteins still remains a problem. Membrane proteins in Gram-negative bacteria include outer membrane proteins, inner membrane proteins (cytoplsamic membrane), many of which can be integral membrane proteins or membrane-associated proteins. Previous experience with LC-MS/MS analysis of membrane proteins suggested complications in the detection of hydrophobic peptides obtained from transmembrane domains (57). Membrane proteins were also identified by adopting different cleavage strategies by different proteolysis enzymes to improve their identification (58). Wolff et al. have also used different membrane proteome preparation methods to identify as many integral membrane proteins as possible (9). The efficiency of the preparation of membrane proteins depends on the composition and nature of the lipids present in the membranes, the efficiency of the procedure used and steps/the sequence used in the preparation. In the current study using different membrane protein preparation methods and using different ionization methods a total of 112 outer membrane proteins of P. syringae Lz4W were identified. This is important as the results posted on the web site (http://beta.psort.org) the number of outer membrane proteins of different mesophilic P. syringae sp. varied from 110–126 as predicted by PSORTb V.3 results. Because, it is a well known fact that microorganisms do not express their entire set of proteins under one single growth conditions the 112 outer membrane proteins of P. syringae, which were identified in this study represent most of the outer membrane proteins. Most importantly, this has been possible despite the fact that that genome sequence of P. syringae Lz4W is not known.
In order to enrich and identify as many as membrane proteins possible, we examined different membrane protein preparation methods and compared their efficiency. Fig. 6 shows that sucrose gradient method is more efficient for the preparation of outer membrane proteins. However, many ribosomal proteins appeared among the membrane proteins by this method (data not shown).
Validation of the peptide sequences is as important as identifying the large number of proteins. MALDI TOF/TOF studies were mainly carried out for this purpose. We have used two-dimensional electrophoresis MS approaches to validate the sequence of the peptides and identification of proteins using carbonate extraction method, an efficient protocol for the preparation and separation of outer membrane proteins as described earlier (39).
In this study we have also identified about 170 hypothetical proteins from this bacterium, which may be because of the reason that the genome sequence of the bacterium is not yet known. Alternatively, it may also be possible that this cold adapted bacterium may have some unique proteins, whose functional significance is not known. Earlier reports have shed light on the role of structural adaptation of enzymes, the function of heat- shock and cold- shock proteins in cold adaptation (35, 38, 59). Several of these enzymes and proteins have been identified with confidence in this study (see supplemental Table S1). In our earlier study, using N-terminal sequencing and proteomics approaches the proteins of the degradosome complex of this bacterium have been identified which were later found to be associated with membranes (38, 56).
In conclusion, we have identified 112 outer membrane proteins of the Antarctic bacterium P. syringae which might represent most of the proteins of the outer membrane. We also found the sucrose density gradient method was most efficient for the outer membrane proteins. The successful identification of large numbers of proteins (a total of 1293) has been feasible because of the use of a combination of ionization methods, different database search tools. The results are significant, as the genome sequence of the bacterium is not known.
Acknowledgments
Heramb M. Kulkarni is a recipient of CSIR research fellowship. We thank Ms. Aparna Jagannath, Ms. Y. Pratyusha for their technical support. I thank Dr M. K. Ray and Dr C. Sivakama Sundari for the comments and criticism on the manuscript.
Footnotes
* This research work was supported by a grant from Department of Biotechnology (BT/PR7383/BRB/10/474/2006), New Delhi. Ehab Abou-Eladab worked at CCMB on a TWAS-CSIR postdoctoral fellowship.
 This article contains supplemental Fig. S1 and Tables S1 to S3.
This article contains supplemental Fig. S1 and Tables S1 to S3.
1 The abbreviations used are:
- LC-ESI
- liquid chromatography-electrospray ionization
- DTT
- dithiothreitol
- ACN
- acetonitrile
- IEF
- Isoelectric focusing
- ASB
- amidosulfobetaine.
REFERENCES
- 1. Graham R. L., Pollock C. E., O'Loughlin S. N., Ternan N. G., Weatherly D. B., Tarleton R. L., McMullan G. (2007) Multidimensional analysis of the insoluble sub-proteome of Oceanobacillus iheyensis HTE831, an alkaliphilic and halotolerant deep-sea bacterium isolated from the Iheya ridge. Proteomics 7, 82–91 [DOI] [PubMed] [Google Scholar]
- 2. Ahram M., Springer D. L. (2004) Large-scale proteomic analysis of membrane proteins. Expert Rev. Proteomics 1, 293–302 [DOI] [PubMed] [Google Scholar]
- 3. Santoni V., Molloy M., Rabilloud T. (2000) Membrane proteins and proteomics: un amour impossible? Electrophoresis 21, 1054–1070 [DOI] [PubMed] [Google Scholar]
- 4. Andersen J. S., Mann M. (2006) Organellar proteomics: Turning inventions into insights. EMBO Rep. 7, 874–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yates J. R., 3rd, Gilchrist A., Howell K. E., Bergeron J. J. (2005) Proteomics of organelles and large cellular structures. Nat. Rev. Mol. Cell Biol. 6, 702–714 [DOI] [PubMed] [Google Scholar]
- 6. Niederweis M., Danilchanka O., Huff J., Hoffmann C., Engelhardt H. (2010) Mycobacterial outer membranes: in search of proteins. Trends Microbiol. 18, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li H., Wang B. C., Xu W. J., Lin X. M., Peng X. X. (2008) Identification and network of outer membrane proteins regulating streptomysin resistance in Escherichia coli. J. Proteome Res. 7, 4040–4049 [DOI] [PubMed] [Google Scholar]
- 8. Wu C. C., Yates J. R., 3rd (2003) The application of mass spectrometry to membrane proteomics. Nat. Biotechnol. 21, 262–267 [DOI] [PubMed] [Google Scholar]
- 9. Wolff S., Hahne H., Hecker M., Becher D. (2008) Complementary analysis of the vegetative membrane proteome of the human pathogen Staphylococcus aureus. Mol. Cell Proteomics 7, 1460–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Görg A., Weiss W., Dunn M. J. (2004) Current two-dimensional electrophoresis technology for proteomics. Proteomics 4, 3665–3685 [DOI] [PubMed] [Google Scholar]
- 11. Yan W., Aebersold R., Raines E. W. (2009) Evolution of organelle-associated protein profiling. J. Proteomics 72, 4–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 13. Eng J. K., McCormack A. L., Yates J. R., 3rd. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 14. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 15. Field H. I., Fenyö D., Beavis R. C. (2002) RADARS, a bioinformatics solution that automates proteome mass spectral analysis, optimises protein identification, and archives data in a relational database. Proteomics 2, 36–47 [PubMed] [Google Scholar]
- 16. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 17. Reiter L., Claassen M., Schrimpf S. P., Jovanovic M., Schmidt A., Buhmann J. M., Hengartner M. O., Aebersold R. (2009) Protein identification false discovery rates for very large proteomics data sets generated by tandem mass spectrometry. Mol. Cell Proteomics 8, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lam H., Aebersold R. (2010) Spectral library searching for peptide identification via tandem MS. Methods Mol. Biol. 604, 95–103 [DOI] [PubMed] [Google Scholar]
- 19. Kolker E., Higdon R., Hogan J. M. (2006) Protein identification and expression analysis using mass spectrometry. Trends Microbiol. 14, 229–235 [DOI] [PubMed] [Google Scholar]
- 20. Cargile B. J., Bundy J. L., Stephenson J. L., Jr. (2004) Potential for false positive identifications from large databases through tandem mass spectrometry. J. Proteome Res. 3, 1082–1085 [DOI] [PubMed] [Google Scholar]
- 21. Carr S., Aebersold R., Baldwin M., Burlingame A., Clauser K., Nesvizhskii A. (2004) The need for guidelines in publication of peptide and protein identification data: Working Group on Publication Guidelines for Peptide and Protein Identification Data. Mol. Cell Proteomics 3, 531–533 [DOI] [PubMed] [Google Scholar]
- 22. Domon B., Aebersold R. (2006) Challenges and opportunities in proteomics data analysis. Mol. Cell Proteomics 5, 1921–1926 [DOI] [PubMed] [Google Scholar]
- 23. Higdon R., Kolker E. (2007) A predictive model for identifying proteins by a single peptide match. Bioinformatics 23, 277–280 [DOI] [PubMed] [Google Scholar]
- 24. Gupta N., Pevzner P. A. (2009) False discovery rates of protein identifications: a strike against the two-peptide rule. J. Proteome Res. 8, 4173–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noga M. J., Lewandowski J. J., Suder P., Silberring J. (2005) An enhanced method for peptides sequencing by N-terminal derivatization and MS. Proteomics 5, 4367–4375 [DOI] [PubMed] [Google Scholar]
- 26. Liu N., Chan W., Lee K. C., Cai Z. (2009) A method to enhance a1 ions and application for peptide sequencing and protein identification. J. Am. Soc. Mass Spectrom. 20, 1214–1223 [DOI] [PubMed] [Google Scholar]
- 27. Kulkarni H. M., Ramesh V., Srinivas R., Jagannadham M. V. (2010) Acetylating tryptic peptides enhances b ion intensity in MALDI-TOF/TOF: Implications in peptide sequencing and identification of proteins in an Antarctic bacterium Pseudomonas syringae. Proteomics Insights 3, 1–16 [Google Scholar]
- 28. Grossmann J., Roos F. F., Cieliebak M., Lipták Z., Mathis L. K., Müller M., Gruissem W., Baginsky S. (2005) AUDENS: a tool for automated peptide de novo sequencing. J. Proteome Res. 4, 1768–1774 [DOI] [PubMed] [Google Scholar]
- 29. Liska A. J., Shevchenko A. (2003) Expanding the organismal scope of proteomics: cross-species protein identification by mass spectrometry and its implications. Proteomics 3, 19–28 [DOI] [PubMed] [Google Scholar]
- 30. Frank A., Tanner S., Bafna V., Pevzner P. (2005) Peptide sequence tags for fast database search in mass-spectrometry. J. Proteome Res. 4, 1287–1295 [DOI] [PubMed] [Google Scholar]
- 31. Samgina T. Y., Kovalev S. V., Gorshkov V. A., Artemenko K. A., Poljakov N. B., Lebedev A. T. (2010) N-terminal tagging strategy for de novo sequencing of short peptides by ESI-MS/MS and MALDI-MS/MS. J. Am. Soc. Mass Spectrom. 21, 104–111 [DOI] [PubMed] [Google Scholar]
- 32. Ray M. K., Sridhar S. S., Srinivas U. K. (Eds), Cold stress response of low temperature adopted bacteria, Research Sign post, Trivandrum, India, 2006, 1–23 [Google Scholar]
- 33. Feller G., Gerday C. (2003) Psychrophilic enzymes: Hot topics in cold adaptation. Nat. Rev. Microbiol. 1, 200–208 [DOI] [PubMed] [Google Scholar]
- 34. Chattopadhyay M. K., Jagannadham M. V. (2001) Maintenance of membrane fluidity in Antarctic bacteria. Polar Biol. 24, 386–388 [Google Scholar]
- 35. Chattopadhyay M. K. (2006) Mechanism of bacterial adaptation to low temperature. J. Biosci. 31, 157–165 [DOI] [PubMed] [Google Scholar]
- 36. Strocchi M., Ferrer M., Timmis K. N., Golyshin P. N. (2006) Low temperature-induced systems failure in Escherichia coli: insights from rescue by cold-adapted chaperones. Proteomics 6, 193–206 [DOI] [PubMed] [Google Scholar]
- 37. Shivaji S., Rao N. S., Saisree L., Sheth V., Reddy G. S., Bhargava P. M. (1989) Isolation and identification of Pseudomonas spp. from Schirmacher Oasis, Antarctica. Appl. Environ. Microbiol. 55, 767–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jagannadham M. V. (2008) Identification of proteins from membrane preparations by a combination of MALDI TOF-TOF and LC-coupled linear ion trap MS analysis of an Antarctic bacterium Pseudomonas syringae Lz4W, a strain with unsequenced genome. Electrophoresis 29, 4341–4350 [DOI] [PubMed] [Google Scholar]
- 39. Molloy M. P., Herbert B. R., Slade M. B., Rabilloud T., Nouwens A. S., Williams K. L., Gooley A. A. (2000) Proteomic analysis of the Escherichia coli outer membrane. Eur. J. Biochem. 267, 2871–2881 [DOI] [PubMed] [Google Scholar]
- 40. Gotoh N., White N. J., Chaowagul W., Woods D. E. (1994) Isolation and characterization of the outer-membrane proteins of Burkholderia (Pseudomonas) pseudomallei. Microbiology 140 (Pt 4), 797–805 [DOI] [PubMed] [Google Scholar]
- 41. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 42. Luche S., Santoni V., Rabilloud T. (2003) Evaluation of nonionic and zwitterionic detergents as membrane protein solubilizers in two-dimensional electrophoresis. Proteomics 3, 249–253 [DOI] [PubMed] [Google Scholar]
- 43. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 44. Vizcaíno J. A., Côté R., Reisinger F., Foster J. M., Mueller M., Rameseder J., Hermjakob H., Martens L. (2009) A guide to the Proteomics Identifications Database proteomics data repository. Proteomics 9, 4276–4283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 46. Xie H., Griffin T. J. (2006) Trade-off between high sensitivity and increased potential for false positive peptide sequence matches using a two-dimensional linear ion trap for tandem mass spectrometry-based proteomics. J. Proteome Res. 5, 1003–1009 [DOI] [PubMed] [Google Scholar]
- 47. Jiang X., Jiang X., Han G., Ye M., Zou H. (2007) Optimization of filtering criterion for SEQUEST database searching to improve proteome coverage in shotgun proteomics. BMC Bioinformatics 8, 323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peng J., Elias J. E., Thoreen C. C., Licklider L. J., Gygi S. P. (2003) Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2, 43–50 [DOI] [PubMed] [Google Scholar]
- 49. Yates J. R., 3rd, Eng J. K., Clauser K. R., Burlingame A. (1996) , Search of sequence databases with uninterpreted high energy collision induced dissociation spectra of peptides. J. Am. Soc. Mass Spectrom. 7, 1089–1098 [DOI] [PubMed] [Google Scholar]
- 50. Tabb D. L., McDonald W. H., Yates J. R., 3rd (2002) DTA Select and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rey S., Acab M., Gardy J. L., Laird M. R., deFays K., Lambert C., Brinkman F. S. (2005) PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 33, D164–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu N. Y., Wagner J. R., Laird M. R., Melli G., Rey S., Lo R., Dao P., Sahinalp S. C., Ester M., Foster L. J., Brinkman F. S. (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Han Y., Ma B., Zhang K. (2005) SPIDER: software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 55. Jagannadham M. V. (2009) Identifying the sequence and distinguishing the oxidised methionine from phenylalanine peptides by MALDI-TOF/TOF mass spectrometry in an Antarctic bacterium Pseudomonas syringae. Proteomics Insights 2, 27–31 [Google Scholar]
- 56. Purusharth R. I., Klein F., Sulthana S., Jáger S., Jagannadham M. V., Evguenieva-Hackenberg E., Ray M. K., Klug G. (2005) Exoribonuclease R interacts with endoribonuclease E and an RNA helicase in the psychrotrophic bacterium Pseudomonas syringae Lz4W. J. Biol. Chem. 280, 14572–14578 [DOI] [PubMed] [Google Scholar]
- 57. Speers A. E., Wu C. C. (2007) Proteomics of integral membrane proteins–theory and application. Chem. Rev. 107, 3687–3714 [DOI] [PubMed] [Google Scholar]
- 58. Fischer F., Poetsch A. (2006) Protein cleavage strategies for an improved analysis of the membrane proteome. Proteome Sci 4, 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sundareswaran V. R., Singh A. K., Dube S., Shivaji S. (2010) Aspartate aminotransferase is involved in cold adaptation in psychrophilic Pseudomonas syringae. Arch. Microbiol. 192, 663–672 [DOI] [PubMed] [Google Scholar]