

Abstract
This randomized, open-label study of patients in India with visceral leishmaniasis (VL) investigated the effect of food on sitamaquine and desethyl-sitamaquine pharmacokinetics. Patients were randomized to receive oral sitamaquine, 2 mg/kg/day, once a day for 21 days across four cohorts (n = 41) (fasted/fed, fed/fasted, fed/fed, and fasted/fasted) over two periods (days 1−10 and 11−21), or intravenous amphotericin B (AmB), 1 mg/kg every other day for 30 days (n = 20). Mean day 21 pharmacokinetics across the four cohorts were sitamaquine, area under curve (AUC)(0−τ) = 6,627−8,903 ng.hr/mL, AUC(0−16) = 4,859−6,633 ng.hr/mL, maximum plasma concentration (Cmax) = 401−570 ng/mL, apparent terminal half-life (t1/2) = 18.3−22.8 hr, time to reach Cmax (tmax) = 3.5−6 hr; and desethyl-sitamaquine, AUC(0−τ) = 2,307−3,163 ng.hr/mL, Cmax = 109−154 ng/mL, t1/2 = 23.0−27.9 hr, tmax = 2−10 hr, with no significant food effect. On-therapy adverse events were observed for sitamaquine in 4 (10%) of 41 patients and for AmB in 17 (85%) of 20 patients. The final clinical cure (day 180) was 85% (95% confidence interval = 70.8–94.4%) for sitamaquine and 95% (95% confidence interval = 75.1–99.9) for AmB. Sitamaquine can be taken regardless of food intake, was generally well tolerated, and showed potential efficacy in patients with visceral leishmaniasis.
Introduction
Visceral leishmaniasis (VL, kala-azar) is the most serious infection caused by the protozoan parasite Leishmania spp. Transmitted by the bites of sand flies, promastigotes are transformed rapidly into amastigotes that invade macrophages mainly in liver, spleen, and bone marrow. The infection may be asymptomatic and symptoms, including fever, weight loss, and hepatosplenomegaly, may appear months after the initial infection. Symptomatic VL is usually fatal if untreated.
In India during 2000–2002, there were 18,472 recorded cases of VL, mostly in the Bihar region.1 However, disease incidence is probably underestimated by at least eight-fold.2,3 In Bihar, clinical resistance to pentavalent antimony generates treatment failure rates in excess of 50%.4–6 Consequently, amphotericin B (AmB) is the standard of care in expert centers in India. Although highly effective, AmB requires intravenous administration over 30 days, has significant toxic effects, and treatment costs are relatively high.4,7–9 Liposomal AmB has improved tolerability, but is too expensive for most patients in Bihar.10 Intramuscular paromomycin was approved for treatment of patients with VL in India in 2006. It has the same efficacy as AmB,7 a 21-day treatment duration, and a low purchase cost, although parenteral therapy may limit its usefulness in a public health setting. Miltefosine was the first oral therapy available for VL and a significant breakthrough.11–13 However, a four-week treatment schedule and a long-half life may make it vulnerable to resistance development if given as monotherapy, and teratogenicity is an issue for its use in women.14 The recent emphasis on Leishmania elimination in India, Nepal and Bangladesh,3,15 sustains a need for alternative, affordable, well-tolerated, and effective oral therapies.
Sitamaquine (WR6026) is an 8-aminoquinoline under investigation as an oral VL therapy. Two initial phase II clinical studies of 1.5−3.0 mg/kg/day of sitamaquine for 28 days in India and Africa showed maximal efficacy with 1.75 or 2 mg/kg/day.16,17 Clinical symptoms resolved rapidly in both studies.16,17 In the study in India, day 14 splenic aspirates were parasite negative in 96 (82%) of 117 patients overall and in 26 (93%) of 28 patients in the cohort receiving 2.0 mg/kg/day.16 These data suggested that a shorter therapy course may be possible.16,17 Some renal adverse events were reported; most occurred after day 21, generally in the groups receiving higher doses (> 2.0 mg/kg/day).16,17 A shorter treatment course could potentially enhance compliance with out-patient oral therapy, improve tolerability, and reduce costs if adequate efficacy could be maintained, particularly given a possible use in combination therapy.
The purpose of our phase IIb, open-label, randomized, clinical trial in VL patients was to determine the multiple-dose pharmacokinetic profile of sitamaquine and its metabolite desethyl-sitamaquine after administration of 2 mg/kg/day of sitamaquine with or without food for 21 days. This study also carefully investigated sitamaquine safety results from previous sitamaquine studies16–20 and compared the safety and efficacy of a 21-day course of oral sitamaquine with a 30-day course of AmB for the treatment of VL.
Patients and Methods
This prospective, open-label, randomized clinical study was conducted during August 2006–September 2007 at three centers in Bihar, India. Study procedures followed Good Clinical Practice and the Declaration of Helsinki. The protocol was reviewed and approved by the independent ethics committee for each study site, the Drug Controller General of India, and the Indian Council of Medical Research. All study participants or their guardians provided written informed consent; assent was required from children able to understand the study.
Study participants.
Eligible patients were men or women 16−50 years of age with VL symptoms or signs (fever, weight loss, hepatosplenomegaly) and Leishmania amastigotes in splenic aspirates or bone marrow. Exclusion criteria were renal, hepatic or biliary disease; renal or hepatic impairment; cardiac disease, arrhythmia, or conduction abnormalities; clinically relevant electrocardiogram (ECG) results or laboratory values; serious underlying disease or infection; glucose-6-phosphate dehydrogenase deficiency (based on phenotype testing); positive results for antibodies against human immunodeficiency virus, hepatitis B surface antigen, or antibodies to hepatitis C virus; contraindication to splenic or bone marrow aspiration; hypersensitivity to study treatments; treatment with an established anti-leishmania drug within 30 days or 5 half-lives (whichever was longer) of the start of the study; or treatment with prohibited medication. Pregnant or nursing women were excluded; a negative urine pregnancy test result was required from female patients at screening and before dosing, plus an agreement to use contraception for two weeks after the last treatment dose.
Study design and interventions.
No formal sample size calculation was performed. Target enrollment was 60 patients to achieve 32 evaluable patients in the sitamaquine group (8 per cohort) and 16 in the AmB cohort, assuming 20% discontinuation. Patients were randomized into blocks of 12 in the ratio 1:1:1:1:2 to one of four sitamaquine cohorts or AmB (Figure 1). Treatment was allocated by using the GlaxoSmithKline Registration and Medication Ordering System (RAMOS).
Figure 1.

Study design.
Randomized patients received either oral sitamaquine, 2 mg/kg/day, once a day, for 21 days (GlaxoSmithKline, Harlow, United Kingdom), or intravenous AmB given initially as 1 mg over 30 minutes and titrated over the first few treatment days to 1 mg/kg, every other day, for 30 days. Until January 2007, 12 (60%) of 20 patients received AmB as generic Fungicin® (Swiss Parenterals, Kerala, India). Subsequently, 8 (40%) of 20 patients received generic Fungizone® (Ambalal Sarabhai Enterprises Ltd., Vadodra, India). Patients in sitamaquine cohorts 1, 2 and 4 fasted for 8 hours (overnight) before dosing for all or part of the treatment period (Figure 1). Vomiting within one hour of dosing resulted in re-dosing. Vomiting within one hour of re-dosing led to withdrawal from the study and administration of rescue medication (liposomal amphotericin B). Therapy interruptions were allowed for AmB but not for sitamaquine. All patients were hospitalized during treatment, plus three days for those treated with sitamaquine or 2 days for those treated with AmB. Follow-up continued for 180 days post-study start with scheduled visits at days 49, 90, and 180 for those treated with sitamaquine and days 58, 90, and 180 for those treated with AmB.
Analytical methods.
Venous blood samples (3 mL) for pharmacokinetic analysis were collected on days 1, 10, and 21 immediately before dosing and at 1, 2, 3, 4, 6, 10, 16, and 24 hours post-dose. Additional samples were obtained on days 23 and 24 to approximate 48 hours and 72 hours after the last sitamaquine dose. After solid-phase extraction from human plasma, sitamaquine and desethyl-sitamaquine concentrations were determined at a central laboratory (Aptuit Inc., Edinburgh, Scotland) by using high-performance liquid chromatography with tandem mass spectrometric detection, a TurbolonSpray® interface (Applied Biosystems, Foster City, CA) and multiple reaction monitoring. The method was validated over the range 1−1000 ng/mL for both analytes using 50 μL of human plasma. Spiked standards and quality control (QC) samples were extracted daily. Within-run precision and bias, calculated by using interpolated concentrations of QC samples (prepared at 2.5, 500, 800, and 5,000 ng/mL of sitamaquine and desethyl-sitamaquine), were less than 10% across all QC levels, demonstrating acceptable performance.
Pharmacokinetic assessment.
Pharmacokinetic analysis of sitamaquine and desethyl-sitamaquine concentration–time data was conducted by using a non-compartmental Model 200 in WinNonlin Professional Edition (Version 5.2; Pharsight Corp. Mountain View, CA). Actual elapsed time after dosing from the individual concentration–time profiles for evaluable patients was used to derive the following pharmacokinetic parameters: area under the concentration−time curve over the dosing interval (AUC(0−τ)); maximum observed plasma concentration (Cmax); time to reach Cmax (tmax); terminal phase elimination rate constant (λz); apparent terminal half-life (t1/2); and observed accumulation ratio (Ro). For AUC(0−τ), where τ = 24 hours, i.e. AUC(0−24), physiologically unfeasible or missed concentrations at the 24-hour time point for 17 patients required substitution with AUC(0−16). To express pharmacokinetic parameter variability, pooled between-subject coefficient of variation and within-subject co-efficient of variation were calculated by using the relevant residual variance.
Safety assessment.
Physical examinations were performed and vital signs and adverse events were assessed at screening, baseline, throughout treatment, and at follow-up visits. Adverse events were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events v3.0,21 or if not listed, the Division of Microbiology and Infectious Diseases Adult Toxicity Table.22 Grade 3 or higher toxicities were deemed clinically relevant. Dose-limiting toxicity required study therapy to be stopped and rescue medication initiated. Serious adverse events were defined as those resulting in death (or life-threatening), hospitalization or prolonged hospitalization, disability or incapacity, or a congenital anomaly or birth defect, or were otherwise medically significant.
Venous blood samples (3 mL) for clinical laboratory analyses and urine samples for urinalysis were obtained at screening, baseline and days 5, 10, 16, 21, 90, and 180 for both study treatments and at day 49 for sitamaquine and days 30 and 58 for AmB. Clinical laboratory tests and urinalysis were performed at a central laboratory in Mumbai, India.
Duplicate, full 12-lead ECGs (ELI 150; Mortara Instruments, Milwaukee, WI) were recorded at screening, baseline (within 24 hours before dosing on day 1), and days 4, 9, 15, plus days 22 and 31 for sitamaquine-treated patients and days 49 and 58 for AmB-treated patients. On-treatment ECGs were recorded four hours after dosing. Transthoracic two-dimensional echocardiograms were performed at screening, on days 22 and 49 for sitamaquine-treated patients, and days 31 and 58 for AmB-treated patients. Ejection fraction was estimated by using the modified Simpson's rule. ECGs and two-dimensional echocardiograms were reviewed locally within 24 hours, but the safety analysis used readings by an independent cardiologist blinded to treatment and recording time.
Efficacy assessment.
Spleen size was measured by using ultrasound on day 1 and at end of therapy. Splenic aspirate (or bone marrow aspirate if splenic aspirate could not be obtained) was conducted at screening, end of therapy, day 90, and day 180 if clinical symptoms indicated relapse and on study withdrawal if the patient had received study therapy for at least 14 days. Samples were stained with Giemsa and parasite load was quantified by using the Leishmania index by a microscopist blinded to study treatment.23 At end of therapy, if the Leishmania index was +1, splenic aspirate was repeated 28 days later; if the Leishmania index was ≥ +2, the patient was classified as a treatment failure, withdrawn from the study, and given rescue medication. Efficacy endpoints were initial parasitologic cure (parasite-negative splenic aspirate at end of therapy or, if the Leishmania index was +1, a parasite-negative splenic aspirate 28 days later), and final clinical cure (initial parasitologic cure and no evidence of relapse at day 180).
Statistical analysis.
Primary pharmacokinetic endpoints were plasma AUC(0−τ), AUC(0–16), Cmax, tmax, and Ro; t1/2 was a secondary endpoint. All statistical analysis was performed by using SAS Version 8.2 (SAS Institute Inc., Cary, NC).
Food effect.
The effect of food on sitamaquine plasma AUC(0–τ), AUC(0–16), and Cmax and on desethyl-sitamaquine AUC(0–τ) and Cmax, was evaluated at day 10 and day 21. After loge transformation, a linear mixed-effects model was used, fitting day and treatment (fasted or fed) as fixed effects and patient as a random effect. Point estimates and associated 90% confidence intervals (CIs) for the difference in each pharmacokinetic parameter between fed and fasted treatment regimens were calculated. These values were exponentially back transformed to provide point estimates and 90% CIs for the fed:fasted ratio. The standard 90% CI bioequivalence range of 0.80−1.25 was used to interpret results. Additional effects such as weight and treatment dose were explored as covariates. For tmax, untransformed data were analyzed non-parametrically by using the Wilcoxon matched pairs method,24 and point estimates and 90% CIs were constructed for the estimated median difference fed–fasted.
Accumulation ratio.
Sitamaquine Ro was calculated by statistical analysis of sitamaquine and desethyl-sitamaquine AUC(0–τ). After loge transformation, a linear mixed-effects model was fitted and included the day, food status (fasted or fed), and their interaction as fixed effects and patient as a random effect. Within each food status, Ro was estimated by comparing values at day 10 or day 21 with values at day 1. Point estimates and associated 90% CIs for day 10 or day 21 versus day 1 for each food state were exponentially back transformed into their original scale. In addition, a combined estimate for Ro over fasted and fed treatments was determined. Additional effects such as weight and treatment dose were explored as covariates.
Secondary outcomes.
All other pharmacokinetic, safety, and efficacy comparisons were investigated by using descriptive statistics. For safety and efficacy evaluations, combined sitamaquine cohorts were compared with AmB.
Results
Patients.
The study design is shown in Figure 1. Of 176 patients screened, 61 were randomized; 41 received sitamaquine and 20 received AmB. There were four early withdrawals in the sitamaquine group (two adverse events, one lack of efficacy, and one relapse) and one in the AmB group (adverse event). Baseline characteristics are shown in Table 1; these were similar across cohorts, except for sex. All patients were Asian. The Leishmania index was 1 or 2 in 80% of patients. Mean (SD) compliance with study medication was 97.8% (11.5%) for sitamaquine and 96.3% (14.9%) for AmB.
Table 1.
Baseline clinical and demographic characteristics of patients receiving sitamaquine and amphotericin B for treatment of visceral leishmaniasis, Bihar, India*
| Characteristic | Sitamaquine (n = 41) | Amphotericin B (n = 20) |
|---|---|---|
| Age, years | 27.5 (9.7), 16–50 | 29.9 (9.3), 19–47 |
| Weight, kg | 44.2 (7.9), 30–69 | 46.8 (6.2), 35–58 |
| Height, cm | 158.8 (7.6) | 163.2 (7.0) |
| Male sex, no. (%) | 23 (56) | 13 (65) |
| Leishmaniasis index | 1.9 (0.8) | 2.0 (0.7) |
| No. (%) patients with following Leishmania index | ||
| 1 | 13 (32) | 5 (25) |
| 2 | 20 (49) | 11 (55) |
| 3 | 7 (17) | 4 (20) |
| 4 | 1 (2) | 0 |
| Spleen size, cm | 15.1 (2.2) | 14.4 (3.0) |
| Temperature, °C | 37.7 (0.9) | 37.0 (0.7) |
| Patients febrile up to day (≥ 38°C), no. positive/no. tested (%) | 23/41 (56) | 10/20 (50) |
Values are mean (SD), range unless otherwise indicated.
Pharmacokinetic outcomes.
Sitamaquine and desethyl-sitamaquine day 21 mean plasma concentration−time curves are shown in Figure 2. Sitamaquine and desethyl-sitamaquine pharmacokinetic parameters by cohort and day are shown in Table 2. Sitamaquine was slowly metabolized to desethyl-sitamaquine, and AUC(0−τ) and Cmax values were higher after multiple dosing at day 10 compared with day 1. For sitamaquine and desethyl-sitamaquine, AUC(0−τ) was greatest at day 10 in all cohorts.
Figure 2.
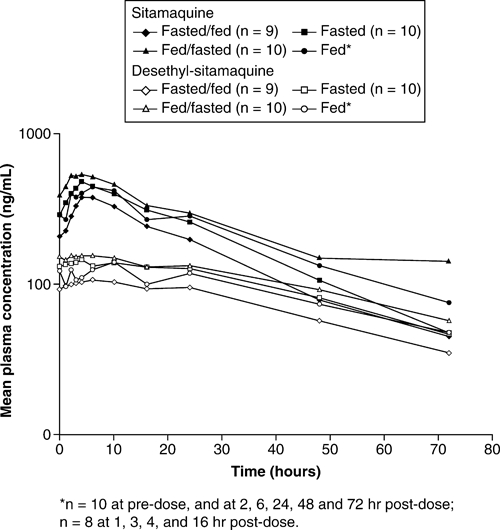
Day 21 mean plasma concentration−time curves for sitamaquine and desethyl-sitamaquine on a log−linear scale.
Table 2.
Key plasma sitamaquine and desethyl-sitamaquine pharmacokinetic parameters for patients with visceral leishmaniasis, Bihar, India*
| Parameter | Day | Sitamaquine | Desethyl-sitamaquine | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 (n = 11) | Cohort 2 (n = 10) | Cohort 3 (n = 10) | Cohort 4 (n = 10) | Cohort 1 (n = 11) | Cohort 2 (n = 10) | Cohort 3 (n = 10) | Cohort 4 (n = 10) | ||
| AUC(0–∞), ng.hr/mL | 1 | 8,120 (73.0) | 9,143 (54.3) | 8,574 (50.3)† | 7,147 (85.3) | 572 (90.4)‡ | 500 (54.8) | 573 (55.9)† | 484 (80.2) |
| 10 | 10,416 (51.3)‡ | 14,382 (50.8) | 10,616 (49.1) | 10,696 (37.2) | 3,125 (34.4)‡ | 4,316 (57.9) | 3,694 (56.0) | 3,512 (40.4) | |
| 21 | 6,627 (21.9)§ | 8,903 (56.2) | 7,723 (59.1) | 8,350 (27.7) | 2,307 (28.7)§ | 3,163 (49.4) | 2,667 (58.7) | 3,083 (26.4) | |
| AUC(0–16), ng.hr/mL | 1 | 5,820 (84.8) | 6,161 (65.8) | 5,830 (70.0)† | 5,003 (80.1) | NC | NC | NC | NC |
| 10 | 7,468 (48.8)‡ | 9,843 (52.4) | 7,779 (48.4) | 7,814 (32.2) | NC | NC | NC | NC | |
| 21 | 4,859 (27.0)§ | 6,633 (55.3) | 5,731 (53.8) | 6,125 (27.9) | NC | NC | NC | NC | |
| Cmax, ng/mL | 1 | 626 (81.3) | 718 (56.4) | 639 (50.7) | 577 (97.8) | 36.1 (75.8) | 33.3 (49.0) | 40.8 (34.1)¶ | 30.3 (80.2) |
| 10 | 582 (50.0)‡ | 810 (43.8) | 659 (47.7) | 637 (31.5) | 148 (33.2)‡ | 214 (49.3) | 183 (56.8) | 174 (41.7) | |
| 21 | 401 (39.6)§ | 570 (53.8) | 451 (44.4) | 503 (28.4) | 109 (34.5)§ | 154 (55.2) | 127 (58.4) | 146 (21.8) | |
| t1/2, hr# | 21 | 18.9 (28.5)† | 22.8 (31.9)† | 19.3 (24.3)§ | 18.3 (24.3) | 27.1 (25.8)† | 23.0 (36.9)** | 25.5 (33.4)¶ | 27.9 (35.6)¶ |
| tmax, hr | 1 | 3.00 [1.00–24.0] | 6.00 [1.00–24.0] | 6.00 [20.0–24.0]† | 5.00 [2.00–24.0] | 23.8 (10.0–24.0) | 23.7 (16.0–24.0) | 23.8 (16.0–24.0) | 19.9 (16.0–24.0) |
| 10 | 5.96 [0–10.0]‡ | 6.00 [3.00–23.8] | 3.50 [1.75–10.0] | 4.00 [3.00–23.8] | 5.09 (0–16.0)‡ | 7.00 (3.00–23.8) | 2.42 (0.75–10.0) | 4.96 (1.00–16.0) | |
| 21 | 6.00 [3.00–10.0]§ | 4.00 [0–6.00] | 3.50 [1.97–10.0] | 4.00 [0–10.0] | 3.83 (0–16.0)§ | 2.00 (0–4.00) | 10.0 (0–16.0) | 3.50 (0–10.0) | |
Values are geometric mean (between-subject coefficient of variation), except for tmax, which is median [range]. AUC = area under curve; NC = not calculated; Cmax = maximum plasma concentration; t1/2 = apparent terminal half-life; tmax, = time to reach Cmax.
n = 8.
n = 10.
n = 9.
n = 7.
t1/2 was not calculated at days 1 and 10 because of the lack of adequate elimination phase concentration−time data.
n = 4.
Food effect.
Within-subject crossover comparison of cohorts 1 and 2 are shown in Table 3. Point estimates close to unity indicated no food effect on sitamaquine AUC(0−τ), AUC(0−16), or Cmax, or on desethyl-sitamaquine AUC(0−τ) or Cmax. Intra-subject variability for AUC(0−τ) and Cmax was moderate to high for sitamaquine and desethyl-sitamaquine (maximum 29.6% and 24.0%, respectively). Adding body weight and dose to the model as covariate effects showed similar results. In cohorts 1 and 2, a within-subject crossover comparison indicated that tmax was 1.52 hours longer (90% CI = 0–3.92) for sitamaquine and 3.00 hours longer (90% CI = −1.00 to 7.50) for desethyl-sitamaquine in a fed versus fasted state; these differences were not significant, as indicated by the 90% CIs including 0. Between-subject parallel-group comparison of cohorts 3 and 4 showed sitamaquine tmax was 0.17 hours shorter for the fed versus fasted state on day 10 (90% CI = −2.25 to 2.00) and no difference for day 21 (90% CI = −1.03 to 4.00). For desethyl-sitamaquine, tmax was 1.25 hours shorter for the fed versus fasted state on day 10 (90% CI = −4.92 to 1.00) and 6 hours longer in a fed versus fasted state at day 21 (90% CI = 0–8.00). However, for all between-subject comparisons of tmax, confidence intervals were wide and included 0, indicating that none of the differences were significant.
Table 3.
Within-subject assessment of food effect on sitamaquine and desethyl-sitamaquine plasma pharmacokinetics on days 10 and 21 in patients with visceral leishmaniasis after repeated once a day oral administration of sitamaquine, 2 mg/kg/day (cohorts 1 and 2), Bihar, India*
| Analyte | Parameter | Comparison | Point estimate† | 90% CI | CVw% |
|---|---|---|---|---|---|
| Sitamaquine | AUC(0−τ) | Fed:fasted | 1.01 | (0.88–1.16) | 29.6 |
| AUC(0−16) | Fed:fasted | 0.99 | (0.86–1.12) | 28.3 | |
| Cmax | Fed:fasted | 0.99 | (0.87–1.13) | 28.7 | |
| Desethyl-sitamaquine | AUC(0−τ) | Fed:fasted | 0.99 | (0.88–1.11) | 23.2 |
| Cmax | Fed:fasted | 1.00 | (0.89–1.12) | 24.0 |
CI = confidence interval; CVw% = within-subject coefficient of variation; AUC = area under curve; Cmax = maximum plasma concentration.
Point estimate is the ratio of adjusted geometric means between fed and fasted states.
Accumulation ratio.
Results of assessment of Ro for sitamaquine and desethyl-sitamaquine are shown in Table 4. For combined sitamaquine cohorts, AUC(0−τ) values for fasted and fed patients increased 1.4-fold on day 10 and decreased marginally on day 21 compared with day 1. The extent of accumulation was greater for desethyl-sitamaquine; combined AUC(0−τ) increased from day 1 to day 10 by 6.8-fold and to day 21 by 5.2-fold. Adding body weight and dose as covariates to the model showed similar results.
Table 4.
Assessment of Ro for sitamaquine and desethyl-sitamaquine plasma pharmacokinetic parameters in patients with visceral leishmaniasis, Bihar, India*
| Analyte | Comparison | Point estimate† | 90% CI | CVw% |
|---|---|---|---|---|
| Sitamaquine | Fed day 10:day 1 | 1.35 | (1.12–1.63) | 35.5 |
| Fed day 21:day 1 | 0.82 | (0.66–1.01) | ||
| Fasted day 10:day 1 | 1.38 | (1.16–1.66) | ||
| Fasted day 21:day 1 | 1.08 | (0.89–1.32) | ||
| All patients day 10:day 1 | 1.37 | (1.20–1.56) | ||
| All patients day 21:day 1 | 0.94 | (0.82–1.07) | ||
| Desethyl-sitamaquine | Fed day 10:day 1 | 7.25 | (5.92–8.88) | 38.5 |
| Fed day 21:day 1 | 4.53 | (3.62–5.66) | ||
| Fasted day 10:day 1 | 6.30 | (5.18–7.66) | ||
| Fasted day 21:day 1 | 5.85 | (4.72–7.24) | ||
| All patients day 10:day 1 | 6.75 | (5.87–7.78) | ||
| All patients day 21:day 1 | 5.15 | (4.46–5.93) |
CI = confidence interval; CVw% = within-subject coefficient of variation.
Point estimate is the ratio of adjusted geometric means between repeat dosing days (days 21 or 10) and single dose day 1.
Safety outcomes.
Adverse events.
During therapy, 4 (10%) of 41 patients in the sitamaquine group experienced an adverse event versus 17 (85%) of 20 patients in the AmB group (Table 5). No serious adverse events occurred during treatment and two occurred during follow-up. Both patients received sitamaquine, neither event was considered drug-related by the investigator: acute gastroenteritis four months after completing therapy, resulting in death and study withdrawal; and cholelithiasis. Adverse events led to study withdrawal for two other patients: one received sitamaquine (grade 4 toxicity: low neutrophil count at day 7, grade 3 toxicity at baseline), and one received AmB (low creatinine clearance at day 16); neither was considered drug related. Two patients stopped treatment prematurely but completed follow-up: one received sitamaquine (increased protein:creatinine ratio at day 16) and one AmB (request of the patient at day 15).
Table 5.
Frequency of adverse events occurring during treatment of patients with visceral leishmaniasis with sitamaquine or amphotericin B of any cause, Bihar, India
| Adverse event | Sitamaquine (n = 41), no. (%) | Amphotericin B (n = 20), no. (%) |
|---|---|---|
| At least one adverse event | 4 (10) | 17 (85) |
| Chills (rigors) | 0 | 16 (80) |
| Vomiting | 0 | 5 (25) |
| Gastritis | 0 | 3 (15) |
| Pyrexia | 0 | 2 (10) |
| Peripheral edema | 0 | 1 (5) |
| Urine protein:creatinine ratio increased | 2 (5) | 0 |
| Headache | 1 (2) | 1 (5) |
| Diarrhea | 0 | 1 (5) |
| Creatinine clearance decreased | 0 | 1 (5) |
| Urticaria | 0 | 1 (5) |
| Urinary tract infection | 0 | 1 (5) |
| Neutrophil count decreased | 1 (2) | 0 |
Hematologic results.
Baseline, end of therapy, and change from baseline values for key hematologic parameters are shown in Table 6. For both treatment groups, mean hemoglobin level decreased initially, followed by a gradual recovery after day 5. In sitamaquine-treated patients, methemoglobin levels increased after day 5 (Figure 3A), peaking between days 16 and 21, but returning to baseline by day 49 in all patients. Four patients had methemoglobin levels of 5−9.9% and one patient had a value > 10% (maximum = 10.8%). No clinical signs or symptoms were associated with increased methemoglobin levels and no treatment was necessary.
Table 6.
Mean (SD) values for key laboratory indices at baseline, the end of therapy, and change from baseline at the end of therapy, in patients treated with sitamaquine and amphotericin B for visceral leishmaniasis, Bihar, India*
| Parameter | Baseline | At end of therapy† | Change from baseline | |||
|---|---|---|---|---|---|---|
| Sitamaquine (n = 40) | Amphotericin B (n = 18) | Sitamaquine (n = 40) | Amphotericin B (n = 18) | Sitamaquine (n = 40) | Amphotericin B (n = 18) | |
| Hemoglobin, g/L | 96.4 (19.6) | 104.2 (27.0) | 108.0 (16.2) | 103.7 (17.3) | 11.4 (11.3) | −0.9 (22.8) |
| Methemoglobin, % | 0.89 (0.28) | 0.88 (0.30) | 1.89 (2.48) | 0.77 (0.15) | 1.00 (2.53) | −0.10 (0.35) |
| Total neutrophils, GI/L | 1.8 (0.8) | 2.1 (1.2) | 2.4 (1.2) | 4.5 (2.9) | 0.54 (1.51) | 2.26 (3.22) |
| Platelet count, GI/L | 154.4 (81.7) | 161.7 (73.8) | 249.9 (97.8) | 259.8 (110.2)‡ | 95.4 (90.0) | 93.5 (103.1)‡ |
| Leukocyte count, GI/L | 4.3 (2.1) | 4.3 (2.1) | 5.7 (1.9) | 8.1 (3.7) | 1.4 (2.2) | 3.5 (4.5) |
| ALP, IU/L | 110.1 (116.6) | 79.0 (45.7) | 90.6 (33.7) | 72.5 (20.2) | −20.7 (105.2) | −10.3 (53.2) |
| ALT, IU/L | 44.8 (29.9) | 46.4 (34.5) | 35.4 (22.8) | 31.3 (16.2) | −10.1 (36.6) | −18.6 (36.8) |
| AST, IU/L | 52.6 (28.1) | 42.5 (23.4) | 34.4 (19.8) | 24.2 (8.5) | −18.9 (36.6) | −19.9 (26.8) |
| Total bilirubin, μmol/L | 11.9 (9.9) | 13.8 (9.5) | 12.2 (5.7) | 13.8 (9.8) | 0.23 (6.73) | −0.27 (6.37) |
| Serum creatinine, μmol/L | 69.9 (13.1) | 71.2 (12.3) | 68.7 (10.5) | 90.9 (35.2) | −1.3 (8.9) | 22.1 (37.0) |
| Creatinine clearance, mL/minute | 83.0 (18.5) | 86.7 (17.9) | 84.5 (16.8) | 70.8 (21.1) | 1.2 (10.8) | −17.2 (25.3) |
| Urine total protein:creatinine ratio | 0.31 (0.29) | 0.24 (0.24) | 2.58 (7.22) | 0.36 (0.28)‡ | NA | NA |
| Creatine kinase MB, μg/L | 4.4 (4.7) | 2.6 (1.8) | 3.2 (3.2) | 2.4 (1.3) | −0.99 (4.94) | −0.25 (2.25) |
| Troponin I, μg/L | 0.51 (1.41) | 0.30 (0.22) | 0.21 (0.03) | 0.20 (0.01)§ | −0.31 (1.43) | −0.10 (0.24)§ |
ALP = alkaline phosphatase; ALT = alanine aminotransferase; AST = aspartate aminotransferase; NA = not available (change from baseline not calculated because this is a ratio).
Day 21 for sitamaquine, day 30 for AmB, except for troponin I where day 21 values are used for both treatment groups (not tested at day 30); paired data were missing for one patient in the sitamaquine group and for two patients in the amphotericin B group.
n = 17.
n = 16.
Figure 3.
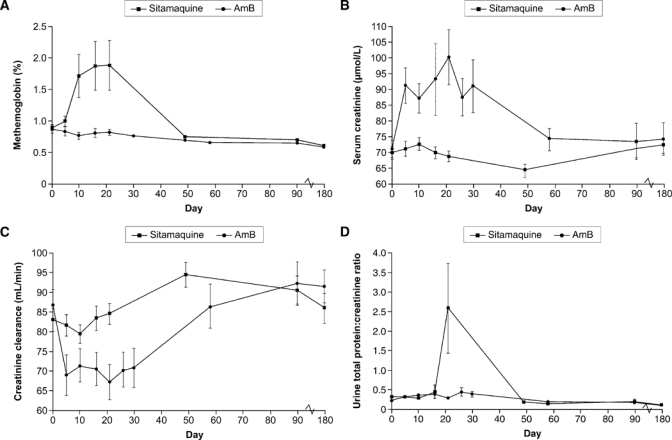
Laboratory parameters throughout the study for which there were clinically important differences between treatment groups: A, methemoglobin level (mean ± SE); B, serum creatinine level (mean ± SE); C, creatinine clearance (mean ± SE); and D, urine total protein:creatinine ratio (mean ± SE).
Clinical chemistry results.
Baseline, end of therapy, and change from baseline values for key clinical chemistry parameters are shown in Table 6. Mean values for alkaline phosphatase, alanine aminotransferase, and aspartate aminotransferase (AST) levels were increased at baseline, although toxicities were grade 2 or less. For both study treatments, hepatic enzyme levels generally decreased during treatment and remained stable after the end of therapy. One AST grade 3 toxicity occurred at day 58 in the AmB group. Mean total bilirubin level was similar between treatment groups with no post-baseline grade 3 or 4 toxicities. Mean serum creatinine level remained stable in the sitamaquine group throughout the study, but increased in the AmB group, peaking at 100 μmol/L on day 21 (Figure 3B). There were no grade 3 or 4 toxicities for serum creatinine in either group. Ten (50%) patients in the AmB group had grade 1 or 2 toxicities during therapy, which were reversible in all cases. At day 90, one patient in each treatment group had a grade 1 toxicity, which was considered unrelated to study treatment. Mean creatinine clearance decreased in patients treated with AmB until day 5, then recovered slowly, with another decrease at approximately day 21 (Figure 3C and Table 7). In the sitamaquine group, creatinine clearance remained generally stable.
Table 7.
Creatinine clearance grade 1 and 2 toxicities in patients treated with sitamaquine and amphotericin B for visceral leishmaniasis, Bihar, India
| Time point | Sitamaquine | Amphotericin B | ||||
|---|---|---|---|---|---|---|
| No. | Grade 1, no. (%) | Grade 2, no. (%) | No. | Grade 1, no. (%) | Grade 2, no. (%) | |
| Baseline | 41 | 3 (7) | 0 | 20 | 1 (5) | 0 |
| Day 5 | 41 | 4 (10) | 0 | 20 | 7 (35) | 2 (10) |
| Day 10 | 37 | 4 (11) | 0 | 19 | 5 (26) | 1 (5) |
| Day 16 | 40 | 2 (5) | 0 | 19 | 4 (21) | 1 (5) |
| Day 21 | 40 | 4 (10) | 0 | 16 | 5 (31) | 1 (6) |
| Day 26 | 40 | 1 (3) | 0 | 17 | 7 (41) | 0 |
| Day 30 | – | – | – | 18 | 3 (17) | 2 (11) |
| Day 58 | – | – | – | 19 | 1 (5) | 0 |
| Day 90 | 38 | 2 (5) | 0 | 19 | 2 (11) | 0 |
| Day 180 | 33 | 2 (6) | 0 | 18 | 0 | 0 |
Urinalysis.
The urine protein:creatinine ratio was > 1.0 in 7 (17%) of 41 patients during sitamaquine treatment; all had normal baseline values (Table 6 and Figure 3D). Six patients had increases of > 3.5 during treatment (mostly after day 16) and maximum changes occurred at approximately day 21. Proteinuria was asymptomatic and reversible and returned to normal/baseline values within 20 days or by the next scheduled visit, except for one patient where the protein:creatinine ratio improved but remained increased at day 180. Plasma sitamaquine and desethyl-sitamaquine AUC(0−τ) and Cmax for the seven patients with protein:creatinine ratios > 1.0 during treatment were within the range of those for patients with normal protein:creatinine ratios. There were no clinically relevant changes in protein:creatinine ratio for AmB-treated patients. Hematuria (> 3 erythrocytes/high-power [HP] field) was observed during treatment in 10 (24%) of 41 patients treated with sitamaquine (maximum value = 10–12 erythrocytes/HP field) and 3 (15%) of 20 patients treated with AmB (maximum value = 20–25 erythroctes/HP field) (Figure 4). Hematuria resolved after therapy completion in both treatment groups. Two patients (both women) in the sitamaquine-treated group had unexplained hematuria (> 100 erythrocytes/HP field) at day 180 and no other findings on renal function or urinalysis. Increased urine leukocyte count was observed for two patients in the AmB-treated group and none in the sitamaquine-treated group.
Figure 4.
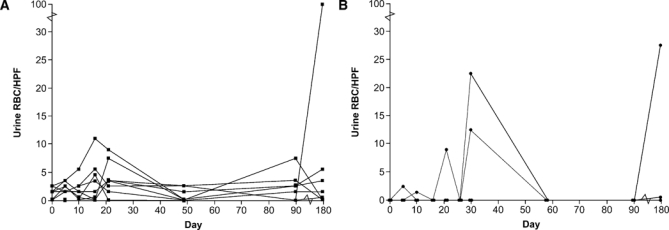
Erythrocytes per high-power field in urine of patients (individual patient profiles) for A, sitamaquine and B, amphotericin B.
Cardiac safety.
One patient in each treatment group had abnormal ECGs at baseline (flat T waves) and end of therapy. Abnormal end of therapy ECGs were recorded for three other patients receiving sitamaquine (flat T waves, sinus tachycardia, first-degree atrioventricular block), and two receiving AmB (ST depression, left anterior hemiblock). There were no differences in QTc changes from baseline between treatment groups. Maximum changes from baseline (≥ 60 msec) were observed in one (3%, QTcB) or two (5%, QTcF) sitamaquine-treated patients, and one (5%, QTcB or QTcF) AmB-treated patient on day 15. In the AmB group, further changes ≥ 60 msec from baseline were observed: two (11%) on day 4 and one (6%) on day 58 (QTcB, QTcF). Maximum values for individual patients were 478 msec on day 22 for sitamaquine and 483 msec on day 58 for AmB. None of the patients with ECG changes had any cardiologic adverse events noted. For two-dimensional echocardiogram, there was no clinically relevant difference between treatment groups in mean left ventricular ejection fraction at end of therapy or follow-up (day 49 for sitamaquine and day 58 for AmB). There were no clinically relevant changes from baseline in creatine kinase muscle–brain in either treatment group (Table 6). Troponin I values were unremarkable during treatment (Table 6), except for one patient receiving sitamaquine with a normal baseline value, but a Day 10 value of 4.1 μg/L. This patient was asymptomatic and had a normal ECG and normal troponin I values at all subsequent tests. Two patients receiving sitamaquine had increased troponin I levels at baseline with no symptoms and no ECG abnormalities; results of troponin I tests two days later and throughout the study were normal. One patient who received AmB had an increased troponin I level at baseline.
Efficacy outcomes.
Initial parasitologic cure was observed in 39 (95%) of 41 patients (95% CI = 83.5–99.4) receiving sitamaquine versus 19 (95%) of 20 patients (95% CI = 75.1–99.9) receiving AmB. Final clinical cure at day 180 was achieved in 35 (85%) of 41 patients (95% CI = 70.8–94.4) receiving sitamaquine versus 19 (95%) of 20 patients (95% CI = 75.1–99.9) receiving AmB. Individual and median sitamaquine and desethyl-sitamaquine AUC(0−τ) and Cmax for the six patients who showed treatment failure for sitamaquine at day 180 were within the range of those patients who achieved final clinical cure. The mean (SD) change in spleen size at end of therapy versus baseline (Table 1) was similar for the two treatment groups: −2.58 cm (1.4 cm) for sitamaquine (day 22) and −2.38 cm (1.8 cm) for AmB (day 31). When we adjusted for treatment, baseline spleen size and center produced similar results: −2.52 cm for sitamaquine, −2.70 cm for AmB (95% CI for difference = −0.549 to 0.924). The mean (SD) change in body weight from baseline (Table 1) to the end of therapy was 0.53 kg (0.8 kg) for sitamaquine and 1.5 kg (1.2 kg) for AmB. Approximately half of all patients were febrile at baseline (Table 1). One-fourth of the febrile patients had a temperature < 38°C by day 1 (95% CI = 0.1–7.2) in the sitamaquine-treated group versus day 8 (95% CI = 1.0–2.0) in the AmB-treated group. All patients were afebrile by day 36 in the sitamaquine group and day 15 in the AmB group.
Discussion
This phase IIb, randomized, open-label, prospective study found no effect of food on sitamaquine or desethyl-sitamaquine pharmacokinetics in VL patients. These data suggest that sitamaquine can be dosed regardless of food intake.
Drug accumulation was expected to be between 1-fold and 2-fold for sitamaquine and its desethyl metabolite on the basis of the estimated t1/2 of 18−28 hours, i.e. expected Ro was 1.44 (t1/2/dosing interval). Sitamaquine Ro, assessed relative to day 1 was 1.37 at day 10 and 0.94 at day 21, which were somewhat lower than expected. Ro for desethyl-sitamaquine, relative to day 1 was 7.0 at day 10 and 5.75 at day 21, which were considerably higher than expected. These results could be explained by sitamaquine induction of its own metabolism after repeat dosing. Another possible explanation may be the improving health of the patients during the 21-day dosing period. In particular, sitamaquine is metabolized by several cytochrome P450 pathways and improved liver function, as shown by decreases in hepatic enzymes, may have resulted in increased sitamaquine metabolism. However, further investigations are required to better understand these findings.
Treatment with sitamaquine was generally well tolerated; 10% of patients reported adverse events versus 85% with AmB. This difference was mostly because of infusion reactions with AmB. These adverse events are broadly consistent with the known safety profile for AmB.7,25,26 This study also carefully investigated hematologic, hepatic, renal, and cardiac safety signals identified in pre-clinical and previous clinical sitamaquine studies.16–20
Increased methemoglobin levels are a known class effect of 8-aminoquinolines; all patients are at risk regardless of glucose-6-phosphate dehydrogenase status. In the previous phase II study in India (sitamaquine, 1.5−2.5 mg/kg for 28 days), laboratory reports showed 40 (33%) of 120 patients with methemoglobin level increases ≥ 10% (maximum = 22.5%, dose = 1.75 mg/kg/day); six patients were symptomatic.16 In the current study, methemoglobin levels were 5−9.9% for four patients and ≥ 10% in one patient (maximum = 10.8%). Methemoglobin level increases were reversible after the end of therapy and asymptomatic. Sitamaquine caused methemoglobin level increases from day 5. Therefore, it is unlikely that the shorter sitamaquine treatment duration in the current study explains the lower effect on methemoglobin levels than in the previous phase II study in India and these findings remain unexplained.
No hepatic adverse events were reported for either treatment in this study, and there was no evidence of hepatotoxicity. Hepatic function was generally abnormal at baseline and tended to normalize during therapy in both treatment groups. Results of previous studies are generally consistent with these findings. In the phase II sitamaquine study in Kenya, 8 (8%) of 97 patients had increased AST levels as an adverse event, although six of eight patients had increased AST levels at baseline.17 There were no reports of hepatic enzyme adverse events in the previous phase II study in India.16
Nephrotoxicity is a known safety issue with AmB,8 and serum creatinine levels were increased and creatinine clearance decreased versus baseline. Renal function was generally stable with sitamaquine. Two sitamaquine-treated patients had a transient decrease in creatinine clearance versus baseline (25% and 46%); values returned to baseline within two weeks after completion of therapy. Two patients had asymptomatic hematuria (normalized by day 90 and day 180). Proteinuria was the most significant renal safety finding with sitamaquine and was observed in 7 of 41 patients, 6 with a protein:creatinine ratio increased from baseline by > 3.5. Proteinuria was observed after day 16 and maximum changes were observed at approximately day 21. These changes were reversible after drug discontinuation. One patient with an increased urine protein:creatinine ratio withdrew from the study on day 16. All patients with proteinuria were asymptomatic and none showed development of nephrotic syndrome or glomerulonephritis. There were no serious renal adverse events. This finding is in contrast to previous clinical studies with sitamaquine in which glomerulonephritis, nephrotic syndrome, and acute renal failure were reported.16–20 However, most of these events occurred at sitamaquine doses > 2–3.5 mg/kg/day after day 21 of a 28-day therapy course.16–20 Our findings suggest that sitamaquine, 2 mg/kg/day for 21 days, has reduced the incidence and severity of renal adverse events.
Cardiovascular effects had been seen in pre-clinical sitamaquine studies in dogs, although these were not corroborated in subsequent telemetered dog safety studies. Cardiomyocyte degeneration had also been noted in a monkey dose-ranging study. Previous clinical studies reported bradycardia and some non-specific ECG changes, but data were limited.16,17 In this study, non-specific ECG abnormalities were noted for patients in both treatment groups; there were no clinical symptoms or correlation with cardiac enzymes. There were no instances of bradycardia. Results for two-dimensional echocardiogram and cardiac enzymes provided no evidence of cardiotoxicity for either treatment group.
For sitamaquine, the final cure rate was 85% after 21 days of therapy, and these results are consistent with final cure rates from previous trials of sitamaquine, 2 mg/kg/day for 28 days, for patients from India (n = 28) and Africa (n = 61): 100% (95% CI = 85.2–100%) and 80.3% (95% CI = 68.2–89.4%), respectively.16,17 The small number of patients in our study and previous sitamaquine studies means that 95% CIs are broad (68.2–100%). Sitamaquine efficacy in VL remains to be proven in larger studies, possibly in combination with other drugs. Clinical efficacy with AmB was consistent with recent reports of VL patients in India.25,26
This study showed that sitamaquine can be administered regardless of food intake. Sitamaquine, 2.0 mg/kg/day for 21 days, was generally well tolerated, and although renal adverse events were evident, these events appeared to be reversible and manageable. The 21-day regimen had final cure rates similar to those of previous studies with 28-day therapy. These data indicate the potential usefulness of sitamaquine for treatment of patients with VL. The pharmacokinetic and safety findings are of particular use should the drug be used in a combination therapy.
ACKNOWLEDGMENTS
We thank Dr. Jaya Chakravarty, Dr. Madhukar Rai, Dr. Anshul Kumar Gupta (Kala-azar Medical Research Centre, Institute of Medical Sciences, Banaras Hindu University, Varanasi, India), R. B. Verma, S. B. Burman, Dr. Arun Kr. Singh, Dr. Nawin Kumar, Naresh Kumar Sinha, and Umesh Kumar (Rajendra Memorial Research Institute of Medical Sciences, Agamkuan Patna Bihar, India) for technical assistance. We also thank Naomi Richardson (Magenta Communications Ltd.) for developing a first draft of this paper from the approved study report and collating author contributions. The study team consisted of the following co-investigators: Dr. Nawin Kumar, Dr. V. N. R. Das, Dr. Krishna Pandey, Dr. Neena Verma (Rajendra Memorial Research Institute of Medical Sciences, Agamkuan Patna, Bihar, India), Dr. Anshul Kumar Gupta, Dr. Deepak Verma, Dr. Shahnawaj Alam, and Dr. Poonam Kumari (Kala-azar Medical Research Centre, Institute of Medical Sciences, Banaras Hindu University, Varanasi, India). This study is registered in the public domain at ClinicalTrials.gov with the identifier NCT00381394. The study has the GlaxoSmithKline identifier STQ105938.
Footnotes
Financial support: Sitamaquine development to date has been supported by GlaxoSmithKline PLC.
Disclosure: GlaxoSmithKline conducted the study and collected and analyzed data. All authors had access to the primary data and take responsibility for data reporting accuracy and completeness. The corresponding authors had responsibility for the final decision to submit for publication. Shyam Sundar has received support for clinical trials and travel funds to attend scientific meetings from Paladin Labs, Institute for One World Health, GlaxoSmithKline, Bharat Serum and Vaccine Ltd., Drugs for Neglected Diseases Initiative, National Institute of Allergy and Infectious Diseases, National Institutes of Health, World Health Organization, and the European Commission. Prabhat K. Sinha has received support from the Institute for One World Health, GlaxoSmithKline, Drugs for Neglected Diseases Initiative, World Health Organization, Médecins Sans Frontières (Barcelona) for clinical trials and travel funds to attend scientific meetings. Susan A. Dixon, Renata Buckley, Ann K. Miller, and Khadeeja Mohamed are employees of GlaxoSmithKline as noted in the affiliations. Mahir Al-Banna is a contractor employed by GlaxoSmithKline as noted in the affiliations.
Authors' addresses: Shyam Sundar, Kala-azar Medical Research Centre, Institute of Medical Sciences, Banaras Hindu University, Varanasi, India, E-mail: drshyamsundar@hotmail.com. Prabhat K. Sinha, Rajendra Memorial Research Institute of Medical Sciences, Indian Council of Medical Research, Agamkuan Patna, Bihar 800 007, India, E-mail: pksinha18@yahoo.com. Susan A. Dixon, Infectious Diseases–Medicines Development Centre, Diseases of the Developing World, GlaxoSmithKline Research and Development, Stockley Park West, Uxbridge, Middlesex UB11 1BT, United Kingdom, E-mail: susan.x.dixon@gsk.com. Renata Buckley, Global Clinical Safety and Pharmacovigilance, GlaxoSmithKline Research and Development, Stockley Park West, Uxbridge, Middlesex UB11 1BT, United Kingdom, E-mail: renata.2.buckley@gsk.com. Ann K. Miller, Clinical Pharmacology, Modelling and Simulation, Quantitative Sciences, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, PA 19406, E-mail: ann.k.miller@gsk.com. Khadeeja Mohamed, Infectious Diseases–Medicines Development Centre, Biomedical Data Sciences, GlaxoSmithKline Research and Development, Stockley Park West, Uxbridge, Middlesex, United Kingdom, E-mail: khadeeja.m.mohamed@gsk.com. Mahir Al-Banna, Al-Banna Consulting, LLC, c/o GlaxoSmithKline Biostatistics and Programming Management, GlaxoSmithKline, 1250 South Collegeville Road, PO Box 5089, Collegeville, PA 19426-0989, E-mail: mahir.2.al-banna@gsk.com.
References
- 1.World Health Organization . Regional Strategic Framework for Elimination of Kala-azar from the South-East Asia Region (2005–2015) New Delhi: World Health Organization; 2005. http://www.searo.who.int/LinkFiles/Kala_azar_VBC-85_Rev_1.pdf Available at. Accessed April 27, 2010. [Google Scholar]
- 2.Singh SP, Reddy DC, Rai M, Sundar S. Serious underreporting of visceral leishmaniasis through passive case reporting in Bihar, India. Trop Med Int Health. 2006;11:899–905. doi: 10.1111/j.1365-3156.2006.01647.x. [DOI] [PubMed] [Google Scholar]
- 3.Joshi A, Narain JP, Prasittisuk C, Bhatia R, Hashim G, Jorge A, Banjara M, Kroeger A. Can visceral leishmaniasis be eliminated from Asia? J Vector Borne Dis. 2008;45:105–111. [PubMed] [Google Scholar]
- 4.Thakur CP, Narayan S, Ranjan A. Epidemiological, clinical and pharmacological study of antimony-resistant visceral leishmaniasis in Bihar, India. Indian J Med Res. 2004;120:166–172. [PubMed] [Google Scholar]
- 5.Das VN, Ranjan A, Bimal S, Siddique NA, Pandey K, Kumar N, Verma N, Singh VP, Sinha PK, Bhattacharya SK. Magnitude of unresponsiveness to sodium stibogluconate in the treatment of visceral leishmaniasis in Bihar. Natl Med J India. 2005;18:131–133. [PubMed] [Google Scholar]
- 6.Croft SL, Sundar S, Fairlamb AH. Drug resistance in leishmaniasis. Clin Microbiol Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundar S, Jha TK, Thakur CP, Sinha PK, Bhattacharya SK. Injectable paromomycin for visceral leishmaniasis in India. N Engl J Med. 2007;356:2571–2581. doi: 10.1056/NEJMoa066536. [DOI] [PubMed] [Google Scholar]
- 8.Deray G. Amphotericin B nephrotoxicity. J Antimicrob Chemother. 2002;49((Suppl 1)):37–41. doi: 10.1093/jac/49.suppl_1.37. [DOI] [PubMed] [Google Scholar]
- 9.Meheus F, Boelaert M, Baltussen R, Sundar S. Costs of patient management of visceral leishmaniasis in Muzaffarpur, Bihar, India. Trop Med Int Health. 2006;11:1715–1724. doi: 10.1111/j.1365-3156.2006.01732.x. [DOI] [PubMed] [Google Scholar]
- 10.Barratt G, Legrand P. Comparison of the efficacy and pharmacology of formulations of amphotericin B used in treatment of leishmaniasis. Curr Opin Infect Dis. 2005;18:527–530. doi: 10.1097/01.qco.0000191508.48481.f4. [DOI] [PubMed] [Google Scholar]
- 11.More B, Bhatt H, Kukreja V, Ainapure S. Miltefosine: great expectations against visceral leishmaniasis. J Postgrad Med. 2003;49:101–103. doi: 10.4103/0022-3859.911. [DOI] [PubMed] [Google Scholar]
- 12.Sundar S, Gupta LB, Makharia MK, Singh MK, Voss A, Rosenkaimer F, Engel J, Murray HW. Oral treatment of visceral leishmaniasis with miltefosine. Ann Trop Med Parasitol. 1999;93:589–597. doi: 10.1080/00034989958096. [DOI] [PubMed] [Google Scholar]
- 13.Jha TK, Sundar S, Thakur CP, Bachmann P, Karbwang J, Fisher C, Voss A, Berman J. Miltefosine, an oral agent, for the treatment of Indian visceral leishmaniasis. N Engl J Med. 1999;341:1795–1800. doi: 10.1056/NEJM199912093412403. [DOI] [PubMed] [Google Scholar]
- 14.Prasad R, Kumar R, Jaiswal BP, Singh UK. Miltefosine: an oral drug for visceral leishmaniasis. Indian J Pediatr. 2004;71:143–144. doi: 10.1007/BF02723096. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharya SK, Sur D, Sinha PK, Karbwang J. Elimination of leishmaniasis (kala-azar) from the Indian subcontinent is technically feasible and operationally achievable. Indian J Med Res. 2006;123:195–196. [PubMed] [Google Scholar]
- 16.Jha TK, Sundar S, Thakur CP, Felton JM, Sabin AJ, Horton J. A phase II dose-ranging study of sitamaquine for the treatment of visceral leishmaniasis in India. Am J Trop Med Hyg. 2005;73:1005–1011. [PubMed] [Google Scholar]
- 17.Wasunna MK, Rashid JR, Mbui J, Kirigi G, Kinoti D, Lodenyo H, Felton JM, Sabin AJ, Albert MJ, Horton J. A phase II dose-increasing study of sitamaquine for the treatment of visceral leishmaniasis in Kenya. Am J Trop Med Hyg. 2005;73:871–876. [PubMed] [Google Scholar]
- 18.Dietze R, Carvalho SF, Valli LC, Berman J, Brewer T, Milhous W, Sanchez J, Schuster B, Grogl M. Phase 2 trial of WR6026, an orally administered 8-aminoquinoline, in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am J Trop Med Hyg. 2001;65:685–689. doi: 10.4269/ajtmh.2001.65.685. [DOI] [PubMed] [Google Scholar]
- 19.Sherwood JA, Gachihi GS, Muigai RK, Skillman DR, Mugo M, Rashid JR, Wasunna KM, Were JB, Kasili SK, Mbugua JM, Kirigi G, Shaefer KU, Oster CN, Fleckenstein LL, Berman JD, Brewer TG, Roberts CR, Johnson AJ, Schuster BG. Phase 2 efficacy trial of an oral 8-aminoquinoline (WR6026) for treatment of visceral leishmaniasis. Clin Infect Dis. 1994;19:1034–1039. doi: 10.1093/clinids/19.6.1034. [DOI] [PubMed] [Google Scholar]
- 20.Yeates C. Sitamaquine (GlaxoSmithKline/Walter Reed Army Institute) Curr Opin Investig Drugs. 2002;3:1446–1452. [PubMed] [Google Scholar]
- 21.National Cancer Institute . Common Terminology Criteria for Adverse Events v3.0 (CTCAE) Bethesda, MD: National Cancer Institute; 2003. http://ctep.cancer.gov/reporting/ctc.html Available at. Accessed April 27, 2010. [Google Scholar]
- 22.National Institute of Allergy and Infectious Diseases (NIAID), US, Division of Microbiology and Infectious Diseases Adult Toxicity Table 2001 [Google Scholar]
- 23.Chulay JD, Bryceson AD. Quantitation of amastigotes of Leishmania donovani in smears of splenic aspirates from patients with visceral leishmaniasis. Am J Trop Med Hyg. 1983;32:475–479. doi: 10.4269/ajtmh.1983.32.475. [DOI] [PubMed] [Google Scholar]
- 24.Steinijans VW, Diletti E. Statistical analysis of bioavailability studies: parametric and nonparametric confidence intervals. Eur J Clin Pharmacol. 1983;24:127–136. doi: 10.1007/BF00613939. [DOI] [PubMed] [Google Scholar]
- 25.Sundar S, Chakravarty J, Rai VK, Agrawal N, Singh SP, Chauhan V, Murray HW. Amphotericin B treatment for Indian visceral leishmaniasis: response to 15 daily versus alternate-day infusions. Clin Infect Dis. 2007;45:556–561. doi: 10.1086/520665. [DOI] [PubMed] [Google Scholar]
- 26.Thakur CP, Narayan S. A comparative evaluation of amphotericin B and sodium antimony gluconate, as first-line drugs in the treatment of Indian visceral leishmaniasis. Ann Trop Med Parasitol. 2004;98:129–138. doi: 10.1179/000349804225003154. [DOI] [PubMed] [Google Scholar]