

Abstract
In this paper we have investigated the hypothesis that neural activity causes rapid activation of TrkB neurotrophin receptors in the adult mammalian CNS. These studies demonstrate that kainic acid-induced seizures led to a rapid and transient activation of TrkB receptors in the cortex. Subcellular fractionation demonstrated that these activated Trk receptors were preferentially enriched in the synaptosomal membrane fraction that also contained postsynaptic glutamate receptors. The fast activation of synaptic TrkB receptors could be duplicated in isolated cortical synaptosomes with KCl, presumably as a consequence of depolarization-induced BDNF release. Importantly, TrkB activation was also observed following pharmacological activation of brain-stem noradrenergic neurons, which synthesize and anterogradely transport BDNF; treatment with yohimbine led to activation of cortical TrkB receptors within 30 min. Pharmacological blockade of the postsynaptic α1-adrenergic receptors with prazosin only partially inhibited this effect, suggesting that the TrkB activation was partially due to a direct effect on postsynaptic cortical neurons. Together, these data support the hypothesis that activity causes release of BDNF from presynaptic terminals, resulting in a rapid activation of postsynaptic TrkB receptors. This activity-dependent TrkB activation could play a major role in morphological growth and remodelling in both the developing and mature nervous systems.
The neurotrophic factor hypothesis, which is largely based on the neurotrophin family of growth factors (for review, see Levi-Montalcini 1987; Thoenen and Barde 1989; Snider 1994), postulates that trophic factors produced and released by target neurons regulate the survival, differentiation, and morphological growth of their innervating neurons (for review, see Oppenheim 1991; Majdan and Miller 1999). However, recent studies suggest that at least one member of the neurotrophin family, brain-derived neurotrophic factor (BDNF) (Barde et al. 1982; Leibrock et al. 1989), acts as an anterograde trophic factor that is derived from afferent neurons (von Bartheld et al. 1996; Altar et al. 1997; Conner et al. 1997; Fawcett et al. 1998). In particular, BDNF has been localized to both axons and terminals of peripheral (Zhou and Rush 1996; Michael et al. 1997) and central neurons (Conner et al. 1997; Fawcett et al. 1997, 1998), and the TrkB/BDNF receptor (Klein et al. 1991; Soppet et al. 1991) has been localized to neuronal dendrites in both the hippocampus and cortex (Fryer et al. 1996; Yan et al. 1997a), where at least a subpopulation of these receptors is present in postsynaptic densities (Wu et al. 1996; Lin et al. 1998). Moreover, this anterogradely trafficked BDNF has the potential to affect the survival and differentiation of target CNS neurons, at least during development (Fawcett et al. 1998).
A number of recent studies also indicate that BDNF is localized to vesicles in presynaptic terminals in vivo (Fawcett et al. 1997; Michael et al. 1997), that it may be released in an activity-dependent fashion (Goodman et al. 1996; Mowla et al. 1999), and that following intense neural activity such as during kindling, Trk receptors are autophosphorylated (Binder et al. 1999), raising the interesting possibility that BDNF secretion in the mature nervous system could be regulated in a manner similar to neuropeptides (Mowla et al. 1999). The consequences of activity-dependent release of BDNF in the mature nervous system might be several. First, BDNF could play a more traditional role in regulating the morphology and, potentially, the survival of mature target neurons, a role analogous to that proposed for anterogradely transported BDNF during development (Fawcett et al. 1998). Second, BDNF could play a novel role for a trophic factor, modulating neuronal excitability either directly and/or by modification of the phosphorylation state of postsynaptic neurotransmitter receptors (Jarvis et al. 1997; Suen et al. 1997; Lin et al. 1998). Finally, postsynaptic signaling events resulting from BDNF-mediated TrkB receptor activation could synergize with signaling events caused by neurotransmitter receptor activation and/or calcium influx (Meyer-Franke et al. 1995; McAllister et al. 1996; Vaillant et al. 1999), raising the possibility that presynaptic corelease of a neurotransmitter and BDNF could have more dramatic effects on the postsynaptic neuron than the release of either of these stimuli alone. Such activity-dependent release of BDNF at central synapses could play an essential role both during development and in the adult. For example, during development, appropriate formation of ocular dominance columns is absolutely dependent on appropriate afferent activity (for review, see McAllister et al. 1999), and either application of exogenous BDNF (Cabelli et al. 1995) or disruption of endogenous BDNF (Cabelli et al. 1997) is sufficient to perturb this developmental process. Moreover, in the mature hippocampus, BDNF can modulate the strength of synaptic transmission at both the presynaptic and postsynaptic neuron (Kang and Schuman 1995, 1996; Levine et al. 1995, 1998; Gottschalk et al. 1998), and elimination of one BDNF allele in the BDNF+/− mice is sufficient to dampen long-term potentiation (LTP), an effect that can be rescued by the addition of exogenous BDNF (Korte et al. 1996; Patterson et al. 1996).
On the basis of these considerations, we have hypothesized that BDNF may function, at least in part, by activity-dependent release from presynaptic terminals and subsequent activation of postsynaptic TrkB receptors. In this paper we have tested this hypothesis and demonstrate that elevated activity leads to rapid activation of synaptic TrkB receptors in the cortex and that pharmacological activation of brain-stem noradrenergic neurons, which synthesize and anterogradely traffic BDNF, is sufficient to cause rapid TrkB receptor activation on postsynaptic cortical neurons.
Materials and Methods
ANIMAL STUDIES
Animals were treated with kainic acid as we have described previously (Fawcett et al. 1997). Briefly, adult female Sprague-Dawley rats (250–300 grams) (Charles River, Montreal, Canada) were injected subcutaneously with kainic acid (10 mg/kg) or with vehicle according to an animal care protocol meeting the standards of the Canadian Council on animal care. Only those animals that achieved status epilepticus and whose behavior was similar to kindled stage 5 seizures (Racine 1972) were used for these experiments; all animals showed head bobbing and “wet-dog shakes” at 20 min, and seizures lasted for ∼3 hr. Alternatively, CD1 adult mice (20–25 grams) were injected subcutaneously with yohimbine (2.5 mg/kg) and/or prazosin (1 mg/kg), or vehicle alone. After the different treatments, the animals were sacrificed by rapid decapitation at time periods ranging from 20 min to 3 days after injection. The sensory and motor cortices were then dissected and analyzed biochemically.
TISSUE ANALYSIS AND SUBCELLULAR FRACTIONATION
After dissection, tissue was homogenized in cold Tris-lysis buffer (0.1 ml/1 gram tissue), containing 137 mm NaCl, 20 mm Tris (pH 8.0), 1% (vol/vol) NP-40, 10% (vol/vol) glycerol, 1 mm PMSF, 10 mg/ml aprotinin, 0.2 mg/ml leupeptin, and 1.5 mm sodium vanadate. Following homogenization in a polytron and incubation for 20 min at 4°C, the lysate was centrifuged at 13,200 rpm for 15 min. The resulting supernatant was then recentrifuged (13,200 rpm, 15 min) to provide a supernatant for analysis. Supernatants were subjected to protein determination using the BSA standards from Pierce Biotechnology.
Subcellular fractionation was performed by differential centrifugation as described previously (Huttner et al. 1983; Fawcett et al. 1997). Briefly, the cortex was removed and homogenized in 10 mm HEPES (pH 7.3), 0.3 m sucrose, 1 mm PMSF, 10 mg/ml aprotinin, 0.2 mg/ml leupeptin, and 1.5 mm sodium orthovanadate. Crude synaptosomes (P2) were separated from cytosol (S) and microsomes (P3) by differential centrifugation of a postnuclear supernatant. Crude synaptosomes (P2) were ruptured by osmotic shock and quickly returned to osmotic balance with the addition of 1 m HEPES (pH 7.4). Enriched synaptosomal membranes (LP1) were separated from a microsomal (LS1) fraction by centrifugation at 160,000 rpm using an SS-34 Sorval rotor. To obtain the synaptic microsome (LP2) fraction, the LS1 fraction was spun for 2 hr at 260,000g. Small aliquots from each fraction were kept for protein determination using the BSA kit from Pierce biotechnology.
ANTIBODIES, IMMUNOPRECIPITATION, AND WESTERN BLOT ANALYSIS
The following, previously described antibodies were used for the biochemical studies described here: anti-TrkBin, which specifically recognizes the intracellular domain of TrkB (Fryer et al. 1996); anti-TrkBout, which specifically recognizes the extracellular domain of full-length and truncated TrkB (Knusel et al. 1994); anti-TrkCin, which recognizes the intracellular domain of TrkC (Belliveau et al. 1997); anti-panTrk 203, which recognizes an intracellular portion of the kinase domain common to all Trk receptors (Hempstead et al. 1992); anti-BDNF (Santa Cruz) (Causing et al. 1997; Fawcett et al. 1997, 1998); anti-phosphotyrosine 4G10 (UBI); anti-synaptotagmin (StressGen Biotechnologies); anti-synaptophysin (Boehringer Mannheim); anti-TGN 38 (the kind gift of Dr. Katherine Howell, University of Colorado); anti-GluR1; and anti-TrkA (RTA; the kind gift of Dr. Louis Reichardt, University of California at San Francisco) (Clary et al. 1994).
Western blot analysis of TrkB and BDNF proteins were essentially performed as described (Fawcett et al. 1997, 1998). Total Trk protein was immunoprecipitated from tissue lysates using either 3 μl of anti-panTrk 203 or 50 μl of wheat germ agglutinin (Pharmacia), which binds to glycosylated proteins, including the full-length and truncated forms of all Trk receptors. The anti-panTrk immunoprecipitates were collected with protein A–Sepharose (Pharmacia) for 1.5 hr at 4°C followed by centrifugation.
For Western blot analysis, immunoprecipitates were washed three times with cold lysis buffer, boiled in sample buffer (2% SDS, 100 mm dithiothreitol, 10% glycerol, and 0.05% bromophenol blue) for 5 min, and electrophoresed on 7.5% SDS–polyacrylamide minigels for all proteins except BDNF, which was analyzed on 15% gels. After electrophoresis, proteins were transferred to 0.2-mm nitrocellulose (Bio-Rad) for 1.5 hr at 0.6 amps, and the membrane was washed 2× 10 min in TBS. For all antibodies except anti-phosphotyrosine, for which membranes were blocked in 2% BSA (Sigma Chemical Co), membranes were blocked in 5% nonfat dry milk, in TBS with 0.1% Tween 20 (TBST) for 1.5 hr. Membranes were then washed 2 × 10 min in TBST, and the primary antibodies were used overnight at 4°C at dilutions of 1:10,000 for anti-phosphotyrosine 4G10 (UBI), 1:2000 for anti-panTrk 203, 1:5000 for anti-TrkBout, anti-TrkBin, and anti-TrkCin, and 1:5000 for anti-BDNF (Santa Cruz). Blots were washed 3 × 10 min in TBST. Secondary antibodies were incubated for 1.5 hr at room temperature and were used at dilutions of 1:10,000 for a goat anti-mouse HRP antibody and 1:5000 for a goat anti-rabbit HRP antibody (Boehringer Mannheim Biochemicals). Detection was performed using enhanced chemiluminescence (Amersham) and XAR X-ray film (Kodak). Results were quantitated by image analysis from at least three independent experiments. Western blot replicas were scanned using a flatbed scanner (Umax, Astra 600S) at 600 dpi, and saved into Adobe Photoshop 4.0. Scanned images were then opened in NIH Image (Release V1.61, Wayne Rasband, National Institutes of Health, Bethesda, MD). Areas of interest were defined, and mean pixel densities of these areas were determined. Statistical significance was determined using Student’s t-test for pairwise comparisons and ANOVA for group comparisons, with a Bonferroni post hoc test.
ANALYSIS OF SYNAPTOSOMES
To generate synaptosomes for stimulation experiments, we followed the protocol in McPherson et al. (1994), with the following modifications: Two rat brain cortices were used for each experiment. The cortex was removed and homogenized in 10 mm HEPES (pH 7.4), 0.3 m sucrose (1:10 wt/vol). The resulting homogenate was centrifuged for 5 min at 2600 rpm using a SS-34 Sorval rotor. This gave rise to a postnuclear supernatant (PNS). The PNS was then spun at 10,000 rpm for 15 min to give a crude synaptosomal preparation (P2). This synaptosomal preparation (P2) was then washed by resuspending it in 20 ml of fresh homogenization buffer. This was then spun at 11,000 rpm for 15 min. Only the beige pellet was taken. This cleaned synaptosomal preparation was then diluted 1.6-fold in a control buffer containing 20 mm HEPES (pH 7.4), 128 mm NaCl, 3 mm KCl, 1.2 mm MgCl2, 1 mm CaCl2, and 11 mm glucose. The resulting suspension was then equally divided into 1-ml aliquots and washed three times with the control buffer. After the third wash, the synaptosomes were left to rest for 1 hr at 37°C.
For BDNF experiments, BDNF (Preprotech) was added at a final concentration of 100 ng/ml, for 7 min. After stimulation, the synaptosomes were quickly spun down, the supernatant was removed, and tubes were snap frozen in liquid nitrogen. For KCl depolarization, the washed synaptosomes were incubated in a depolarization buffer containing 20 mm HEPES, 16.5 mm NaCl, 117 mm KCl, 1.2 mm MgCl2, 1 mm CaCl2, and 11 mm glucose for 10 min at 37°C. After stimulation the synaptosomes were spun down, supernatant was removed, and the synaptosomes were snap frozen in liquid nitrogen and stored for further analysis.
The frozen synaptosomes were resuspended in Tris–lysis buffer to give a final concentration of 137 mm NaCl, 20 mm Tris (pH 8.0), 1% (vol/vol) NP-40, 10% (vol/vol) glycerol, 1 mm PMSF, 10 μg/ml aprotinin, 0.2 μg/ml leupeptin, and 1.5 mm sodium vanadate. Protein was extracted, and Trk protein was immunoprecipitated from an equal amount of protein from each aliquot. The immunoprecipitated Trk was then run on SDS-PAGE and transferred and probed with anti-phosphotyrosine or anti-TrkBout, as outlined above. The resulting Western blot replicas were then scanned and analyzed as described above.
Results
BOTH PRECURSOR AND MATURE BDNF ARE INCREASED IN THE CORTEX FOLLOWING KAINIC ACID-INDUCED SEIZURE ACTIVITY
To test the hypothesis that neuronal activity leads to neurotrophin secretion and subsequent Trk receptor activation in the mature brain, we used kainic acid, which causes widespread neuronal activity and which is known to induce synthesis of neurotrophins, including BDNF (Dugich-Djordjevic et al. 1992). Specifically, we treated rats with 10 mg/kg kainic acid for time points ranging from 20 min to 3 days and analyzed the cortex for alterations in BDNF levels and concurrent activation of TrkB. Only those animals that showed motor seizure activity were analyzed further. We chose the sensory and motor cortex, because there is little cell loss in this region following kainic acid treatment for up to 12 hr, although some cortical cell loss is observed at 3 days (Dugich-Djordjevic et al. 1992; Kasof et al. 1995). We also focused on BDNF and TrkB because (1) TrkB is partially localized to dendrites in the cortex (Fryer et al. 1996; Yan et al. 1997b), where it is the predominant Trk receptor, and (2) BDNF is axonally transported in CNS neurons (von Bartheld et al. 1996; Altar et al. 1997; Conner et al. 1997; Fawcett et al. 1998) and is packaged into vesicles (Fawcett et al. 1997), making it a prime candidate for activity-dependent release.
Initially, we measured levels of BDNF following kainic acid, using an antibody we have shown previously to be specific for BDNF (Causing et al. 1997; Fawcett et al. 1997, 1998). Western blot analysis of equal amounts of protein isolated from the cortex of individual treated animals revealed that 20 min following kainic acid treatment, levels of mature BDNF were decreased ∼40% (Fig. 1C). By 2 hr, BDNF levels had recovered, and levels continued to increase until 12 hr postkainic acid, at which point they were approximately sevenfold higher than controls (Fig. 1A,C). By 3 days, levels of mature BDNF had decreased and were not significantly elevated relative to controls (Fig. 1B,C).
Figure 1.
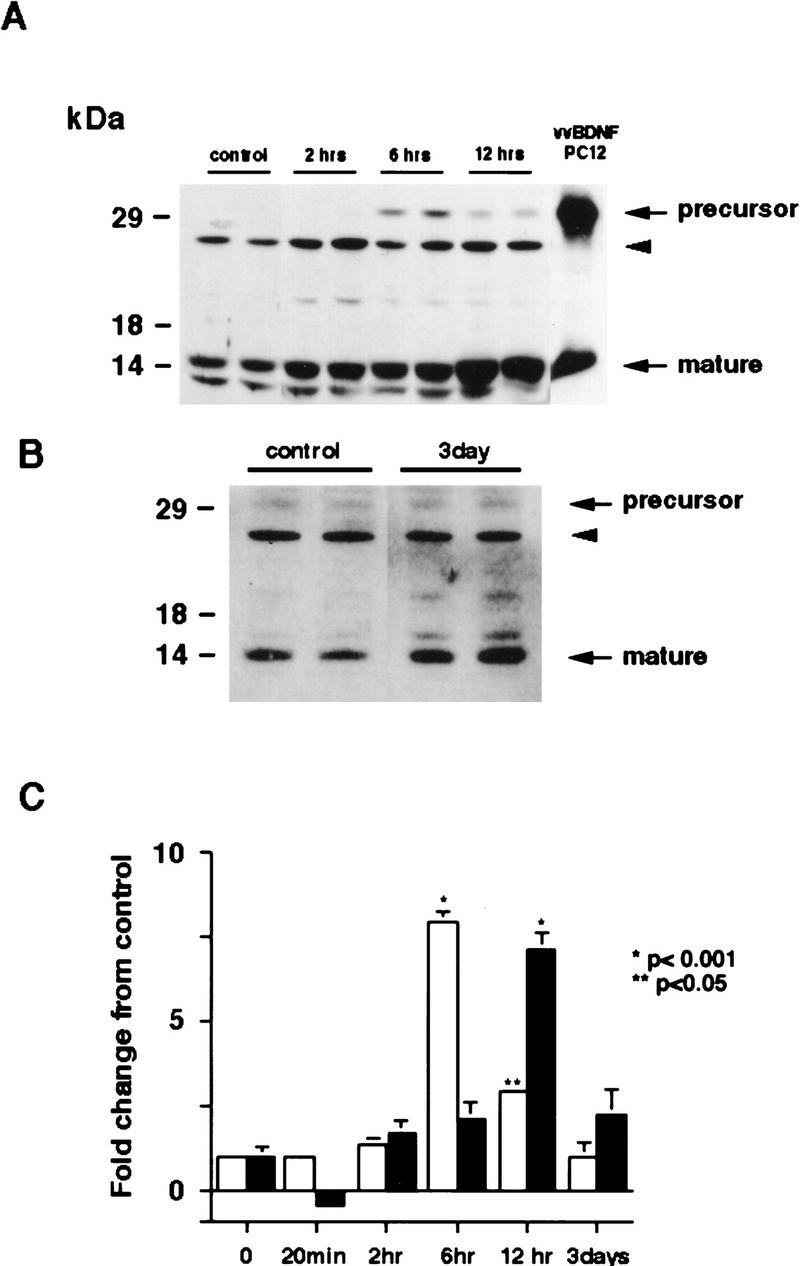
Increased synthesis of BDNF in the cortex of kainic acid-treated animals. (A,B) Western blot analysis of equal amounts of protein derived from the cortex of individual control animals or of animals 2 hr (2 hrs), 6 hr (6 hrs), 12 hr (12 hrs) (A), or 3 days (B) following kainic acid treatment. As a control, lysates of PC12 cells infected with a vaccinia virus encoding BDNF (vv BDNF PC12) (Fawcett et al. 1997; Mowla et al. 1999) were also run. The numbers at left indicate the molecular mass markers; the arrows at right indicate the 32-kD BDNF precursor and mature, processed BDNF. The arrowhead indicates a nonspecific, cross-reactive band (see text). (C) Quantitation of experiments similar to those shown in A and B. Western blots were scanned, the intensity of the bands quantitated by image analysis, and these numbers were then normalized to the numbers obtained from control animals analyzed on the same Western blots. The graph represents the mean fold change from controls at time points ranging from 20 min to 3 days, and the error bars indicate the s.e.m. (Open bars) BDNF precursor; (solid bars) mature BDNF. For each time point, n = 4 animals. Statistical significance was determined using ANOVA, and results that are significantly different from controls are indicated by the asterisks (*) P < 0.001; (**) P < 0.05.
Interestingly, a second BDNF-immunoreactive band of 32 kD, the size of BDNF precursor protein (Mowla et al. 1999), was also observed in these experiments (Fig. 1A,B). This precursor was first detected at 2 hr following kainic acid treatment and reached highest levels at 6 hr (Fig. 1A,C). By 12 hr, levels of BDNF precursor started to decrease and by 3 days were again similar to controls (Fig. 1A–C). This time course is consistent with the previously described increase in BDNF mRNA in the occipital cortex, which commences at 1 hr and reaches a maximum at 4 hr (Dugich-Djordjevic et al. 1992). The other immunoreactive bands running at 13 kD and 25 kD have been reported previously (Fawcett et al. 1997) and are nonspecific cross-reactive bands recognized by the BDNF antibody.
CORTICAL TRK RECEPTORS ARE ACTIVATED 20 MIN AFTER KAINIC ACID INJECTION
The fast decrease in BDNF protein observed 20 min following kainic acid suggested that BDNF was being secreted in an activity-dependent fashion and subsequently degraded. If this hypothesis is true, then we would predict a similarly quick activation of the BDNF receptor, TrkB, in response to this secreted BDNF. To test this prediction, we initially confirmed that the vast majority of Trk receptors in the cortex were TrkB. Specifically, cortical lysates from control rats were precipitated with wheat germ agluttinin (WGA), which binds to all glycosylated proteins, or with an antibody that recognizes the kinase domain of all Trk receptors (anti-panTrk), and these precipitates were then probed on Western blots with antibodies to the extracellular domains of TrkA, TrkB, and TrkC (Fig. 2A, left panel). Precipitation with WGA demonstrated that the predominant Trk receptor in the cortex is TrkB, with minor amounts of TrkA and no detectable TrkC (Fig. 2A). Precipitation with anti-panTrk confirmed these results and also demonstrated that TrkC was present in the adult cortex, albeit at low levels (Fig. 2A, right panel).
Figure 2.



Rapid activation of cortical Trk receptors following kainic acid treatment. (A) Western blot analysis of cortical tissue derived from adult rats precipitated with either WGA (lectin) or anti-panTrk (pan-Trk) and then probed with antibodies specific to TrkA, TrkB [both full-length (FL-TrkB) and truncated (TrkB-T) forms], or TrkC. Note that TrkB is the most abundant Trk in the adult cortex and that although TrkC is not detectable in the WGA precipitates, it can be detected at low levels when cortical tissue is immunoprecipitated with anti-panTrk. The numbers at left of the blots indicate the molecular mass markers. (B) Western blot analysis of equal amounts of protein isolated from the cortex of naive, control animals (0) or of adult rats at 20 min and 6 hr following kainic acid treatment. In the case of BDNF (top), total lysates were probed with a BDNF-specific antibody. The same samples were also immunoprecipitated with anti-panTrk, which recognizes all Trk receptors, and the immunoprecipitates were probed with anti-phosphotyrosine (anti-pTyr) (middle) or with an antibody specific for the full-length form of TrkB (anti-trkBin). In both cases, the arrows indicate the size of full-lengthTrkB, whereas in the middle panel, the arrowhead indicates a tyrosine phosphorylated protein that coimmunoprecipitates with Trk and whose identity is currently unknown. The numbers at left of the blots indicate the molecular mass markers. Note that although similar amounts of TrkB are present at all time points, the level of Trk receptor autophosphorylation is increased at 20 min following kainic acid treatment. (C) Quantitation of three separate experiments similar to that shown in B. Western blots were scanned, the intensity of the bands quantitated by image analysis, and the data were expressed as the ratio of phosphoTrk to full-length TrkB levels. These ratios were then normalized to the ratio for control animals analyzed on the same blots. The graph represents the mean fold change from controls at 20 min, 2 hr, and 6 hr, and the error bars indicate the s.e.m. For each time point, n = 4 animals. Asterisks (*) indicate those points that are statistically different from the controls using ANOVA (P < 0.01).
We then determined whether TrkB receptors were activated following kainic acid treatment, by examining receptor autophosphorylation. Specifically, cortical lysates were immunoprecipitated with an antibody that recognizes all full-length Trk receptors, and these immunoprecipitates were analyzed by Western blot analysis with anti-phosphotyrosine (Fig. 2B). Western blots were then reprobed for full-length TrkB (Fig. 2B), to allow normalization of the level of Trk receptor tyrosine phosphorylation to the total level of TrkB receptors. This analysis revealed a statistically significant two- to threefold increase in the ratio of Trk autophosphorylation to TrkB levels at 20 min following kainic acid treatment (Fig. 2B,C). By 6 hr, this ratio had again decreased to control levels (Fig. 2B,C). A second, phosphotyrosine-positive protein of ∼90 kD was also observed on these blots (Fig. 2B); this protein, whose identity is currently unknown, coprecipitated with Trk receptors in the anti-panTrk immunoprecipitations. Interestingly, the increase in Trk receptor activation corresponds to the time point when BDNF levels are decreased (Figs. 1C and 2B), presumably as a consequence of secretion in response to kainic acid. We interpret this specific increase in TrkB autophosphorylation as reflecting an acute activation event in response to activity-dependent BDNF secretion.
ACTIVATED TRK RECEPTORS ARE ENRICHED AT SYNAPSES
If activity-dependent secretion of presynaptic BDNF causes rapid TrkB receptor activation, then the tyrosine phosphorylated TrkB that we observe should be localized to synapses. To test this prediction, we used subcellular fractionation to determine TrkB localization; the cortices of control or kainic acid-treated rats were fractionated according to well-characterized procedures (Huttner et al. 1983; Fawcett et al. 1997), and the fractions were then analyzed by Western blot analysis. Initially, we characterized the subcellular fractions using the following proteins whose enrichment has previously been demonstrated (Fig. 3A): TGN38, a marker for the trans-Golgi apparatus (Luzio et al. 1990); synaptotagmin (Walch-Solimena et al. 1993) and synaptophysin (Fletcher et al. 1991), two proteins associated with large dense core and synaptic vesicles; and the NMDA receptor subunit GluR1 (Ehlers et al. 1995; Garcia et al. 1995), a postsynaptic neurotransmitter receptor found in the synaptic membrane fraction (Fig. 3A). This analysis demonstrated that TGN 38 was most enriched in the P3 microsomal fraction, synaptotagmin and synaptophysin in the LP2 fraction, which contains synaptic vesicles, and GluR1 in the LP1 fraction, which is composed of enriched pre- and postsynaptic membranes.
Figure 3.




Activated Trk is enriched in synaptic fractions following kainic acid treatment. (A) Adult cortical tissue was fractionated, and Western blot analysis was performed for a variety of marker proteins to ensure the accuracy of the subcellular fractionation. Equal amounts of protein from each fraction were probed with antibodies specific for TGN 38, a marker for the Golgi apparatus that is enriched in P3, for synaptotagmin and synaptophysin, two synaptic vesicle markers that are enriched in LP2, and for GluR1, the postsynaptic glutamate receptor that is enriched in LP1 (a fraction containing synaptic membranes) and in P2 (the fraction from which LP1 and LP2 are derived). H indicates the original tissue homogenate, and S is a cytosolic fraction. (B) Equal amounts of protein from the cortex of animals that were kainic acid treated for 20 min were immunoprecipitated with anti-panTrk, and the immunoprecipitates were analyzed by Western blot analysis with anti-phosphotyrosine (top) or anti-TrkBout (bottom). The arrows at right of both panels indicate the size of full-length TrkB, and the numbers at left indicate the molecular mass markers. Note that although TrkB is present in most fractions, tyrosine phosphorylated Trk is most enriched in LP1. (C) Equal amounts of protein from subcellular fractions derived from the cortex of animals that were kainic acid treated for 20 min were precipitated with WGA (lectin) and then probed with antibodies against phosphotyrosine (top), against all forms of TrkB (middle), or against synaptotagmin, a marker for synaptic vesicles (bottom). (Top) The arrow indicates a tyrosine phosphorylated band the size of full-length TrkB. (Middle) One arrow indicates the full-length TrkB receptor (FL TrkB), whereas the second indicates the truncated TrkB receptor (TrkB-T). For all panels, numbers at left of the blots indicate the molecular mass markers. (D) Equal amounts of protein from subcellular fractions of the cortex of control animals or of animals that were treated with kainic acid for 12 hr were precipitated with WGA (lectin) and then probed with antibodies against phosphotyrosine (top) or against the TrkB receptor (bottom). At top, the arrow indicates a tyrosine phosphorylated protein of the same size as full-length TrkB; at bottom, the arrows indicate both the full-length (FL TrkB) and truncated (TrkB-T) forms of TrkB.
We then characterized the distribution of phosphorylated Trk receptors in these same subcellular fractions. Specifically, subcellular fractions were prepared from the cortex 20 min (Fig. 3B,C) or 12 hr (Fig. 3D) following kainic acid treatment and were precipitated with anti-panTrk (Fig. 3B) or with WGA (Fig. 3C,D). These precipitates were probed with anti-phosphotyrosine to visualize Trk activation levels and then reprobed with anti-TrkB or anti-panTrk to measure total Trk levels in each fraction (Fig. 3B–D). This analysis demonstrated that, in control animals, autophosphorylated Trk receptors were slightly enriched in both LP1 and LP2 fractions (Fig. 3D), although levels of Trk autophosphorylation were low. Reprobing of these same samples for TrkB revealed that both the full-length and truncated forms of TrkB were present at the highest levels in P3 and in LP1; the TrkB seen in the P3 fraction presumably includes newly synthesized TrkB receptors being trafficked via the Golgi apparatus, whereas the relative enrichment in LP1 indicates that much of the TrkB is present at the synapse itself.
Twenty minutes following kainic acid treatment, immunoprecipitation with anti-panTrk followed by Western blot analysis revealed that autophosphorylated Trk receptors were highly enriched in LP1 and, to a lesser degree, in LP2 and P2 (the latter being the fraction from which LP1 and LP2 are derived) (Fig. 3B). Reprobing of the same blots with anti-TrkBout revealed that full-length TrkB receptors were now present in all subcellular fractions (Fig. 3B), with some degree of enrichment in P2, LP1, and LP2. Confirmation of these results was obtained using WGA precipitations (Fig. 3C). As seen following anti-panTrk immunoprecipitation, a tyrosine phosphorylated band of the same size as full-length TrkB was most enriched in LP1, with some degree of enrichment in LP2 and P2 (Fig. 3C). Reprobing of this blot with anti-TrkBout revealed that both full-length and truncated TrkB were distributed throughout the fractions, with some enrichment in LP1 (Fig. 3C). In contrast to activated Trk, synaptotagmin was highly enriched in LP2 in these samples (Fig. 3C), as seen in control animals (Fig. 3A).
Twelve hours following kainic acid treatment, activated Trk receptors were still dramatically enriched in LP1 and, to some degree, in P2 (Fig. 3D). However, at this later time point, the relative amount of total full-length and truncated TrkB present in LP1 was relatively reduced, as indicated by reprobing of the same samples with anti-TrkBout (Fig. 3D). This shift in distribution of TrkB protein may be due to a specific down-regulation of total TrkB receptor levels at the synapse, because there is no alteration in levels of total full-length TrkB following kainic acid treatment (Dugich-Djordjevic et al. 1995; Rudge et al. 1998). Thus, following kainic acid treatment, activated Trk receptors are preferentially enriched in the LP1 synaptic membrane fraction, the same fraction that is enriched in the postsynaptic receptor GluR1, consistent with the idea that kainic acid causes rapid release of BDNF from neuronal terminals, resulting in rapid activation of synaptic TrkB receptors.
DEPOLARIZATION-INDUCED ACTIVATION OF TRK RECEPTORS IN SYNAPTOSOMES
To confirm that the increase in synaptic TrkB receptor activation observed in the kainic acid experiments was due to neuronal activity, we turned to cortical synaptosomes. We chose the synaptosomal preparation, because (1) this preparation contains synaptic profiles (Huttner et al. 1983), (2) we have shown previously that BDNF was enriched within this fraction (Fawcett et al. 1997), and (3) depolarization causes regulated release of 125I-labeled BDNF loaded into cortical synaptosomes (Androutsellis-Theotokis et al. 1996). Initially, we confirmed that synaptosomes contained TrkB receptors that were capable of activation by exposing them to exogenous BDNF; synaptosomes were incubated with or without 100 ng/ml BDNF for 7 min, and total Trk receptors were immunoprecipitated using anti-panTrk. Western blot analysis with anti-phosphotyrosine revealed an increase in the level of TrkB autophosphorylation in the presence of BDNF (Fig. 4A), confirming the presence of accessible TrkB receptors in this preparation.
Figure 4.



Depolarization-induced activation of Trk receptors in isolated cortical synaptosomes. (A) Western blot analysis of anti-panTrk immunoprecipitates of equal amounts of protein from synaptosomes treated with (+) or without (−) 100 ng/ml BDNF and probed with anti-phosphotyrosine. The numbers at left of the blot indicate the molecular mass markers, whereas the arrowhead indicates the size of full-length TrkB. (B) Western blot analysis of anti-panTrk immunoprecipitates of equal amounts of protein from synaptosomes treated with (KCL) or without (control) 117 mm KCl for 10 min and probed with anti-phosphotyrosine (top). To ensure that similar amounts of TrkB are present in each sample, the same blot was reprobed with anti-TrkBout (bottom). In both panels, the numbers at left of the blot indicate the molecular mass markers, whereas the arrowhead indicates the size of full-length TrkB. (C) Quantitation of experiments similar to that shown in B (n = 8 for controls, n = 9 for KCl treated). Western blots were scanned, the intensity of the bands quantitated by image analysis, and the data were expressed as a ratio of phosphoTrk to full-length TrkB levels. These ratios were then normalized to the ratio for the control synaptosomes analyzed on the same Western blots. Significance was determined by Student’s t-test (P = 0.0081). (*) P < 0.05.
Our in vivo data suggested that the TrkB receptor autophosphorylation seen in response to kainic acid was due to activity-dependent release of BDNF at the synapse. If this is true, then we would predict that depolarization of synaptosomes would lead to release of BDNF and that this BDNF would cause activation of TrkB receptors. To test this prediction, we incubated synaptosomes in the presence of KCl for 10 min and then examined Trk tyrosine phosphorylation (Fig. 4B). Western blot analysis of KCl-treated synaptosomes immunoprecipitated with anti-panTrk revealed that KCl reproducibly led to an increase in Trk receptor tyrosine phosphorylation (Fig. 4C), presumably as a consequence of depolarization-induced release of endogenous BDNF.
ACTIVATION OF CENTRAL NORADRENERGIC NEURONS LEADS TO AUTOPHOSPHORYLATION OF TRKB RECEPTORS IN THE NEOCORTEX
We have demonstrated previously that central noradrenergic neurons anterogradely transport BDNF and that when noradrenergic neuron-derived BDNF is increased in transgenic mice, activation of postsynaptic cortical TrkB receptors increases (Fawcett et al. 1998). We therefore hypothesized that pharmacological activation of noradrenergic neurons would cause BDNF to be released onto noradrenergic target neurons in the cortex, resulting in TrkB receptor activation. To test this hypothesis, we treated mice with 2.5 mg/kg yohimbine, a drug that, at the dose used, specifically blocks presynaptic α2-adrenergic receptors, thereby causing rapid firing of noradrenergic neurons. Thirty and 60 min after yohimbine treatment, we isolated and lysed the cortical tissue and immunoprecipitated full-length Trk receptors using anti-panTrk. Western blots of these samples were then probed with anti-phosphotyrosine to measure levels of TrkB autophosphorylation and then reprobed to measure full-length TrkB receptor levels. This analysis demonstrated that 30 min following yohimbine treatment, there was a significant threefold increase in the ratio of Trk receptor autophosphorylation to total TrkB levels relative to the control cortex (Fig. 5A,D). This increase was somewhat diminished by 60 min, but Trk autophosphorylation remained higher than controls (Fig. 5A).
Figure 5.




Activation of noradrenergic neurons with yohimbine leads to rapid autophosphorylation of postsynaptic Trk receptors in the cortex. (A) Yohimbine treatment increases tyrosine phosphorylation of TrkB. Cortical lysates from mice treated for 30 or 60 min with 2 mg/kg yohimbine were immunoprecipitated with anti-panTrk, and Western blots of the immunoprecipitates were probed with anti-phosphotyrosine to monitor Trk receptor activation (top) and then reprobed with anti-TrkBin to monitor total TrkB levels (bottom). The numbers at left of the panels indicate the molecular mass markers. (B) BDNF levels in the cortex do not increase after yohimbine treatment. Cortical lysates from individual control and yohimbine-treated (30 min) animals were analyzed by SDS-PAGE, and the resultant Western blot was probed with an antibody specific to BDNF. Each lane represents lysates from one animal. BDNF produced by PC12 cells infected with a BDNF-expressing vaccinia virus (Fawcett et al. 1997) was used as a control. (C) Prazosin partially reverses yohimbine-induced activation of Trk receptors in the cortex. Cortical lysates from mice treated for 30 min with 2.5 mg/kg yohimbine were immunoprecipitated with anti-panTrk and then probed with anti-phosphotyrosine (top) and reprobed with anti-TrkBin (bottom). Some of these animals were pretreated with 1 mg/kg prazosin 40 min prior to yohimbine injection. Each lane represents lysates from one animal. The arrows indicate the size of full-length TrkB, and the numbers at left of the panels indicate the molecular mass markers. Note that although there is some animal-to-animal variation, in all cases the highest levels of Trk tyrosine phosphorylation are observed in the yohimbine-treated animals and that this is somewhat diminished in animals that were also treated with prazosin. (D) Quantitation of experiments similar to those shown in C (n = 3 for controls, n = 4 for yohimbine, and n = 3 for yohimbine + prazosin). Western blots were scanned, the intensity of the bands quantitated by image analysis, and the data were expressed as the ratio of phosphoTrk to full-length TrkB levels. These ratios were then normalized to the ratio for control animals analyzed on the same blots. The graphs represent the mean fold change from controls, and the error bars indicate the s.e.m. Asterisks (*) indicate those values that are significantly different from the controls as assessed using ANOVA (P < 0.01).
These observations suggest that activation of noradrenergic neurons leads to a rapid, localized secretion of BDNF into targets like the neocortex, resulting in activation of postsynaptic TrkB receptors. Alternatively, because BDNF synthesis is regulated by neural activity (Lindholm et al. 1994; for review, see Thoenen 1995), the activation of noradrenergic neurons could lead to a local increase in synthesis of BDNF, thereby leading to increased cortical TrkB receptor activation. To distinguish these two possibilities, we examined the levels of BDNF in the cortex 30 min following yohimbine treatment. Western blot analysis demonstrated that the levels of BDNF were similar in the cortex of control and yohimbine-treated mice (Fig. 5B), indicating that the increased TrkB autophosphorylation was not due to increased local BDNF synthesis.
Because local BDNF synthesis was not increased, it is likely that the increased TrkB activation was due to BDNF released directly from noradrenergic neurons and/or to BDNF released intrinsically in the cortex as a consequence of noradrenergic stimulation. To distinguish these two possibilities, we inhibited the stimulatory postsynaptic α1-adrenergic receptors using the drug prazosin and determined whether yohimbine was still able to cause Trk receptor activation in the cortex. Specifically, adult mice were treated with 1 mg/kg prasozin 40 min prior to yohimbine injection and were then injected with yohimbine, and 30 min later, cortical Trk receptor activation was analyzed. Western blot analysis of anti-panTrk immunoprecipitates with both anti-phosphotyrosine and anti-TrkBin revealed that pretreatment with prazosin partially inhibited the yohimbine-induced increase in the ratio of Trk receptor autophosphorylation to total TrkB levels (Fig. 5C,D) but that relative levels of Trk autophosphorylation were still twofold higher than in controls (Fig. 5D). We interpret these data to indicate that yohimbine is acting (1) to cause activity-dependent BDNF release from noradrenergic neurons, directly causing postsynaptic TrkB receptor autophosphorylation independent of the stimulatory effects of noradrenaline and (2) to stimulate cortical neurons that, in turn, release BDNF in an activity-dependent fashion, thereby causing further TrkB receptor autophosphorylation.
Discussion
In this paper we have tested the hypothesis that neural activity leads to rapid TrkB receptor activation at synapses and that at least in some situations, this activation occurs on postsynaptic neurons. The data presented here supporting this hypothesis are threefold. First, we demonstrate that kainic acid-induced seizures caused Trk receptor activation at synapses 20 min following kainic acid treatment. Second, we demonstrate that depolarization of cortical synaptosomes by treatment with elevated KCl for 10 min also led to Trk receptor activation, presumably as a consequence of depolarization-induced BDNF secretion. Finally and most importantly, pharmacological activation of brain-stem noradrenergic neurons, which synthesize and anterogradely transport BDNF to the cortex (Fawcett et al. 1998), led to rapid and transient activation of cortical Trk receptors. This activation is partially due to activity-dependent secretion of BDNF on postsynaptic neurons and partially due to stimulation of cortical noradrenergic receptors. Together, these data support the hypothesis that activity-dependent secretion of BDNF from incoming afferents causes rapid activation of postsynaptic TrkB receptors and suggest that even physiological levels of neural activity may regulate synaptic TrkB receptor activity, a finding that has important implications for activity-dependent synaptic rearrangement.
Data reported here indicate that kainic acid-induced seizures led to a rapid activation of synaptic TrkB receptors in the cortex. Because TrkB is localized on cell bodies, axons, and dendrites of cortical neurons (Fryer et al. 1996; Yan et al. 1997b) and is found in most subcellular fractions (as shown here), then the preferential tyrosine phosphorylation of synaptic TrkB is presumably due to a preferential increase in secretion of neurotrophins at the synapse. However, the kainic acid data do not distinguish between presynaptic and postsynaptic Trk receptors and do not, therefore, distinguish between models invoking activity-dependent secretion of neurotrophins from dendrites and subsequent activation of presynaptic receptors versus secretion from terminals and postynaptic receptor activation. Moreover, the kainic acid data are complicated by the fact that this treatment not only leads to widespread seizure activity, but it also leads to long-term damage to hippocampal and cortical neurons. Finally, although our studies demonstrate that TrkB is by far the most abundant Trk receptor in the cortex, our immunoprecipitations were performed using an antibody that recognizes all Trk receptors, and our tyrosine phosphorylation studies do not therefore formally rule out the possibility of a small contribution from Trk receptors other than TrkB to our data. Thus, although the kainic acid data support the idea that neural activity causes preferential TrkB receptor activation at cortical synapses, this interpretation is complicated by these other considerations.
To eliminate a number of the variables inherent to the kainic acid experiment, we turned to cortical synaptosome preparations. These fractions are highly enriched in synaptic profiles (Huttner et al. 1983), and BDNF is found in the vesicular fraction of synaptosomes (Fawcett et al. 1997), consistent with its localization to large dense-core vesicles at presynaptic terminals in tissue sections (Michael et al. 1997). Data presented here indicate that, as predicted by this localization of BDNF, KCl-induced depolarization led to increased Trk receptor activation in synaptosomes, presumably as a consequence of activity-dependent secretion of BDNF (Androutsellis-Theotokis et al. 1996). Thus, these experiments strongly support the idea that neural activity directly causes TrkB receptor activation at synapses, although, like the kainic acid data, they do not distinguish between presynaptic and postsynaptic Trk receptors.
The argument that such activity-dependent activation of synaptic TrkB receptors might also occur under more physiological conditions derives from the experiments reported here with noradrenergic neurons. We have demonstrated previously that brain-stem noradrenergic neurons synthesize BDNF and that this BDNF is anterogradely transported into axons and terminals of noradrenergic neurons. Moreover, when BDNF was overexpressed two- to fourfold in noradrenergic neurons of transgenic animals (Causing et al. 1997), this resulted in (1) increased Trk receptor activation in noradrenergic targets such as the cortex, (2) disruption of cortical structure during development, and (3) rescue of another noradrenergic neuron target, facial motoneurons, from axotomy-induced death (Fawcett et al. 1998). Thus, anterogradely transported BDNF from noradrenergic neurons was able to modify the development and survival of target neuron populations in vivo. In this paper we have extended these findings and have demonstrated that pharmacological activation of the same neurons was sufficient to cause TrkB activation in the cortex within 30 min. In addition, we have demonstrated that, although a portion of this TrkB activation required coincident activation of postsynaptic α1-adrenergic receptors, it was also partially due to direct effects on the postsynaptic neuron. These data therefore support two important conclusions. First, they indicate that more physiological levels of neural activity lead to synaptic TrkB receptor activation in vivo. Second, they indicate that at least some of the activity-dependent TrkB tyrosine phosphorylation at synapses is likely due to activation of postsynaptic TrkB receptors as a consequence of secretion of BDNF from presynaptic terminals. This final conclusion is tempered somewhat by the possibility that BDNF secreted from noradrenergic terminals in the cortex may also cause activation of presynaptic TrkB receptors on the noradrenergic axons themselves, but because TrkB receptors have never been localized to noradrenergic axons but have been localized to postsynaptic cortical neuron dendrites, we feel that such a contribution would be minor, if it occurs at all. Thus, BDNF derived from afferents is likely to play a key role in modulating the biology of target neurons in the mature CNS, as we have demonstrated previously during development (Fawcett et al. 1998). Moreover, the secretion of this BDNF and activation of postsynaptic Trk receptors is apparently regulated in an activity-dependent manner, thereby providing a novel mechanism for coordinating the effects of trophic factors and neural activity on synaptic plasticity.
What are the physiological implications of these data? For the developing nervous system, the coordinate secretion of neurotransmitters and BDNF from incoming afferents may play a major role in determining neuronal survival and in the selection of appropriate connections. It is clear that both neurotrophins (Snider and Lichtman 1996; Cabelli et al. 1995, 1997; Causing et al. 1997; McAllister et al. 1997) and neural activity (Galli-Resta et al. 1993; Linden 1994; Sherrard and Bower 1998) are essential for the establishment of a correctly wired nervous system, and a number of recent studies indicate that the two act together to determine neuronal survival and differentiation in both the peripheral (Wang et al. 1995; Lu and Figurov 1997) and central nervous systems (Cohen-Cory et al. 1991; Ghosh et al. 1994; Meyer-Franke et al. 1995, 1998; Morrison and Mason 1998). The coordinate activation of neurotransmitter receptors and growth factor receptors could lead to crosstalk both at the levels of the receptors themselves (Meyer-Franke et al. 1995; Morrison and Mason 1998) and at the level of intracellular signaling pathways (Blair et al. 1999; Vaillant et al. 1999). Thus, a strong synaptic input from an appropriate afferent may (1) confer a significant advantage on the target neuron in terms of survival during naturally occurring cell death (Catsicas et al. 1992; Linden et al. 1994; Sherrard and Bower 1998), and (2) may locally regulate the generation and maintenance of postsynaptic spines and specializations (Morrison and Mason 1998; Shimada et al. 1998). Conversely, the coincident activation of postsynaptic receptors for both neurotransmitters and neurotrophins may well lead to the generation of a strong retrograde signal that itself stabilizes appropriate afferent synapses. A prediction of such a model is that activity and neurotrophins synergize to regulate neuronal survival and morphological growth, a prediction supported by a number of recent findings (Cohen-Cory and Fraser 1995; McAllister et al. 1996; Lu and Figurov 1997).
Similar implications hold for the mature nervous system. Although afferent innervation is not apparently essential for survival of mature neurons (Linden 1994), lesion of afferents leads to well-documented changes in soma size, dendritic complexity (Cook et al. 1951; Matthews et al. 1960; Tierney et al. 1997) and neuropeptide phenotype (J.P. Fawcett, M. Alonso-Vanegas, S.J. Morris, F.D. Miller, A. Sadikot, and R.A. Murphy, unpubl.), reflecting at least some role of afferent trophic support in the maintenance of cellular phenotype and morphology. Such a role may become increasingly important in the traumatized or diseased nervous system. For example, trophic support derived from noradrenergic afferents is thought to play an essential role both during development (Maeda et al. 1974; Felten et al. 1982) and in the maintenance of neuronal survival in neurodegenerative disorders such as Parkinson’s disease (Colpaert 1994). Finally, BDNF, acting through the TrkB receptor, is known to directly regulate synaptic innervation density (Causing et al. 1997; Martinez et al. 1998), suggesting that activation of synaptic Trk receptors in response to neural activity may be essential for the maintenance or strengthening of pre-existing synapses and/or for the morphological plasticity and synaptic remodelling that is thought to underly long-term adaptive changes in the mature nervous system.
Acknowledgments
We thank Dr. T. Reader (Université de Montreal) for advice regarding pharmacology of the noradrenergic system, Dr. Peter McPherson (M.N.I., McGill University) for helpful advice regarding synaptosome experiments, and members of the Miller, Kaplan, and Murphy labs for their input. This work was supported by grants from the Medical Research Council of Canada to D.R.K., R.A.M., and F.D.M. During the course of this work, R.A. was supported by a NeuroSciences Network fellowship and J.P.F. by a Rick Hansen Foundation studentship. F.D.M. is a Killam Scholar, and D.R.K. is a recipient of the Harold Johns and Canadian Cancer Society Research Scientist Award.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Barde Y-A, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliveau DJ, Krivko I, Kohn J, Lachance C, Pozniak C, Rusakov D, Kaplan D, Miller FD. NGF and NT-3 both activate TrkA on sympathetic neurons but differentially regulate survival and neuritogenesis. J Cell Biol. 1997;136:374–388. doi: 10.1083/jcb.136.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. 1999;19:4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair LA, Bence-Hanulec KK, Mehta S, Franke T, Kaplan D, Marshall J. Akt-dependent potential of L channels by insulin-like growth factor-1 is required for neuronal survival. J Neurosci. 1999;19:1940–1951. doi: 10.1523/JNEUROSCI.19-06-01940.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- Catsicas M, Pequignot Y, Clarke PG. Rapid onset of neuronal death induced by blockade of either axoplasmic transport or action potentials in afferent fibers during brain development. J Neurosci. 1992;12:4642–4650. doi: 10.1523/JNEUROSCI.12-12-04642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- Clary DO, Weskamp G, Austin LR, Reichardt LF. TrkA cross-linking mimics neuronal responses to nerve growth factor. Mol Biol Cell. 1994;5:549–563. doi: 10.1091/mbc.5.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Dreyfus CF, Black IB. NGF and excitatory neurotransmitters regulate survival and morphogenesis of cultured cerebellar Purkinje cells. J Neurosci. 1991;11:462–471. doi: 10.1523/JNEUROSCI.11-02-00462.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpaert F. Noradrenergic mechanisms in Parkinson’s Disease, Pierre Fabre monograph series. Boca Raton, FL: CRC Press; 1994. pp. 225–254. [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: Evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WH, Walker JH, Barr ML. A cytological study of transneuronal atrophy in the cat and rabbit. J Comp Neurol. 1951;94:267–292. doi: 10.1002/cne.900940207. [DOI] [PubMed] [Google Scholar]
- Dugich-Djordjevic MM, Tocco G, Lapchak PA, Pasinetti GM, Najm I, Baudry M, Hefti F. Regionally specific and rapid increases in brain-derived neurotrophic factor messenger RNA in the adult rat brain following seizures induced by systemic administration of kainic acid. Neuroscience. 1992;47:303–315. doi: 10.1016/0306-4522(92)90246-x. [DOI] [PubMed] [Google Scholar]
- Dugich-Djordjevic MM, Ohsawa F, Okazaki T, Mori N, Day JR, Beck KD, Hefti F. Differential regulation of catalytic and non-catalytic trkB messenger RNAs in the rat hippocampus following seizures induced by systemic administration of kainate. Neuroscience. 1995;66:861–877. doi: 10.1016/0306-4522(94)00631-e. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Tingley WG, Huganir RL. Regulated subcellular distribution of the NR1 subunit of the NMDA receptor. Science. 1995;269:1734–1737. doi: 10.1126/science.7569904. [DOI] [PubMed] [Google Scholar]
- Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PJ, Murphy RA. Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem. 1997;272:8837–8840. doi: 10.1074/jbc.272.14.8837. [DOI] [PubMed] [Google Scholar]
- Fawcett JP, Bamji SX, Causing CG, Aloyz R, Ase AR, Reader TA, McLean JH, Miller FD. Functional evidence that BDNF is an anterograde neuronal trophic factor in the CNS. J Neurosci. 1998;18:2808–2821. doi: 10.1523/JNEUROSCI.18-08-02808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felten DL, Hallman H, Jonsson G. Evidence for a neurotrophic role of noradrenaline neurons in the postnatal development of rat cerebral cortex. J Neurocytol. 1982;11:119–135. doi: 10.1007/BF01258008. [DOI] [PubMed] [Google Scholar]
- Fletcher TL, Cameron P, DeCamilli P, Banker G. The distribution of synapsin I and synaptophysin in hippocampal neurons developing in culture. J Neurosci. 1991;11:1617–1626. doi: 10.1523/JNEUROSCI.11-06-01617.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Galli-Resta L, Ensini M, Fusco E, Gravina A, Margheritti B. Afferent spontaneous electrical activity promotes the survival of target cells in the developing retinotectal system of the rat. J Neurosci. 1993;13:243–250. doi: 10.1523/JNEUROSCI.13-01-00243.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EP, McPherson PS, Chilcote TJ, Takei K, DeCamilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol. 1995;129:105–120. doi: 10.1083/jcb.129.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Goodman LJ, Valverde J, Lim F, Geschwind MD, Federoff HJ, Geller AI, Hefti F. Regulated release and polarized localization of brain-derived neurotrophic factor in hippocampal neurons. Mol Cell Neurosci. 1996;7:222–238. doi: 10.1006/mcne.1996.0017. [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR. Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron. 1992;9:883–896. doi: 10.1016/0896-6273(92)90241-5. [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, DeCamilli P. Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol. 1983;96:1374–1388. doi: 10.1083/jcb.96.5.1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis CR, Xiong ZG, Plant JR, Churchill D, Lu WY, MacVicar BA, MacDonald JF. Neurotrophin modulation of NMDA receptors in cultured murine and isolated rat neurons. J Neurophysiol. 1997;78:2363–2371. doi: 10.1152/jn.1997.78.5.2363. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- ————— A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kasof MK, Mandelzys A, Maika SD, Hammer RE, Curran T, Morgan JI. Kainic acid-induced neuronal death is associated with DNA damage and a unique immediate-early gene response in c-fos-lacZ transgenic rats. J Neurosci. 1995;15:4238–4249. doi: 10.1523/JNEUROSCI.15-06-04238.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knusel B, Rabin SJ, Hefti F, Kaplan DR. Regulated neurotrophin receptor responsiveness during neuronal migration and early differentiation. J Neurosci. 1994;14:1542–1554. doi: 10.1523/JNEUROSCI.14-03-01542.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibrock J, Lottspeich F, Hohn A, Hofer M, Hengerer B, Masiakowski P, Thoenen H, Barde Y-A. Molecular cloning and expression of brain-derived neurotrophic factor. Nature. 1989;341:149–152. doi: 10.1038/341149a0. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-D-aspartic acid receptor activity. Proc Natl Acad Sci. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Linden R. The survival of developing neurons: A review of afferent control. Neuroscience. 1994;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain—Implications for neuronal plasticity. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- Lu B, Figurov A. Role of neurotrophins in synapse development and plasticity. Rev Neurosci. 1997;8:1–12. doi: 10.1515/revneuro.1997.8.1.1. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38) Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Tohyama M, Shimizu N. Modification of postnatal development of neocortex in rat brain with experimental deprivation of locus coeruleus. Brain Res. 1974;70:515–520. doi: 10.1016/0006-8993(74)90261-3. [DOI] [PubMed] [Google Scholar]
- Majdan, M. and F.D. Miller. 1999. Neuronal life and death decisions: Functional antagonism between the Trk and p75 neurotrophin receptors. Int. J. Devl. Neurosci. (in press). [DOI] [PubMed]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E. TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci. 1998;18:7336–7350. doi: 10.1523/JNEUROSCI.18-18-07336.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews MR, Cowan WM, Powell TPS. Transneuronal cell degeneration in the lateral geniculate nucleolus of macaque monkey. J Anat. 1960;94:145–169. [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- ————— Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- ————— Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- McPherson PS, Takei K, Schmid SL, DeCamilli P. p1415, a major Grb2-binding protein in brain, co-localized with dynamin in nerve terminals where it undergoes activity-dependent dephosphorylation. J Biol Chem. 1994;269:30132–30139. [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison ME, Mason CA. Granule neuron regulation of Purkinje cell development: Striking a balance between neurotrophin and glutamate signaling. J Neurosci. 1998;18:3563–3573. doi: 10.1523/JNEUROSCI.18-10-03563.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowla SJ, Pareek S, Farhadi HF, Petrecca K, Fawcett JP, Seidah NG, Morris SJ, Sossin WS, Murphy RA. Differential sorting of nerve growth factor and brain-derived neurotrophic factor in hippocampal neurons. J Neurosci. 1999;19:2069–2080. doi: 10.1523/JNEUROSCI.19-06-02069.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modifications of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- Sherrard RM, Bower AJ. Role of afferents in the development and cell survival of the vertebrate nervous system. Clin Exp Pharmacol Physiol. 1998;25:487–495. doi: 10.1111/j.1440-1681.1998.tb02241.x. [DOI] [PubMed] [Google Scholar]
- Shimada A, Mason CA, Morrison ME. TrkB signaling modulates spine density and morphology independent of dendrite structure in cultured neonatal Purkinje cells. J Neurosci. 1998;18:8559–8570. doi: 10.1523/JNEUROSCI.18-21-08559.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Snider WD, Lichtman JW. Are neurotrophins synaptotrophins? Mol Cell Neurosci. 1996;7:433–442. doi: 10.1006/mcne.1996.0031. [DOI] [PubMed] [Google Scholar]
- Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, Burton LE, Stanton BR, Kaplan DR, Hunter T, Nikolics K, Parada LF. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell. 1991;65:895–903. doi: 10.1016/0092-8674(91)90396-g. [DOI] [PubMed] [Google Scholar]
- Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-D-aspartate receptor subunit 1. Proc Natl Acad Sci. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Barde Y-A. Physiology of nerve growth factor. Physiol Rev. 1989;60:1284–1335. doi: 10.1152/physrev.1980.60.4.1284. [DOI] [PubMed] [Google Scholar]
- Tierney TS, Russell FA, Moore DR. Susceptibility of developing cochlear nucleus neurons to deafferentation-induced death abruptly ends just before the onset of hearing. J Comp Neurol. 1997;378:295–306. doi: 10.1002/(sici)1096-9861(19970210)378:2<295::aid-cne11>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Vaillant, A., I. Mazzoni, C. Tudan, M. Boudreau, D.R. Kaplan, and F.D. Miller. 1999. Depolarization and neurotrophins converge on the PI3-kinase-Akt pathway to synergistically regulate neuronal survival. J. Cell Biol. (in press). [DOI] [PMC free article] [PubMed]
- von Bartheld CS, Byers MR, Williams R, Bothwell M. Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature. 1996;379:830–833. doi: 10.1038/379830a0. [DOI] [PubMed] [Google Scholar]
- Walch-Solimena C, Takei K, Marek ML, Midyett K, Sudhof TC, DeCamilli P, Jahn R. Synaptotagmin: A membrane constituent of neuropeptide-containing large dense-core vesicles. J Neurosci. 1993;13:3895–3903. doi: 10.1523/JNEUROSCI.13-09-03895.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K, Xu JL, Suen PC, Levine E, Huang YY, Mount HT, Lin SY, Black IB. Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Brain Res Mol Brain Res. 1996;43:286–290. doi: 10.1016/s0169-328x(96)00211-2. [DOI] [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997a;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol. 1997b;378:135–157. [PubMed] [Google Scholar]
- Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience. 1996;74:945–953. doi: 10.1016/0306-4522(96)00237-0. [DOI] [PubMed] [Google Scholar]