
Fig. 3.
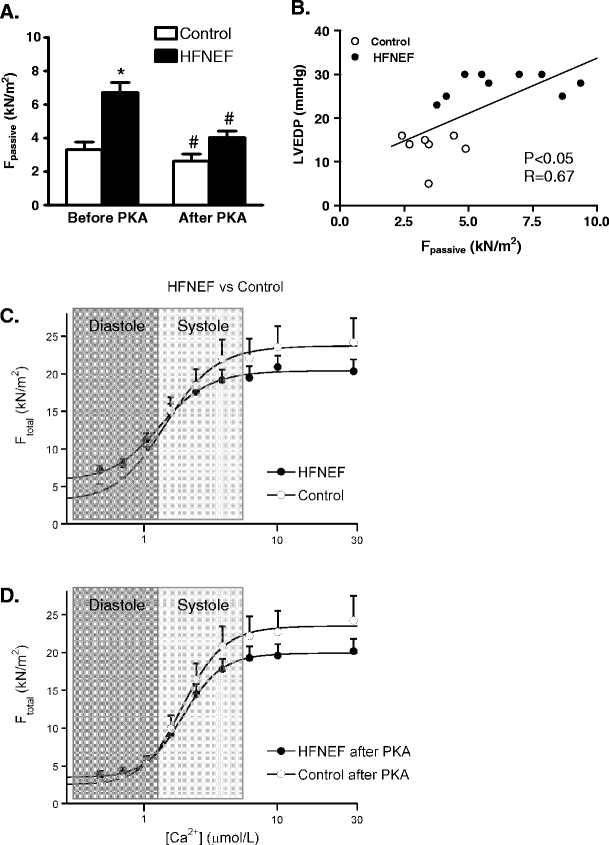
a Isolation of single Triton-permeabilized cardiomyocytes from biopsies taken during cardiac catheterization from patients with a diastolic dysfunction evident from increased left ventricular (LV) end-diastolic pressures (>16 mmHg: heart failure patients with normal (or preserved) LV ejection fraction, HFNEF) revealed increased passive stiffness compared to control cells from patients with normal LV ejection fraction and normal LV end-diastolic pressure [11]. The high passive stiffness was corrected to control values upon treatment with protein kinase A (PKA), suggesting that the high passive force is largely caused by hypophosphorylation of a sarcomeric protein (most likely titin [7]). b A significant relation was found between in vivo LV end-diastolic pressure and cardiomyocyte stiffness, indicating that the in vivo diastolic dysfunction in HFNEF patients is at least partly due to an intrinsic defect of the myofilaments. c Illustrates that the increased myofilament passive stiffness would impair diastolic function of the heart of HFNEF patients compared to controls, while the force-generating capacity of the myofilaments during systole may be somewhat lower compared to controls. d PKA treatment corrected myofilament diastolic dysfunction to values observed in controls