

Abstract
Signal-processing molecules inside cells are often present at low copy number, which necessitates probabilistic models to account for intrinsic noise. Probability distributions have traditionally been found using simulation-based approaches which then require estimating the distributions from many samples. Here we present in detail an alternative method for directly calculating a probability distribution by expanding in the natural eigenfunctions of the governing equation, which is linear. We apply the resulting spectral method to three general models of stochastic gene expression: a single gene with multiple expression states (often used as a model of bursting in the limit of two states), a gene regulatory cascade, and a combined model of bursting and regulation. In all cases we find either analytic results or numerical prescriptions that greatly outperform simulations in efficiency and accuracy. In the last case, we show that bimodal response in the limit of slow switching is not only possible but optimal in terms of information transmission.
I. INTRODUCTION
Signals are processed in cells using networks of interacting components, including proteins, mRNAs, and small signaling molecules. These components are usually present in low numbers [1-6], which means the size of the fluctuations in their copy counts is comparable to the copy counts themselves. Noise in gene networks has been shown to propagate [7] and therefore explicitly accounting for the stochastic nature of gene expression appears important when predicting the properties of real biological networks.
Although summary statistics such as mean and variance are sometimes sufficient for answering questions of biological interest [8], calculating certain quantities, such as information transmission [9-14], requires knowing the full probability distribution. Full knowledge of the probability distribution can also be used to discern different molecular models of the noise sources based on recent exact measurements of probability distributions [15-18].
Much analytical and purely computational effort has gone into detailed models of noise in small genetic switches [8,19-21]. The most general description is based on the master equation describing the time evolution of the joint probability distribution over all copy counts [22]. Some progress has been made by applying approximations to the master equation [24-26]. For example, a wide class of approximations focuses on limits of large concentrations or small switches [8,19,23]. More often, modelers resort to stochastic simulation techniques, the most common of which is based on the varying-step Monte Carlo method [27,28]. This method requires a computational challenge (generating many sample trajectories) followed by an even more difficult statistical challenge (parametrizing or otherwise estimating the probability distribution from which the samples are drawn) [29]. Recently there have been significant advances in simulation-based methods to circumvent these problems [20,21,30]. Simulation techniques are especially useful for more detailed studies of experimentally well-characterized systems, including those incorporating DNA looping, non-specific binding [30], and explicit spatial effects [20,21]. However, it is often beneficial to first gain intuition from simplified analytical models.
In a recent paper [31] we introduce an alternative method for calculating the steady-state distributions of chemical reactants, which we call the spectral method. The procedure relies on exploiting the natural basis of a simpler problem from the same class. The full problem is then solved numerically as an expansion in the natural basis [32]. In the spectral method we use the analytical guidance of a simple birth-death problem to reduce the master equation for a cascade to a set of algebraic equations. We break the problem into two parts: a parameter-independent preprocessing step and the parameter-dependent step of obtaining the actual probability distributions. The spectral method allows huge computational gains with respect to simulations. In prior work [31] we illustrate the method in the example of gene regulatory cascades. We combine the spectral method with a Markov approximation, which exploits the observation that the behavior of a given species should depend only weakly on distant nodes given the proximal nodes.
In this paper we expand upon the application of the spectral method to more biologically realistic models of regulation: (i) a model of bursting in gene expression and (ii) a model that includes both bursts and explicit regulation by binding of transcription factor proteins. In both cases we demonstrate how the spectral method gives either analytic results or reduced algebraic expressions that can be solved numerically in orders of magnitude less time than stochastic simulations.
We note that although information propagation in biological networks is impeded by numerous mechanisms collectively modeled as noise, here we focus on the inescapable or “intrinsic” noise resulting from the finite copy number of the molecules. One should consider these results, then, as an upper bound on information propagation, further hampered by, for example, cell division [33]; spatial effects [20,21]; active degradation of the constituents (here modeled via a constant degradation rate); and other complicating mechanisms particular to specific systems. Additionally, here we focus on steady-state solutions, but extensions to dynamics are straightforward and currently being pursued.
We begin with a model of a multistate birth-death process, a special case of which has been used to describe transcriptional bursting [17,34]. We illustrate how the spectral method reduces the model to a simple iterative algebraic equation, and in the appropriate limiting case recovers the known analytic results. We also use this section to introduce the basic notation used throughout the paper. Next we explore the problem of gene regulation in detail. The main idea behind the spectral method is the exploitation of an underlying natural basis for a problem which we can solve exactly. We explore four different spectral representations of the regulation model used in previous work [31] that arise from four natural choices of eigenbasis in which to expand the solution (cf. Sec. II A and Fig. 2). All representations reduce the master equation to a set of linear algebraic equations, and one admits an analytic solution by virtue of the tridiagonal matrix algorithm. We compare the efficiencies of the representations’ numerical implementations and show that all outperform simulation. Lastly, we apply the spectral method to a model that combines bursting and regulation. We obtain a linear algebraic expression that permits large speedup over simulation and thus admits optimization of information transmission. Optimization reveals two types of solutions: a unimodal response when the rates of switching between expression states are comparable to degradation rates and a bimodal response when switching rates are much slower than degradation rates.
FIG. 2.
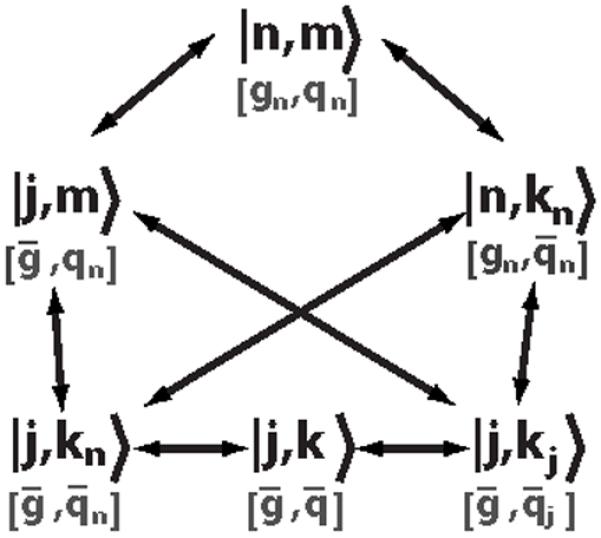
Summary of the bases discussed in Sec. II A (black) and their gauge freedoms (barred parameters in gray). In the top row, neither the m nor the n sector is expanded in an eigenbasis; in the middle row, one sector is expanded; and in the bottom row, both sectors are expanded. The |j,k〉 basis can be viewed as a special case of the |j,kj〉 or |j,kn〉 basis with q‾j=q‾ or q‾n=q‾, respectively. The |j,m〉 basis is not discussed as it is not useful in simplifying the problem.
II. BURSTS OF GENE EXPRESSION
We first consider a model of gene expression in which a gene exists in one of Z stochastic “states,” i.e., protein production obeys a simple birth-death process but with a state-dependent birth rate. In the special case of Z=2, this corresponds to a gene existing in an on or an off state due, for example, to the binding and unbinding of the RNA polymerase. Such a model has been used to describe transcriptional bursting [17,34], and we specialize to this case in Sec. II C.
For a general Z-state process, the master equation
| (1) |
describes the time evolution of the joint probability distribution , where z specifies the state (1≤z≤Z), n is the number of proteins, gz is the production rate in state z, Ωzz’ is a stochastic matrix of transition rates between states, and denotes differentiation of the probability distribution with respect to time. Time and all rates have been nondimensionalized by the protein degradation rate. Note that conservation of probability requires
| (2) |
The relationship between the transition rates in Ωzz’ and the probabilities of being in the zth state can be seen by summing Eq. (1) over n; at steady state one obtains
| (3) |
and normalization requires
| (4) |
In the following sections, we introduce the spectral method and demonstrate how it can be used to solve for the full joint distribution .
A. Notation and definitions
We begin our solution of Eq. (1) by defining the generating function [22] over complex variable x [35] (note that superscript z is an index, while superscript n on xn is a power). It will prove more convenient to rewrite the generating function in a more abstract representation using states indexed by protein number |n〉,
| (5) |
with inverse transform
| (6) |
The more familiar form can be recovered by projecting the position space 〈x| onto Eq. (5), with the provision that
| (7) |
Concurrent choices of conjugate state
| (8) |
and inner product
| (9) |
ensure orthonormality of the states, 〈n|n’〉=δnn’, as can be verified using Cauchy’s theorem,
| (10) |
where the convention θ(0)=0 is used for the Heaviside function.
With these definitions, summing Eq. (1) over n against |n〉 yields
| (11) |
where the operators â+ and â− raise and lower protein number, respectively, i.e.,
| (12) |
| (13) |
with adjoint operations
| (14) |
| (15) |
As in the operator treatment of the simple harmonic oscillator in quantum mechanics, the raising and lower operators satisfy the commutation relation [â−,â+]=1 and â+â− is a number operator, i.e., â+â−|n〉=n|n〉 [36]. This operator formalism for the generating function was introduced in the context of diffusion independently by Doi [37] and Zel’dovich [38] and developed by Peliti [39]. We note that all results can equivalently be obtained by remaining in x space (for example, â+↔x and â−↔∂x). However, the raising and lowering operators define an extremely simple algebra which allows us to calculate all projections without explicitly computing overlap integrals (as shown, for example, in Appendix A). A review by Mattis and Glasser [40] introduces and discusses the applications of the formalism for diffusion.
The factorized form of the birth-death operator in Eq. (11) suggests the definition of shifted raising and lowering operators
| (16) |
| (17) |
making Eq. (11)
| (18) |
Since b̂+ and are simply shifted from â+ and â− by constants, is a new number operator, and its eigenvalues are nonnegative integers j, i.e.,
| (19) |
where j indexes (z-dependent) eigenfunctions |jz〉. In position space, b̂+ and are x−1 and ∂x−gz, respectively. Projecting 〈x| onto Eq. (19) therefore yields a first-order ordinary differential equation whose solution (up to normalization) is
| (20) |
with conjugate
| (21) |
such that orthonormality is satisfied under the inner product in Eq. (9). Note that b̂+ and raise and lower eigenstates |jz〉 as in Eqs. (12)–(15), i.e.,
| (22) |
| (23) |
| (24) |
| (25) |
As we will see in this and subsequent sections, going between the protein number basis |n〉 and the eigenbasis |jz〉 requires the mixed product 〈n|jz〉 or its conjugate 〈jz|n〉. There are several ways of computing these objects, as described in Appendix A. Notable special cases are
| (26) |
| (27) |
where the latter is the Poisson distribution.
B. Spectral method
We now demonstrate how the spectral method exploits the eigenfunctions |jz〉 to decompose and simplify the equation of motion. Expanding the generating function in the eigenbasis,
| (28) |
and projecting the conjugate state 〈jz| onto Eq. (18) yields the equation of motion
| (29) |
for the expansion coefficients [where the dummy index j in Eq. (28) has been changed to j’ in Eq. (29)]. Using Eqs. (9), (10), (20), and (21), the product evaluates to
| (30) |
where Δzz’=gz−gz’. Noting that 〈jz|jz’〉=1 and that for j’<j, Eq. (29) becomes
| (31) |
The last term in Eq. (31) makes clear that each jth term is slaved to terms with j’<j, allowing the to be computed iteratively in j. The lower-triangular structure of the equation is a consequence of rotating to the eigenspace of the birth-death operator; this structure was not present in the original master equation.
At steady state, obeys
| (32) |
from which it is clear that can be computed successively in j. Since
| (33) |
[cf. Eq. (26)], the computation is initialized using Eqs. (3) and (4), i.e.,
| (34) |
| (35) |
Recalling Eqs. (6) and (28), the probability distribution is retrieved via
| (36) |
where the mixed product 〈n|jz〉 can be computed as described in Appendix A.
There is an alternative way to decompose the master equation spectrally. Instead of expanding the generating function in eigenfunctions |jz〉, which depend on the production rates gz in each state, we may expand in eigenfunctions parametrized by a single rate g‾, i.e.,
| (37) |
where
| (38) |
| (39) |
The parameter g‾ is arbitrary and thus acts as a “gauge” freedom.
We may now partition the birth-death operator as
| (40) |
where b‾−=â−−g‾ such that the |j〉 are the eigenstates of b̂+b‾−, i.e.,
| (41) |
and Γz=g‾−gz describes the deviation of each state’s production rate from the constant g‾.
Projecting the conjugate state 〈j| onto Eq. (18) and using Eq. (37) (with dummy index j changed to j’) gives
| (42) |
Recalling Eq. (24), Eq. (42) at steady state becomes
| (43) |
Equation (43) is subdiagonal in j, meaning computation of the jth term requires only the previous (j−1)th term and the inversion of the Z-by-Z matrix (Ω−jI) (where I is the identity matrix). It is initialized with Eqs. (34) and (35) and solved successively in j. The probability distribution is retrieved via
| (44) |
where 〈n|j〉 is computed as described in Appendix A.
As an example of a simple computation employing the spectral method, Fig. 1 shows probability distributions for the case of Z=3 states, corresponding to a gene that is either off, producing proteins at a low rate, or producing proteins at a high rate. For simplicity we set the rates of switching among all states equal to a constant ω, making the stochastic matrix
| (45) |
As seen in Fig. 1, when ω≪1 (corresponding to a switching rate much slower than the degradation rate) the dwell time in each expression state lengthens. The slow switching gives the protein copy number time to equilibrate in any of the three expression states, resulting in a trimodal marginal distribution pn. When ω≫1 (corresponding to a switching rate much faster than the degradation rate), the system switches frequently among the three expression states, resulting in an average production rate. In this limit, the expression state equilibrates on a faster time scale than the protein number state.
FIG. 1.
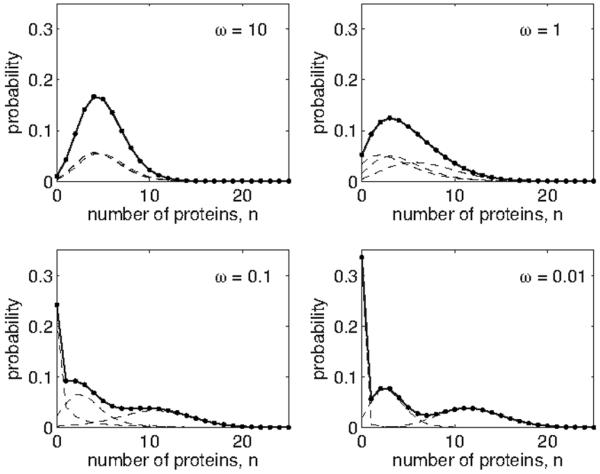
An example of the spectral method for Z=3 states. Dashed curves show the joint distribution for each of z=1, 2, and 3, and solid curves show the marginal distribution pn. The stochastic transition matrix is given in Eq. (45), and the setting of ω in each panel is indicated in the upper-right corner. Production rates are g1=0, g2=3, and g3=12 for all panels. Distributions are calculated via the spectral decomposition in Eq. (43) with g‾=〈gz〉=5.
C. On/off gene
For the special case of Z=2 states, as when a gene is either “on” (z=+) or “off” (z=−), it is useful to demonstrate how the spectral method reproduces known analytic results [17,34]. The probability distribution can be written in vector form as
| (46) |
and defining ω+ and ω− as the transition rates to and from the on state, respectively, the stochastic matrix takes the form
| (47) |
Note that Eq. (3) implies
| (48) |
which makes clear that increasing the rate of transition to either state increases the probability of being in that state.
From Eq. (32) the spectral expansion coefficients obey
| (49) |
where Δ=Δ+−=−Δ−+. Initializing with and computing the first few terms reveals the pattern
| (50) |
| (51) |
where in the second line the products are written in terms of the Gamma function.
For comparison with known results [17,34] we write the total generating function |G〉=Σ±|G±〉 in position space,
| (52) |
| (53) |
| (54) |
where
| (55) |
is the confluent hypergeometric function. As shown in Appendix B, in the limit g−=0, Eq. (54) reduces to
| (56) |
and the marginal pn is given by
| (57) |
in agreement with the results of Iyer-Biswas et al. [34] and Raj et al. [17]. We remind the reader that in addition to reducing to known results in the special case of Z=2 states with a vanishing off-rate, the spectral method is valid for any number of states with arbitrary production rates.
III. GENE REGULATION
Next we consider a two-gene regulatory cascade, in which the production rate of the second gene is a function of the number of proteins of the first gene. As shown in previous work [31], a cascade of any length can be reduced to such a generalized two-dimensional system using the Markov approximation, which asserts that the probability distribution for a given node of the cascade should depend only weakly on the probability distributions of the distant nodes given the proximal nodes.
In the present section, we consider only the generalized two-dimensional equation and explore different approaches to solving it. The equation describes two genes, each with one expression state, with regulation encoded by a functional dependence of the downstream protein production rate on the upstream protein copy number. In Sec. III D we make an explicit connection between the on/off gene discussed in Sec. II C and the case when the functional dependence is a threshold. Finally, in Sec. III we combine the two types of models and consider a system with regulation and bursts.
A. Representations of the master equation
1. |n,m〉 basis
Defining n and m as the numbers of proteins produced by the first and second gene, respectively, the master equation describing the time evolution of the joint probability distribution pnm is [31]
| (58) |
The function qn describes the regulation of the second species by the first, and the function gn describes the effective autoregulation of the first species due either to a non-Poissonian input distribution or to effects further upstream in the case of a longer cascade [31]. Time is rescaled by the first gene’s degradation rate so that each gene’s production rate (gn or qn) is normalized by its respective degradation rate, and ρ is ratio of the second gene’s degradation rate to that of the first. We impose no constraints on the form of gn or qn—they can be arbitrary nonlinear functions.
Summing Eq. (58) over m gives a simple recursion relation between gn and pn at steady state, from which explicit relations are easily identified. If pn is known, gn is found as
| (59) |
If on the other hand gn is known, pn is found as
| (60) |
with p0 set by normalization. Note that if the first species obeys a simple birth-death process, gn=g=constant, and Eq. (60) reduces to the Poisson distribution.
In the current representation [Eq. (58)], which we denote the |n,m〉 basis, finding the steady-state solution for the joint distribution pnm means finding the null space of an infinite (or, effectively for numerical purposes, very large) locally banded tridiagonal matrix. More precisely, defining N as the numerical cutoff in protein number n or m, the problem amounts to inverting an N2-by-N2 matrix, which is computationally taxing even for moderate cutoffs N.
In order to solve Eq. (58) more efficiently we will employ the spectral method. We begin as before by defining the generating function [22] G(x, y)=Σnm pnmxnym over complex variables x and y or, in state notation,
| (61) |
with inverse transform
| (62) |
Summing Eq. (58) over n and m against |n,m〉 and employing the same operator notation as in Eqs. (12)–(15) yields
| (63) |
where
| (64) |
and
| (65) |
| (66) |
| (67) |
| (68) |
Here the regulation functions have been promoted to operators obeying ĝn|n〉=gn|n〉 and q̂n|n〉=qn|n〉, subscripts on operators denote the sector (n or m) on which they operate, and the arguments of and remind us that both are n dependent.
Equation (64) makes clear that if not for the n dependence of the operators the Hamiltonian Ĥ would be diagonalizable, or, equivalently, if gn and qn were constants the master equation would factorize into two individual birth-death processes. We may still, however, partition the full Hamiltonian as
| (69) |
where Ĥ0 is a diagonalizable part (and Ĥ1 is the corresponding deviation from the diagonal form), and expand |G〉 in the eigenbasis of Ĥ0 to exploit the diagonality.
As with the multistate system in Sec. II B, where we expand the solution in two different bases, there are several natural choices of eigenbasis of Ĥ0. Figure 2 summarizes these choices diagrammatically: starting in the |n,m〉 basis (at the top of Fig. 2), one may expand in eigenfunctions either the first species, yielding the |j,m〉 basis (left), or the second species, yielding the |n,kn〉 basis (right; in general we allow the parameter defining the second species’ eigenfunctions to depend on n to reflect the regulation of the second species by the first). From either the |j,m〉 or the |n,kn〉 basis, one may expand in eigenfunctions the remaining species, yielding either the |j,kn〉 basis (bottom left), in which the second species’ eigenfunctions depend on the first species’ copy number n or the |j,kj〉 basis (bottom right), in which the second species’ eigenfunctions depend on the first species’ eigenmode number j. Both bases reduce to the |j,k〉 basis (bottom center) when the parameter of the second species’ eigenfunctions is a constant.
The |n,m〉 and |j,m〉 bases are less numerically useful than the other bases: as discussed above and detailed in Fig. 3, the |n,m〉 basis is numerically inefficient; and the |j,m〉 basis does not exploit the natural structure of the problem, since, unlike the other bases, it neither retains the tridiagonal structure in n nor gains a lower triangular structure in k (see the sections below). Each of the remaining bases, however, has preferable properties in terms of numerical stability and ability to represent the function sparsely yet accurately (either using a few values of n in the |n,kn〉 basis or a few values of j in the |j,k〉, |j,kj〉, or |j,kn〉 bases, for example). Moreover, the equation of motion simplifies differently in each of the different bases. We present the derivations of the equations of motion in the following sections, beginning with the |j,k〉 basis, generalizing to the |j,kj〉 basis, moving to the |n,kn〉 basis, and ending with the |j,kn〉 basis.
FIG. 3.
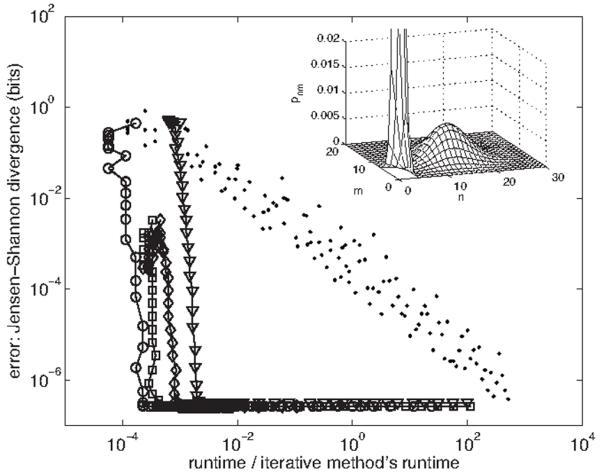
Error vs runtime for the spectral method and stochastic simulation. Error is the Jensen-Shannon divergence [41] between pnm obtained using the |n,m〉 basis (via iterative solution of the original master equation) and that obtained using the |j,k〉 basis [circles; cf. Eq. (80)], the |j,kj〉 basis [triangles; cf. Eq. (90)], the |n,kn〉 basis [squares; cf. Eq. (100)], the |j,kn〉 basis [diamonds; cf. Eq. (114)], or stochastic simulation [28] (dots). Runtimes are scaled by that of the iterative solution, 150 s (in matlab). Spectral basis data is obtained by varying K, the cutoff in the eigenmode number k of the second gene; simulation data is obtained by varying the integration time. The input distribution [from which gn is calculated via Eq. (59)] is a mixture of two Poisson distributions with λ1=0.5, λ2=15, and π1=0.5. The regulation function is a Hill function with q−=1, q+=11, n0=7, and ν=2. The gauge choices used (cf. Fig. 2) are g‾=Σn pngn, q‾=Σn pnqn, q‾n=qn, and q‾j=Σn〈j|n〉qn〈n|j〉. The cutoffs used are J=80 for the eigenmode number j of the first gene and N=50 for the protein numbers n and m. Inset: the joint probability distribution pnm. The peak at low protein number extends to p00≈0.1.
2. |j,k〉 basis
For expository purposes we start by recalling the spectral representation used in previous work [31]. We choose the diagonal part of the Hamiltonian to correspond to two birth-death process with constant production rates g‾ and q‾,
| (70) |
where and . As in Sec. II B, the gauge parameters g‾ and q‾ can be set arbitrarily without affecting the final solution; however, their values can affect the numerical stability of the method. For example, when the regulation qn is a threshold function, a large discontinuity can narrow the range of q‾ for which the method is numerically stable. The nondiagonal part,
| (71) |
captures the deviations Γ̂n=g‾−ĝn and Δ̂n=q‾−q̂n of the regulation functions from the constant rates. We expand the generating function as
| (72) |
where |j,k〉 is the eigenbasis of Ĥ0, i.e.,
| (73) |
The eigenbasis is parametrized by the rates g‾ and q‾, meaning in position space 〈x|j〉 is as in Eq. (38) and similarly for 〈y|k〉 with x→y, j→k, and g‾→q‾.
With Eqs. (70)–(72), projecting the conjugate state 〈j,k| onto Eq. (63) yields
| (74) |
[where the dummy indices j and k in Eq. (72) have been changed to j’ and k’, respectively, in Eq. (74)]. Recalling Eq. (24) and restricting attention to steady state, Eq. (74) becomes
| (75) |
where the deviations have been rotated into the eigenbasis as
| (76) |
| (77) |
Equation (75) is subdiagonal in k and is therefore similar to Eq. (43) in that the last term acts as a source term. The subdiagonality is a consequence of the topology of the two-gene network: the first species regulates only itself (effectively) and the second species. Although the spectral method is fully applicable to systems with feedback, the subdiagonal structure is not preserved.
Equation (75) is initialized using
| (78) |
| (79) |
[cf. Eq. (26)] with known pn [cf. Eq. (60)], then solved at each subsequent k using the result for k−1. Equation (75) can be written in linear algebraic notation as
| (80) |
where is a vector over j, bold denotes matrices, and and are diagonal and subdiagonal matrices, respectively. Equation (80) makes clear that the solution involves only matrix multiplication and the inversion of a J-by-J matrix K times, where J and K are cutoffs in the eigenmode numbers j and k, respectively. In fact, if the first species obeys a simple birth-death process, i.e., gn=g=constant, setting g‾=g makes Γjj’=0, and since Dk is diagonal, the solution involves only matrix multiplication. The decomposition of the master equation into a linear algebraic equation results in huge gains in efficiency over direct solution in the |n,m〉 basis; the efficiency of all bases presented in this section is described in Sec. II B and illustrated in Fig. 3.
Recalling Eqs. (62) and (72), the joint distribution is retrieved from Gjk via the inverse transform
| (81) |
a computation again involving only matrix multiplication. The mixed product matrices 〈n|j〉 and 〈m|k〉 are computed as described in Appendix A.
3. |j,kj〉 basis
The |j,k〉 basis treats both genes similarly by expanding each around a constant production rate. We may instead imagine an eigenbasis that more closely reflects the underlying asymmetry imposed by the regulation and make the basis of the second gene a function of that of the first. That is, we expand the first gene in a basis |j〉 with gauge g‾ as before, but now we expand the second gene in a basis |kj〉 with a j-dependent local gauge q‾j. We write the generating function as
| (82) |
and Eq. (63) at steady state becomes
| (83) |
where we have partitioned the Hamiltonian for each j as
| (84) |
| (85) |
with and Γ̂n=g‾−ĝn as before and now and Δ̂n=q‾j−q̂n. Note that this basis enjoys the eigenvalue equation
| (86) |
Projecting the conjugate state 〈j,kj| onto Eq. (83) yields, after some simplification (cf. Appendix C), the equation of motion
| (87) |
where Γjj’ is as in Eq. (76) and
| (88) |
| (89) |
with Qjj’=q‾j−q‾j’. Equation (87) can be written linear algebraically as
| (90) |
where and are defined as before [cf. Eq. (80)], and * denotes an element-by-element matrix product. Once again, Eq. (90) is lower-triangular in k and requires only matrix multiplication and the inversion of a J-by-J matrix K times. is initialized as in Eq. (79), and the kth term is computed from the previous k’<k terms. The joint distribution is retrieved via the inverse transform
| (91) |
4. |n,kn〉 basis
Expansion in either the |j,k〉 or the |j,kj〉 basis conveniently turns the master equation into a lower-triangular linear algebraic equation and replaces cutoffs in protein number with cutoffs in eigenmode number (which can be smaller with appropriate choices of gauge). However, these bases sacrifice the original tridiagonal structure of the master equation in the copy number of first gene, n. Therefore we now consider a mixed representation, in which the first gene remains in protein number space |n〉, and we expand the second gene in an n-dependent eigenbasis |kn〉,
| (92) |
If the rate parameter of the |kn〉 basis were the regulation function qn, it would be natural to make Ĥ0 the entire Hamiltonian [Eq. (64)]. For generality we will instead allow the rate parameter of the |kn〉 basis to be an arbitrary n-dependent local gauge q‾n, such that Eq. (63) at steady state naturally partitions as
| (93) |
where
| (94) |
| (95) |
with and Δ̂n(n)=q‾n−q̂n. Note that |n,kn〉 is not the eigenbasis of Ĥ0(n) but rather Ĥ0(n)|n,kn〉 retains the original tridiagonal structure in n, i.e.,
| (96) |
where Eqs. (12), (13), (65), and (67) are recalled in applying the first term of Ĥ0(n).
Projecting the conjugate state 〈n,kn| onto Eq. (93) yields, after some simplification (cf. Appendix C), the equation of motion
| (97) |
where
| (98) |
| (99) |
and . Equation (97) can be written linear algebraically as
| (100) |
where XT indicates the transpose of X, * denotes an element-by-element product, are super-(+) and subdiagonal (−) matrices, is a tridiagonal matrix, and , which is G with the columns reversed (i.e., ), is built incrementally in k. As with the |j,k〉 and |j,kj〉 bases, the kth term is slaved to the previous k’<k terms; in total the solution requires K inversions of an N-by-N matrix. However, here the task of inversion is simplified because the matrix to be inverted is tridiagonal. In fact, using the Thomas algorithm [42], we obtain an analytic solution for the case of constant production of the first gene and threshold regulation of the second gene, as described in Sec. II C.
The solution is initialized at k=0 using
| (101) |
[cf. Eq. (26)], where pn is known [cf. Eq. (60)], and the joint distribution is retrieved via the inverse transform
| (102) |
5. |j,kn〉 basis
We now consider a basis which employs both the constant-rate eigenfunctions |j〉 in the n sector and the n-dependent eigenfunctions |kn〉 in the m sector. Expressing the joint distribution directly in terms of the eigenbasis expansion of the generating function [43], we write
| (103) |
where, as in the |j,k〉 and |j,kj〉 bases, the |j〉 are parametrized by the constant rate g‾, and, as in the |n,kn〉 basis, the |kn〉 are parametrized by the arbitrary function q‾n. The inverse of Eq. (103) is
| (104) |
Substituting Eq. (103) into Eq. (58) at steady state gives, after some simplification (cf. Appendix C),
| (105) |
where Ĥ is defined as in Eq. (64), and act as in Eqs. (12)-(15), and
| (106) |
We may now partition Ĥ=Ĥ0+Ĥ1 as
| (107) |
| (108) |
with and Γ̂n=g‾−ĝn as in the |j,k〉 and |j,kj〉 bases and and Δ̂n(n)=q‾n−q̂n as in the |n,kn〉 basis. Noting that |j,kn〉 is the eigenbasis of Ĥ0(n), i.e.,
| (109) |
Equation (105) becomes, after more simplification (cf. Appendix C),
| (110) |
where Γjj’ is as in Eq. (76),
| (111) |
| (112) |
| (113) |
and is as in Eq. (99). Linear algebraically,
| (114) |
with and defined as before [cf. Eq. (80)], revealing once again a lower-triangular equation (i.e., each kth term is slaved to the previous k’<k terms) requiring only matrix multiplication and the inversion of a J-by-J matrix K times. Recalling Eq. (104), the scheme is initialized using
| (115) |
[cf. Eq. (26)] with known pn [cf. Eq. (60)], and the joint distribution is retrieved using Eq. (103).
B. Comparison of the representations
The spectral representations in Sec. II A produce equations of motion with similar levels of numerical complexity. In all cases, the original two-dimensional master equation has been reduced by the lower-triangular structure in the second gene’s eigenmode number k to a hierarchy of evaluations of one-dimensional problems. The bases differ in the rate parameters or equivalently gauge freedoms that one is free to choose: the |j,k〉 basis requires two constants g‾ and q‾; the |j,kj〉 basis requires g‾ and a J-valued vector q‾j; the |n,kn〉 basis requires a N-valued vector q‾n; and the |j,kn〉 basis requires g‾ and q‾n.
The bases also differ in the types of problems for which they are most suitable. For example, the |j,k〉, |j,kj〉, and |j,kn〉 bases, which all expand the parent species in eigenfunctions |j〉, are best when a cutoff in j is most appropriate, such as when the parent distribution is a Poisson. The |n,kn〉 basis, on the other hand, is useful when a cutoff is n is most appropriate, such as when the parent species is concentrated at low protein number. Different bases are more robust to numerical errors for different regulation functions as well: the |n,kn〉 and |j,kn〉 bases, which both rely upon repeated manipulation of the object , are best for smooth regulation functions, for which the differences between q‾n and q‾n+1 are small; the |j,k〉 and |j,kj〉 bases on the other hand, which involve the deviations q‾−qn and q‾j−qn respectively, are less susceptible to numerical error given sharp regulation functions, such as a threshold.
As indicated in Fig. 2, the |j,k〉 basis can be viewed as a special case of either the |j,kj〉 basis with q‾j=q‾ [in which case Eq. (87) reduces to Eq. (75)] or of the |j,kn〉 basis with q‾n=q‾ [in which case Eq. (110) reduces to Eq. (75)]. Although possible in principle, expanding in the |j,m〉 basis does not exploit the natural structure of the problem, since it neither retains the tridiagonal structure in n nor gains the lower triangular structure in k. This example explicitly shows that not all bases are good candidates for simplifying the master equation.
The strength of all the spectral bases discussed in this section and of the proposed spectral method in general is that it allows for a fast and accurate calculation of full steady-state probability distributions of the number of protein molecules in a gene regulatory network. In Fig. 3 we demonstrate this property for the two-gene system by plotting error versus computational runtime for each spectral basis, as well as for a stochastic simulation using a varying step Monte Carlo procedure [28]. For error we use the Jensen-Shannon divergence [41] (a measure in bits between two probability distributions) between the distribution pnm computed in the |n,m〉 basis (via iterative solution of the original master equation) and the distribution computed either via the spectral formulas in this section or by stochastic simulation. We plot this measure against the runtime of each method scaled by the runtime of the iterative solution in the |n,m〉 basis (all numerical experiments are performed in matlab). We find that the computations via the spectral bases achieve accuracy up to machine precision ~103–104 times faster than the iterative method’s runtime and ~107–108 times faster than the runtime necessary for the stochastic simulation to achieve the same accuracy. Computation in the |j,k〉 basis is most efficient since its equation of motion is simplest [cf. Eq. (80)]; the |j,kj〉 and |j,kn〉 bases tend to be slightly less efficient since they require inner loops over ℓ [cf. Eqs. (90) and (114)]. Figure 3 demonstrates the tremendous gain in performance achieved by the joint analytic-numerical spectral method over traditional simulation approaches.
C. Analytic solution
In general, the equations of motion in the spectral representations [Eqs. (75), (87), (97), and (110)] need to be evaluated numerically. In the case of the |n,kn〉 basis, however, we can exploit the tridiagonal structure of Eq. (97) to find an exact analytic solution. Specifically, in the case of a Poisson parent (gn=g=const) and for threshold regulation, i.e.,
| (116) |
setting q‾n=qn makes Eq. (97)
| (117) |
where
| (118) |
| (119) |
Δ=q+−q−, and n1=n0+1. Equation (117) is solved using the tridiagonal matrix algorithm (also called the Thomas algorithm [42]), as described in detail in Appendix D. The result is an analytic expression for the kth column of Gnk in terms of its previous columns (i.e., the matrix inversion has been performed explicitly),
| (120) |
where
| (121) |
| (122) |
| (123) |
| (124) |
| (125) |
and N is the cutoff in protein number n. Along with the analytic form of the mixed product
| (126) |
(cf. Appendix A), Eq. (120) in the limit N → ∞ constitutes an exact analytic solution for the joint distribution pnm as calculated using Eq. (102).
D. Threshold-regulated gene approximates the on/off gene
If a gene is regulated via a threshold function [cf. Eq. (116)], its steady-state protein distribution pm can be well approximated by the two-state process discussed in Sec. II C. To make the connection clear, we first observe that the off-state (z=−) corresponds to the first gene expressing the same or fewer proteins n than the threshold n0, i.e.,
| (127) |
and the on state (z=+) corresponds to the first gene expressing more proteins than the threshold, i.e.,
| (128) |
The dynamics of are then obtained by summing the master equation for two-gene regulation [Eq. (58)] over either all n≤n0 or all n>n0, giving
| (129) |
where n1=n0+1. Making the approximations
| (130) |
| (131) |
where
| (132) |
| (133) |
are the total probabilities of being in the off and on states, respectively, and noting from Eq. (59) that gn0=n1pn1/pn0, Eq. (129) at steady state becomes
| (134) |
with z=± and
| (135) |
where
| (136) |
Equations (134) and (135) have the same form as Eqs. (1) and (47) at steady state with n→m and g→q, and Eq. (136) relates the effective switching rates ω± to input and regulation parameters pn1, π±, and n1, and the ratio ρ of the degradation rate of the second gene to that of the first. Note that Eq. (136) satisfies
| (137) |
in agreement with Eq. (48), and exhibits the intuitive behavior that increasing ρ (i.e., decreasing the relative response rate of the first gene) is equivalent to decreasing the switching rates ω±.
A comparison of the distributions of a threshold-regulated gene with those of an on/off gene for various parameter settings reveals that Eqs. (130) and (131) are a good approximation. Figure 4 shows a demonstration for a threshold-regulated system with a Poisson input distribution. In the first column, the mean g of the input lies above the threshold n0, making the output more likely to be in the on-state, i.e., π+>π−; in the second column, g<n0, making π+<π−. In the first row ρ<1; in the second row ρ>1, corresponding to lower effective switching rates ω± and producing bimodal distributions with peaks near the on/off rates q±. In all examples, the approximation as a two-state process with switching rates given by Eq. (136) agrees well with the actual output from threshold regulation.
FIG. 4.
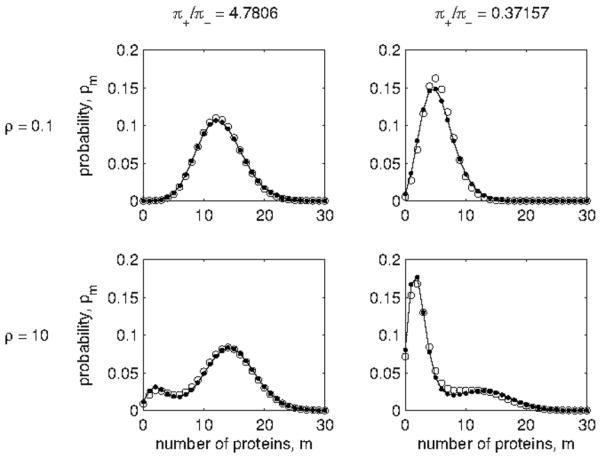
Protein distributions for a gene regulated by a threshold function [dots; calculated via Eq. (75)] and a gene with two stochastic states [circles; calculated via Eq. (43)]. The relationship between regulation parameters and state transition rates is given by Eq. (136). In all panels the input to the regulation is a Poisson distribution with mean g=7, and the regulation rates [cf. Eq. (116)] are q−=2 and q+=15. In the first column the threshold is n0=4 making π+=0.827>π−=0.173; in the second column n0=8 making π+=0.271<π−=0.729. In the first row the ratio of the second gene’s degradation rate to that of the first is ρ=0.1; in the second row ρ=10.
IV. REGULATION WITH BURSTS
The final system we consider combines the multistate process used to model bursts of expression in Sec. I with gene regulation as discussed in Sec. II. Specifically we consider a system of two species, with protein numbers n and m, existing in Z possible states, distinguished by the settings of the two production rates gz and qz respectively, where 1≤z≤Z. Regulation is achieved by allowing the rates of transition among states affecting the production of the second gene to depend on the number n of proteins expressed by the first gene. Recalling Eqs. (1) and (58), the master equation describing the evolution of the joint probability distribution reads
| (138) |
where the dependence of the stochastic matrix (Ωzz’ on n incorporates the regulation.
As with the previously discussed models, Eq. (138) benefits from spectral expansion, and for simplicity we present only the formulation in the |j,k〉 basis, parametrized by constant rates g‾ and q‾ respectively, as in Secs. II B and III A 2. As before the first step is to define the generating function
| (139) |
with which Eq. (138), upon summing over n and m against |n,m〉, becomes
| (140) |
where
| (141) |
| (142) |
| (143) |
| (144) |
| (145) |
and Ω̂zz’ is Ωzz’(n) with every instance of n replaced by the number operator . Defining and , we partition the Hamiltonian as , with
| (146) |
as the operator of which |j,k〉 is the eigenbasis, i.e.,
| (147) |
and
| (148) |
capturing the deviations Γz=g‾−gz and Δz=q‾−qz of the constant rates from the state-dependent rates. Upon expanding the generating function in the eigenbasis,
| (149) |
and taking dummy indices j→j’ and k→k’, projecting the conjugate state 〈j,k| onto Eq. (140) gives
| (150) |
where the components of Ω̂zz’ need only be evaluated in the j sector, not the k sector, because the transition rates depend on only n, not m [cf. Eq. (138)]. Like Eqs. (43) and (75), Eq. (150) is subdiagonal in k and thus far more efficient to solve than the original master equation [Eq. (138)] as we demonstrate for a special case in the next section. The joint distribution is retrieved from via inverse transform,
| (151) |
with the mixed products calculated as in Appendix A.
A. Four-state process
As a simple example of the model in Eq. (138), we consider a system in which each of the two species has an on state and an off state, and the transition rate of the second species to its on state is a function of the number of copies of the first species. This system models both (i) a single gene for which the production of proteins depends on the number of transcripts, and each is produced in on and off states by the binding and unbinding of ribosomes and RNA polymerase, respectively, and (ii) one gene regulating another with each undergoing burstlike expression.
There are a total of Z=4 states, i.e.,
| (152) |
where the first signed index denotes the state of the first gene (with protein count n) and the second signed index denotes the state of the second gene (with protein count m). Defining g± as the production rates of the first species in its on (+) and off states (−), and similarly q± for the second species, the production rates of the Z=4 states are
| (153) |
| (154) |
Defining ω± as the transition rates of the first species to (+) and from (−) its on state, and similarly α± for the second species, the transition matrix takes the form
| (155) |
The simple form α+(n)=cnν for constant c and integer ν corresponds to the first species activating the second as a multimer, with ν the order of the multimerization. In the limit of fast switching this description reduces to a Hill function with cooperativity ν [23]. Recalling that and , the n-dependent terms of 〈j|Ω̂zz’|j’〉 are evaluated as
| (156) |
Since and raise and lower |j’〉 states respectively [cf. Eqs. (22) and (23)], the modified transition matrix 〈j|(Ω̂zz’|j’〉 is nearly diagonal, with nonzero terms only for |j−j’|≤ν.
Equation (150) at steady state,
| (157) |
is solved successively in k, requiring the inversion of a 4J-by-4J matrix K times. It is initialized at k=0 by computing the null space of the left-hand side and normalizing with [cf. Eq. (35)]. The joint distribution is retrieved via inverse transform [Eq. (151)].
With ν=2, a typical solution of Eq. (157) takes a few seconds (in matlab), which, depending on the cutoff N, is ~102–103 times faster than direct solution of the master equation [Eq. (138)] by iteration for equivalent accuracy. The advantage of such a large efficiency gain is that it allows repeated evaluations of the governing equation, necessary for parameter inference or optimization [31]. We demonstrate this possibility in the next section by finding and interpreting the solutions that optimize the information flow from the first to the second species.
B. Information-optimal solution
Cells use regulatory processes to transmit relevant information from one species to the next [44-48]. Information processing is quantified by the mutual information I, which, between the first and second species in the four-state process, is
| (158) |
where the distributions pnm, pn, and pm are obtained from summing the joint distribution [cf. Eq. (151)], and the log is taken with base 2 to give I in bits.
Upon optimization of I for the four-state process, two distinct types of optimal solutions become clear: those in which the distribution pnm has one peak and those in which pnm has two peaks. The former occur when copy number is constrained to be low, and switching rates are constrained to be near the decay rates of both species, producing a single peak at low copy number [see lower left inset of Fig. 5(B)]. As these constraints are lifted, it is optimal for the switching rates of the parent species to become much less than the decay rate. The slow switching produces a second peak whose location is specified by the on rate of each species [see upper right inset of Fig. 5(B)].
FIG. 5.

(A) Mutual information I [cf. Eq. (158)] versus stiffness σ [cf. Eq. (160)] for fixed gain [γ=16, cf. Eq. (159)] obtained by optimizing Eq. (161) for λ values between 10−3 and 101. Squares denote solutions whose joint distribution pnm has one peak (cf. B, lower left inset), and dots denote solutions for which pnm has two peaks (cf. B, upper right inset). Solid lines show the convex hulls of the one- and two-peaked solutions. Dotted lines indicate the stiffness value at which the hulls intersect and the stiffness values of the hull points to the left and right of the intersection. (B) Phase diagram between one- and two-peaked optimal solutions in the gain-stiffness plane. Circles and left and right error bars at each gain are determined by the stiffness values at the intersection of the one- and two-peak convex hulls and at the hull points to the left and right of the intersection, respectively (see dotted lines for the example case in A). Solid line shows a line of best fit. Insets show examples of one-(lower left) and two-peaked (upper right) optimal distributions pnm.
To quantify the transition between the two types of solutions, we numerically optimized mutual information over parameters g+, q+, ω−, ω+, α−, and c (the off-rates g− and q− were fixed at 0; the cooperativity ν was fixed at 2; and the decay rate ratio ρ was fixed at 1). Information may always be trivially optimized by allowing infinite copy number or arbitrary separation of relevant time scales. We limit copy number by constraining the gain
| (159) |
defined as the average of the parent gain 〈=g+−g− and the child gain Δ=q+−q−. Since g−=q−=0, the maximum number of particles is dictated by the on rates g+ and q+ and thus constraining γ limits the copy number. We limit separation between the switching time scales and the decay time scales by constraining the stiffness
| (160) |
where the average 〈nν〉 is taken over pn. Stiffness σ is the average of the absolute deviation of (the logs of) all four switching rates from the (unit) decay rates so constraining σ prevents fast or slow switching. Gain is fixed by varying g+ and q+ such that γ is a constant, and stiffness is constrained by optimizing the objective function
| (161) |
for a given value of the Lagrange multiplier λ.
As shown in Fig. 5(A), one-peaked solutions are more informative at low stiffness, while two-peaked solutions are more informative at high stiffness. We compute the convex hulls of the one- and two-peaked data to remove suboptimal solutions, and the transition occurs at the stiffness value at which the convex hulls intersect [cf. Fig. 5(A)]. Repeating this procedure for many choices of gain allows one to trace out the phase transition shown Fig. 5(B), which makes clear that one-peaked solutions are most informative at low stiffness, two-peaked solutions are most informative at high stiffness, and the critical stiffness decreases weakly with increasing gain.
Examining the two-peaked solutions, which are optimal at high stiffness, we find that the marginal probability to be in the (−,+) state, where the first gene is off and the second gene is on, , is much smaller than the marginal probabilities to be in the other three states [π−,+/πz<0.01, where z={(+,+), (+,−), (−,−)}]. This result states that it is unlikely for the regulated gene to be expressing proteins at a high level, if the regulator protein or mRNA (depending on the interpretation of the model) is not being expressed. Optimizing information, we find rates which result in this intuitive solution.
V. CONCLUSIONS
The presented spectral method exploits the linearity of the master equation to solve for probability distributions directly by expanding in the natural eigenfunctions the linear operator. We demonstrate the method on three models of gene expression: a single gene with multiple expression states, a gene regulatory cascade, and a model that combines multistate expression with explicit regulation through binding of transcription factor proteins.
The spectral method permits huge computational gains over simulation. As demonstrated for all spectral expansions of the two-gene cascade (cf. Fig. 3), directly solving for the distribution via the spectral method is ~107–108 times faster than building the distribution from samples using a simulation technique. This massive speedup makes possible optimization and inference problems requiring full probability distributions that were not computationally feasible previously. For example, by optimizing information flow in a two-gene cascade in which both parent and child undergo two-state production, we reveal a transition from a one-peaked to a two-peaked joint probability distribution when constraints on protein number and time scale separation are relaxed. We emphasize that this optimization would not have been possible without the efficiency of the spectral method.
The spectral method also makes explicit the linear algebraic structure underlying the master equation. In many cases, such as in two-state bursting and the two-gene threshold regulation problem, this leads to analytic solutions. In general, such as shown in the case of the linear cascade, this leads to a set of natural bases for expansion of the generating function and reveals the features of each basis that are better suited to different types of problems. Specifically, bases in which the parent species is expanded in eigenfunctions are best when the parent distribution is Poissonian, and bases in which the parent is left in protein number space are best when the parent distribution is concentrated at low protein number. As well, bases in which the eigenfunctions of the child depend on the number of copies of the parent’s protein are best suited for smooth regulation functions, whereas a basis in which the eigenfunctions of the child are parametrized by a constant is more numerically robust for sharp regulation functions such as thresholds. In all cases the linear algebraic structure of the spectral decomposition yields numerical prescriptions that greatly outperform simulation techniques. We anticipate that the computational speedup of the method, as well as the removal of the statistical obstacle of density estimation inherently limiting simulation-based approaches, will make spectral methods such as those demonstrated here useful in addressing a wide variety of biological questions regarding accurate and efficient modeling of noisy information transmission in biological systems.
ACKNOWLEDGMENTS
A.M. was supported by National Science Foundation Grant No. DGE-0742450. A.M. and C.W. were supported by National Science Foundation Grant No. ECS-0332479. A.M.W. was supported by the Princeton Center for Theoretical Science and by Columbia’s Professional Schools Visiting Scholar Grant. C.W. was supported by National Institutes of Health Grants No. 5PN2EY016586-03 and No. 1U54CA121852-01A1.
APPENDIX A
In this appendix we describe two ways to compute the mixed products 〈n|j〉 and 〈j |n〉 between the protein number states |n〉 and the eigenstates |j〉: by direct evaluation and by recursive updating.
The direct evaluation follows from Eqs. (8)–(10) and (20), and the fact that repeated derivatives of a product follow a binomial expansion. Introducing g as the rate parameter for the |j〉 states,
| (A1) |
| (A2) |
| (A3) |
| (A4) |
| (A5) |
| (A6) |
where
| (A7) |
Similarly, noting Eqs. (7) and (21),
| (A8) |
with ζnj as in Eq. (A7). Equations (A6) and (A8) clearly reduce to Eqs. (26) and (27) for the special case j =0.
It is more computationally efficient to take advantage of the selection rules in Eqs. (12)–(15) and (22)–(25) to compute the mixed products recursively. For example, using Eqs. (14), (16), and (22),
| (A9) |
which can be initialized using 〈n|0〉=e−ggn/n! [cf. Eq. (A6)] and updated recursively in j. Equation (A9) makes clear that in n space the (j +1)th mode is simply the (negative of the) discrete derivative of the jth mode.
Alternatively, Eqs. (15), (17), and (23) give
| (A10) |
which can be initialized using 〈0|j〉=(−1)je−g [cf. Eq. (A6)] and updated recursively in n.
One may similarly derive recursion relations for 〈j |n〉, i.e.,
| (A11) |
| (A12) |
initialized with 〈j |0〉=(−g)j / j! or 〈0|n〉=1, respectively [cf. Eq. (A8)] and updated recursively in n or j, respectively.
One may also use the full birth-death operator b̂+b̂− to derive the recursion relations
| (A13) |
| (A14) |
initialized with 〈0|j〉=(−1)je−g and 〈1|j〉=(−1)je−g(g− j) [cf. Eq. (A6)], and 〈j |0〉=(−g)j / j! and 〈j |1〉=(−g)j(1−j /g)/ j! [cf. Eq. (A8)], respectively, and updated recursively in n.We find Eqs. (A13) and (A14) are more numerically stable than Eqs. (A9)–(A12), as the former are two-term recursion relations while the latter are one-term recursion relations.
APPENDIX B
In the limit g−=0, Eq. (54) reads
| (B1) |
where y=eg+(x−1). Using the fact that [49]
| (B2) |
Eq. (B1) can be written
| (B3) |
or noting Eq. (55) and the fact that Γ(z+1)=zΓ(z) for any z,
| (B4) |
| (B5) |
| (B6) |
| (B7) |
as in Eq. (56).
The marginal pn is given by
| (B8) |
[cf. Eq. (10)]. Using Eq. (56) and the derivative of the confluent hypergeometric function,
| (B9) |
one obtains Eq. (57).
APPENDIX C
In this appendix, we fill in the details of the derivations of the equations of motion for the latter three of the four spectral bases discussed in Sec. II A.
1. |j,kj〉 basis
Projecting the conjugate state 〈j,kj| onto Eq. (83) (in which dummy indices j and k are changed to j’ and k’, respectively) gives
| (C1) |
From the orthonormality of states, the first term of Eq. (C1) simplifies to
| (C2) |
Recalling Eq. (30), the product simplifies to
| (C3) |
with Qjj’=q̂j−q̂j’, whereupon Eq. (C1), separating the part of its second term which is diagonal in k from that which is subdiagonal and applying Eq. (24) to its third term, becomes
| (C4) |
with Γjj’ as in Eq. (76) and
| (C5) |
| (C6) |
| (C7) |
[where the orthonormality of |j〉 states is used in going from Eq. (C5) to Eq. (C6)]. Defining ℓ=k−k’ and
| (C8) |
2. |n,kn〉 basis
Projecting the conjugate state 〈n,kn| onto Eq. (93) (in which dummy indices n and k are changed to n’ and k’, respectively) gives
| (C9) |
where Δn=q‾n−qn. Noting that, as in Eq. (30),
| (C10) |
where , Eq. (C9) becomes
| (C11) |
Separating the parts of the second and third term that are diagonal in k and defining ℓ=k−k’ and
| (C12) |
3. |j,kn〉 basis
Substituting Eq. (103) into Eq. (58) at steady state gives
| (C13) |
or in terms of raising and lowering operators [cf. Eqs. (14)]
| (C14) |
Using the definitions in Eqs. (64)–(68), Eq. (C14) can be written as Eq. (105).
Using Eqs. (107)–(109), Eq. (105) can be written
| (C15) |
where
| (C16) |
and the dummy indices j and k have been changed to j’ and k’ respectively. Using Eq. (C10) to note that
| (C17) |
where , we multiply Eq. (C15) by 〈kn|m〉 and sum over m to obtain
| (C18) |
in which we exploit the completeness of |m〉 states, i.e., Σm|m〉〈m|=1, and is as in Eq. (99). Multiplying Eq. (C18) by 〈j |n〉, summing over n, and exploiting Σn|n〉〈n|=1 for the first two terms, we obtain Eq. (110).
APPENDIX D
In this appendix we explicitly solve for Gnk in Eq. (117) using the tridiagonal matrix or Thomas [42] algorithm. We start by identifying the subdiagonal, diagonal, superdiagonal, and right-hand side elements of Eq. (117), respectively, as
| (D1) |
| (D2) |
| (D3) |
| (D4) |
where N is the cutoff in protein count n and n1=n0+1. Auxiliary variables are defined iteratively as
| (D5) |
| (D6) |
| (D7) |
| (D8) |
and the solution is obtained by backward iteration with
| (D9) |
| (D10) |
(where k has been moved from subscript to superscript for ease of reading).
Computing the first few terms of Eq. (D6) reveals the pattern
| (D11) |
where
| (D12) |
with the convention that if a>b. Note that since , we may also use the integral representation of the Gamma function to write
| (D13) |
| (D14) |
| (D15) |
Using Eq. (D8) it is immediately clear that
| (D16) |
The first nonzero term is
| (D17) |
where we have defined
| (D18) |
Further iteration of Eq. (D8) makes clear that
| (D19) |
| (D20) |
where
| (D21) |
Computing the first few terms of Eq. (D10) reveals that
| (D22) |
| (D23) |
where
| (D24) |
At the threshold Eq. (D10) gives
| (D25) |
and since , the solution is easily completed using Eq. (D10), giving
| (D26) |
| (D27) |
Footnotes
PACS number(s): 87.10.Mn, 82.20.Fd, 87.10.Vg, 02.70.Hm
Contributor Information
Andrew Mugler, Department of Physics, Columbia University, New York, New York 10027, USA.
Aleksandra M. Walczak, Princeton Center for Theoretical Science, Princeton University, Princeton, New Jersey 08544, USA.
Chris H. Wiggins, Department of Applied Physics and Applied Mathematics, Center for Computational Biology and Bioinformatics, Columbia University, New York, New York 10027, USA.
References
- [1].Hooshangi S, Thiberge S, Weiss R. Proc. Natl. Acad. Sci. U.S.A. 2005;102:3581. doi: 10.1073/pnas.0408507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Thattai M, van Oudenaarden A. Biophys. J. 2002;82:2943. doi: 10.1016/S0006-3495(02)75635-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Elowitz MB, Levine AJ, Siggia ED, Swain PS. Science. 2002;297:1183. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- [4].Ozbudak EM, Thattai M, Kurtser I, Grossman AD, van Oudenaarden A. Nat. Genet. 2002;31:69. doi: 10.1038/ng869. [DOI] [PubMed] [Google Scholar]
- [5].Swain PS, Elowitz MB, Siggia ED. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12795. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Paulsson J, Berg OG, Ehrenberg M. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7148. doi: 10.1073/pnas.110057697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pedraza JM, van Oudenaarden A. Science. 2005;307:1965. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- [8].Paulsson J. Nature (London) 2004;427:415. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- [9].Tkačik G, Callan CG, Bialek W. Phys. Rev. E. 2008;78:011910. doi: 10.1103/PhysRevE.78.011910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tkacik G, Callan CG, Jr., Bialek W. Proc. Natl. Acad. Sci. U.S.A. 2008;105:12265. doi: 10.1073/pnas.0806077105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ziv E, Nemenman I, Wiggins CH. PLoS ONE. 2007;2:e1077. doi: 10.1371/journal.pone.0001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mugler A, Ziv E, Nemenman I, Wiggins CH. IET Sys. Bio. 2009;3:379. doi: 10.1049/iet-syb.2008.0165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Emberly E. Phys. Rev. E. 2008;77:041903. doi: 10.1103/PhysRevE.77.041903. [DOI] [PubMed] [Google Scholar]
- [14].Tostevin F, ten Wolde PR. Phys. Rev. Lett. 2009;102:218101. doi: 10.1103/PhysRevLett.102.218101. [DOI] [PubMed] [Google Scholar]
- [15].Elf J, Li G-W, Xie XS. Science. 2007;316:1191. doi: 10.1126/science.1141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Choi PJ, Cai L, Frieda K, Xie XS. Science. 2008;322:442. doi: 10.1126/science.1161427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. PLoS Biol. 2006;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Golding I, Paulsson J, Zawilski SM, Cox EC. Cell. 2005;123:1025. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- [19].Tǎnase-Nicola S, Warren PB, Rein ten Wolde P. Phys. Rev. Lett. 2006;97:068102. doi: 10.1103/PhysRevLett.97.068102. [DOI] [PubMed] [Google Scholar]
- [20].van Zon JS, Morelli MJ, Tǎnase-Nicola S, ten Wolde PR. Biophys. J. 2006;91:4350. doi: 10.1529/biophysj.106.086157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].van Zon JS, ten Wolde PR. J. Chem. Phys. 2005;123:234910. doi: 10.1063/1.2137716. [DOI] [PubMed] [Google Scholar]
- [22].van Kampen NG. Stochastic Processes in Physics and Chemistry. North-Holland; Amsterdam: 1992. [Google Scholar]
- [23].Walczak AM, Sasai M, Wolynes PG. Biophys. J. 2005;88:828. doi: 10.1529/biophysj.104.050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lan Y, Papoian GA. J. Chem. Phys. 2006;125:154901. doi: 10.1063/1.2358342. [DOI] [PubMed] [Google Scholar]
- [25].Lan Y, Wolynes PG, Papoian GA. J. Chem. Phys. 2006;125:124106. doi: 10.1063/1.2353835. [DOI] [PubMed] [Google Scholar]
- [26].Lan Y, Papoian GA. Phys. Rev. Lett. 2007;98:228301. doi: 10.1103/PhysRevLett.98.228301. [DOI] [PubMed] [Google Scholar]
- [27].Bortz AB, Kalos MH, Lebowitz JL. J. Comput. Phys. 1975;17:10. [Google Scholar]
- [28].Gillespie DT. J. Phys. Chem. 1977;81:2340. [Google Scholar]
- [29].Bellman RE. Adaptive Control Processes. Princeton University Press; Princeton, NJ: 1961. [Google Scholar]
- [30].Morelli MJ, ten Wolde PR, Allen RJ. Proc. Natl. Acad. Sci. U.S.A. 2009;106:8101. doi: 10.1073/pnas.0810399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Walczak AM, Mugler A, Wiggins CH. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6529. doi: 10.1073/pnas.0811999106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].A master equation in which the coordinate appears explicitly in the rates [e.g. gn or qn in Eq. (58)] is sometimes mistakenly termed “nonlinear” in the literature, perhaps discouraging calculations which exploit its inherent linear algebraic structure. We remind the reader that the master equation is perfectly linear.
- [33].Klumpp S, Hwa T. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20245. doi: 10.1073/pnas.0804953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Iyer-Biswas S, Hayot F, Jayaprakash C. Phys. Rev. E. 2009;79:031911. doi: 10.1103/PhysRevE.79.031911. [DOI] [PubMed] [Google Scholar]
- [35].Note that setting x=eik makes clear that the generating function is simply the Fourier transform.
- [36].Note here, however, the difference with respect to the normalization convention commonly used in quantum mechanics in the prefactors of the creation and annihilation operations.
- [37].Doi M. J. Phys. A. 1976;9:1465. [Google Scholar]
- [38].Zel’dovich Ya. B., Ovchinnikov AA. JETP. 1978;47:829. [Google Scholar]
- [39].Peliti L. J. Phys. A. 1986;19:L365. [Google Scholar]
- [40].Mattis DC, Glasser ML. Rev. Mod. Phys. 1998;70:979. [Google Scholar]
- [41].Lin J. IEEE Trans. Inf. Theory. 1991;37:145. [Google Scholar]
- [42].Thomas L. Watson Sci. Columbia University; New York: 1949. [Google Scholar]
- [43].Because this basis is a function of three coordinates (j, k, and n), we cannot abstractly define the generating function as in Eq. (61); we must instead work directly with the joint probability distribution.
- [44].Gomperts BD, Kramer IM, Tantham PER. Signal Transduction. Academic Press; San Diego: 2002. [Google Scholar]
- [45].Ting AY, Endy D. Science. 2002;298:1189. doi: 10.1126/science.1079331. [DOI] [PubMed] [Google Scholar]
- [46].Detwiler PB, Ramanathan S, Sengupta A, Shraiman BI. Biophys. J. 2000;79:2801. doi: 10.1016/S0006-3495(00)76519-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bolouri H, Davidson EH. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9371. doi: 10.1073/pnas.1533293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bassler BL. Curr. Opin. Microbiol. 1999;2:582. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- [49].Koepf W. Hypergeometric Summation: An Algorithmic Approach to Summation and Special Function Identities. Vieweg; Braunschweig, Germany: 1998. [Google Scholar]