

Abstract
Motivation: X-ray crystallography-based protein structure determination, which accounts for majority of solved structures, is characterized by relatively low success rates. One solution is to build tools which support selection of targets that are more likely to crystallize. Several in silico methods that predict propensity of diffraction-quality crystallization from protein chains were developed. We show that the quality of their predictions drops when applied to more recent crystallization trails, which calls for new solutions. We propose a novel approach that alleviates drawbacks of the existing methods by using a recent dataset and improved protocol to annotate progress along the crystallization process, by predicting the success of the entire process and steps which result in the failed attempts, and by utilizing a compact and comprehensive set of sequence-derived inputs to generate accurate predictions.
Results: The proposed PPCpred (predictor of protein Production, Purification and Crystallization) predict propensity for production of diffraction-quality crystals, production of crystals, purification and production of the protein material. PPCpred utilizes comprehensive set of inputs based on energy and hydrophobicity indices, composition of certain amino acid types, predicted disorder, secondary structure and solvent accessibility, and content of certain buried and exposed residues. Our method significantly outperforms alignment-based predictions and several modern crystallization propensity predictors. Receiver operating characteristic (ROC) curves show that PPCpred is particularly useful for users who desire high true positive (TP) rates, i.e. low rate of mispredictions for solvable chains. Our model reveals several intuitive factors that influence the success of individual steps and the entire crystallization process, including the content of Cys, buried His and Ser, hydrophobic/hydrophilic segments and the number of predicted disordered segments.
Availability: http://biomine.ece.ualberta.ca/PPCpred/.
Contact: lkurgan@ece.ualberta.ca
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Structural genomics (SG) is an international initiative that aims at solving 3D shapes of important biological macro-molecules, primarily focusing on proteins, and which significantly contributes to the overall protein structure determination efforts (Chandonia and Brenner, 2006). This initiative shifts the focus from one-by-one determination of individual protein structures to protein family-directed structure analyses where a group of proteins is targeted and structure(s) of representative members are determined (Terwilliger et al., 1998). The process of choosing the representative proteins, which is known as target selection, encompasses restricting the candidate proteins to those that are tractable and of unknown structure and prioritizing them according to the expected interest and accessibility (Brenner, 2000). For instance, in case of the protein structure initiative (PSI), target selection concentrates on representatives from large, structurally uncharacterized protein domain families, and from structurally uncharacterized subfamilies in very large and diverse families with incomplete structural coverage (Dessailly et al., 2009). The most recent PSI:Biology phase (PSI-Nature Structural Biology Knowledgebase, 2011), includes four major SG centers that will continue high-throughput structure determination defined through community nomination process. Importantly, these approaches allow for flexibility in the selection of the targets and would benefit from tools that support target selection.
Protein structures are solved using X-ray crystallography, NMR spectroscopy, electron microscopy and (recently) by application of computational approaches, such as homology modeling. The most popular method, which accounts for about 87% of the structures deposited into the Protein Data Bank (PDB), is the X-ray crystallography (Berman et al., 2000). One of the main challenges the SG faces is that only about 2–10% of protein targets pursued yield high-resolution protein structures (Service, 2005). A more recent analysis shows that about 4.6% of targets produce the diffraction-quality crystals (Kurgan and Mizianty, 2009). Moreover, estimates show that >60% of the cost of structure determination is consumed by the failed attempts (Slabinski et al., 2007a), while crystallization is characterized by a significant rate of attrition and is among the most complex and the least understood problems in the structural biology (Hui and Edwards, 2003). This provides motivation for further research in this area. Several strategies have been proposed to improve the success rates including obtaining one representative structure per protein family and working with multiple orthologs (Brenner, 2000; Chandonia and Brenner, 2005; Hui and Edwards, 2003). In spite of the advances made in the context of protein crystallization (McPherson, 2004), the production of high-quality crystals is one of the major bottlenecks in the structure determination pipelines (Biertumpfel et al., 2005; Chayen, 2004; Puesy et al., 2005). This problem is usually tackled using an empirical trial and error approach, where a large number of experiments is brute-forced to find a suitable setup, and through understanding of the fundamental principles that govern crystallization (Chayen, 2004). The latter is used to design new and improved methodologies that produce the high-quality crystals.
One of the steps taken to alleviate difficulties in solving the structures via the X-ray crystallography was to create databases that record information concerning both successful and failed attempts to produce the structures (Rodrigues and Hubbard, 2003). The largest and most comprehensive database, TargetDB (Chen et al., 2004), which was launched July 2001, builds upon the work on the PRESAGE database (Brenner et al., 1999). TargetDB consolidates data from 28 SG centers in USA, Canada, Germany, Isreal, Japan, France and UK, including nine PSI centers. The PepcDB (Protein Expression Purification and Crystallization DataBase) was established around 2004 as an extension to the TargetDB to collect more detailed status information and experimental details for each step in the protein structure production pipelines (Kouranov et al., 2006). This database stores a complete history of the status of the experimental steps in each production trial, the current status and stop conditions, which are collected from 15 SG centers in USA.
The availability of the databases motivated the development of analytical and predictive models that either support or directly predict protein crystallization (Rupp and Wang, 2004). Initial work concentrated on finding sequence-derived factors that are useful to determine crystallization propensity. For instance, using the data from the TargetDB, Goh et al. (2004) found that conservation of the sequence across organisms, composition of charged residues, occurrence of hydrophobic patches in the sequence, number of binding partners and chain length influence the feasibility for the high-throughput structure determination. The isoelectric point calculated from the sequence was used to suggests optimal pH ranges for the crystallization screening (Kantardjieff and Rupp, 2004; Kantardjieff et al., 2004). The Berkeley's SG center utilized several protein features including chain length and predicted transmembrane helices, coiled coils and low-complexity regions to eliminate the intractable targets (Chandonia et al., 2006). A recent study shows that the crystallization propensity can be computed from the knowledge of predicted disordered residues, side-chain entropy of predicted exposed residues and the amount of Phe and predicted buried Gly in the input sequence (Price et al., 2009). These works demonstrate that the crystallization propensity can be successfully predicted from the protein chain. However, these studies usually concern data from a single SG center, and propose simple predictive models (that could be outperformed by more advanced models) that are rarely made available, e.g. via web servers, to the community.
To this end, several computational sequence-based crystallization propensity predictors which utilize the data that span multiple SG centers and more advanced predictive models were proposed in the recent years. They include SECRET (Smialowski et al., 2006), OB-Score (Overton and Barton, 2006), CRYSTALP (Chen et al., 2007), XtalPred (Slabinski et al., 2007a, b), ParCrys (Overton et al., 2008), CRYSTALP2 (Kurgan et al., 2009), MetaPPCP (Mizianty and Kurgan, 2009), PXS (Price et al., 2009), SVMCrys (Kandaswamy et al., 2010) and MCSG-Z score (Babnigg and Joachimiak, 2010). Some of them, including the OB-score and XtalPred, were already used by the SG centers. Details concerning these methods can be found in a recent review (Kurgan and Mizianty, 2009). The above predictors have a few drawbacks, which motivate this work. They are built and tested using outdated data, which results in a relatively poor performance for recent data (we demonstrate that in Fig. 2 in Section 3), the annotation of the chains used in their training database (crystallizable versus crystallization resistant) is based on an incomplete/inaccurate protocol, and they address the prediction of the success of the entire crystallization process without pointing out which of the steps is responsible for the failure. Our objective is to alleviate these drawbacks by proposing a novel predictor that (i) is built using a recent and large dataset, (ii) uses improved annotation protocol, (iii) targets prediction of the success of the entire crystallization process and also predicts which of the steps results in the failed attempts and (iv) uses a compact and comprehensive range of sequence-derived inputs to generate accurate predictions. In collaboration with the TargetDB and PepcDB curators, we formulate a more precise and comprehensive protocol to annotate proteins. Also, our method can be used to predict propensity of a given chain for (i) production of diffraction quality crystals, (ii) production of crystals, (iii) purification and (iv) production of the protein material; the existing methods target only one of these outcomes. The proposed method, called PPCpred (predictor of protein Production, Purification and Crystallization) provides individual predictions for each the four steps, and it also provides an integrated output that predicts whether the chain would produce the diffraction quality crystals, and if not then it predicts which of the steps is most likely to cause the failure.
Fig. 2.

The MCC (A) and AUC (B) values obtained by the considered crystallization propensity predictors with respect with the date of the test trials (x-axis) from the DB_CRYS test dataset. BLAST and SVMCrys provide only binary prediction; their AUC cannot be computed.
2 METHODS
2.1 Current annotation protocols
The existing sequence-based crystallization propensity predictors assign one of the two labels, crystallizable or non-crystallizable, to each protein chain. The only exception is the XtalPred which defines five classes that range between ‘easy to crystallize’ and ‘hard to crystallize’. However, the five classes are equivalent to the probabilities/scores generated by the other methods. The annotations used in SECRET are primarily based on the data from the PDB and they were improved by the subsequent works. Most of the subsequent predictors, including CRYSTALP, OB-Score, ParCrys, CRYSTALP2, MetaPPCP and SVMCrys, utilize data extracted from PepcDB and TargetDB databases using approach described in Overton and Barton (2006) to annotate proteins as crystallizable and non-crystallizable and to derive their training and test datasets. As it was pointed out in Overton and Barton (2006), this annotation protocol has a few shortcomings. The authors of XtalPred use a different approach in terms of how the crystallizable samples were extracted. In XtalPred (Slabinski et al., 2007b) only trials deposited in PDB were marked as the crystallizable trials, whereas in Overton et al. (2008) those trials were excluded. The two remaining predictors use data from only one SG center; PXS is based on data from the Northeast SG Consortium and MCSG-Z score from the Midwest Center for SG.
Our aim is to deliver more comprehensive and precise annotations of the crystallization propensity (and the other steps in the crystallization process). First, we consider only the proteins with the known outcome of the experiment, i.e. completed stop status, as opposed to the previous approaches in which the experiment was considered as finished if there was no update for the specific amount of time or it was annotated as work stopped. We note that there are instances where work on a given target was discontinued for unspecified reasons, not necessarily related to the crystallization protocol, e.g. the targets were re-prioritized and administratively abandoned. This is often done without evaluating/reporting the success or failure at the trial level, and thus it is difficult to associate an interpretation to an unqualified ‘work-stopped’ status. Although the authors of Overton and Barton (2006) were aware of this, a small number of trials with the completed stop status at the time when they developed their method did not allow them to utilize a more accurate annotation protocol. Secondly, we filter our dataset to further improve the quality of annotations for the selected samples; details are explained in Section 2.2. Thirdly, we divide the chains annotated as the non-crystallizable to indicate the reason for the crystallization failure. We hypothesize that success/failure in each of the crystallization process steps may be associated with different protein properties. Fourthly, previous methods used the training and test datasets with substantially reduced sequence similarity (<25%). This removes hard to predict samples, as sometimes even relatively minor changes in the protein sequence (e.g. point mutations and usage of C- and N-terminus tag to ease purification) may change the final outcome of the crystallization. In contrast, we remove similar sequences only within each class (e.g. similar crystallizable chains), but we do not reduce the sequence similarity between the classes (e.g. between crystallizable and non-crystallizable chains). Finally, we use new data (from between 2006 and 2009) to address recent changes in the crystallization protocols; the prior predictors were built on older data from before 2006.
2.2 Annotations and datasets
The protein chains were extracted from the PepcDB (Kouranov et al., 2006). We used PepcDB downloaded on November 17, 2010, which includes 261 572 targets. A target defines the objective of the crystallization attempt for either a single protein or a collection of proteins. Targets in PepcDB can have either one or multiple trials, each trial representing a set of procedures used to crystallize a target. There are 817 099 trials in our PepcDB dataset. Each trial has information about its current status and, in case if work was finished, the stop status, see Table 1. The stop statuses indicate the step at which the work on a given trial was stopped and the reason of the failure (which divides the work covered in the current status into substeps) or the fact that the trial produced a proper outcome. The majority of trials have the stop status field empty, which makes it impossible to deduct the final outcome of the trial; we cannot be sure whether the experiment was finished, abandon or is still in progress. Therefore, we selected trials with the completed stop status field, with the exception for trials with the current status ‘in PDB’ or ‘crystal structure’, as they clearly indicate the successful crystallization attempts. Since each trial may concern more than one sequence, we considered each sequence from each trial as a separate trial.
Table 1.
List of stop statuses and current statuses in PepcDB
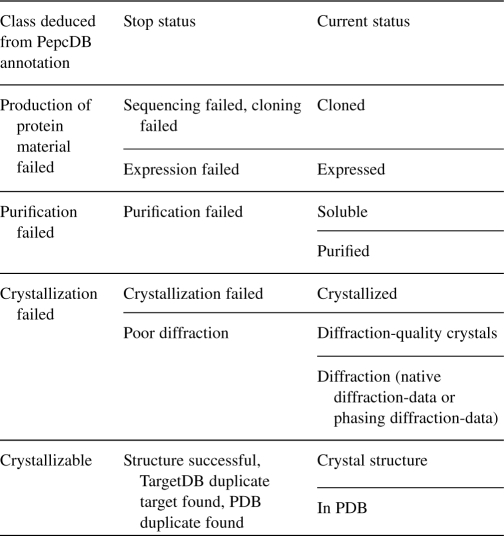 |
The statuses are sorted top-down from steps earlier to further in the crystallization procedure. The current status indicates the current, rather than the completed activity, e.g. for the ‘cloning failed’ stop status, the current status ‘cloned’ does not mean that cloning was successful, but if the current status is ‘expressed’ then cloning can be assumed successful. We disregarded ‘other’, ‘poor NMR’, ‘mass spec failed’ and ‘duplicate target found’ stop statuses and ‘other’, ‘test target’, ‘work stopped’, ‘selected’, ‘mass spec verified’, ‘NMR assigned’, ‘HSQC’, ‘NMR structure’ current statuses.
For the set of the non-crystallizable proteins (NCDB), we considered 45 924 trials with the completed stop status field, which includes any of the following: ‘sequencing failed’, ‘cloning failed’, ‘expression failed’, ‘purification failed’, ‘crystallization failed’ or ‘poor diffraction’.
The set of the crystallizable protein (CDB) was developed using 15 412 trials with the stop status equal to ‘structure successful’, ‘TargetDB duplicate target found’ or ‘PDB duplicate found’ and the trials with the current status ‘crystal structure’ or ‘in PDB’.
We did not use trials corresponding to the NMR structures and we disregarded the trials with the ‘other’ or ‘duplicate target found’ stop status.
We filtered both sets to remove the trials with duplicate sequences, i.e. so far we collected all trials with the complete stop (or current) status irrespective of the sequence. Given two trials with the same sequence, we removed the trial with an earlier stop status (Table 1), e.g. given two trials with the same sequence marked with ‘sequencing failed’ and ‘purification failed’ stop statuses, we removed the former one since another attempt has succeeded with the sequencing step for the same chain. In case of two trials with the same stop status, we removed the older trial.
Next, we filtered all chains in the NCDB set against the CDB set and the chains in the PDB, i.e. we remove a given chain from the NCDB set in case if this sequence occurs in the CDB set or in the PDB.
In the next step, we filtered the chains in the NCDB set against all trials in the PepcDB based on their current status field. We removed each non-crystallizable trial for which there is a trial with the same sequence and the current status further along the crystallization process (Table 1). In this case, the current status indicates that the trial succeeded with the step (stop status) which was used to enter it into the NCDB set.
Next, we removed all trials from before January 1, 2006 and after December 31, 2009. We removed the older samples to accommodate for the latest advances in the crystallization protocols. For example, our analysis of the PepcDB shows that before 2006, i.e. in the first PSI phase, a large number of failures corresponded to problems with cloning, whereas after 2005 the problems with cloning subsided. The samples from 2010 could not be used since some of them may not be yet completed or updated in the database.
We assigned the following classes to the remaining trials: (i) production of the protein material failed (MF) (for all trials with stop status ‘sequencing failed’, ‘cloning failed’ and ‘expression failed’); (ii) purification failed (PF) (for the ‘purification failed’ stop status); (iii) crystallization failed (CF) (for the ‘crystallization failed’ and ‘poor diffraction’); and (iv) CRYStallizable (CRYS) (for the stop statuses ‘structure successful’, ‘TargetDB duplicate target found’ and ‘PDB duplicate found’; or the current statuses ‘crystal structure’ and ‘in PDB’).
Finally, using BLASTCLUST we reduced the sequence identity among chains within the same class, i.e. for each class we kept only the sequences below 25% sequence identity threshold. This is consistent with the threshold used in the prior studies (Overton and Barton, 2006), but we did not reduce the sequence identity between trials from different classes.
We created three datasets to build predictors for each of the non-crystallizable classes (MF, PF and CF). Each of these datasets includes trials which failed to proceed through a given step (as the samples in the negative set), and trails which passed this step (as the positive set). In the dataset for the prediction of the production of the protein material (DB_MF), the negative set contains all trials labeled as MF and the positive set contains the remaining proteins; for the purification dataset (DB_PF), the negative set contains trials marked as PF and the positive set includes trails from the CF and CRYS classes; for the crystallization dataset (DB_CF), the negative set contains trials with the CF class, and the positive set includes trials from the CRYS class. For the DB_PF and DB_CF datasets, we did not include trials from the MF, and the MF and PF classes, respectively, due to the fact that we do not known whether these trials would pass the purification or crystallization steps since they did not pass the previous steps, e.g. we do not know whether the MF trials would purify if they pass the production of the protein material step. We also created the DB_CRYS dataset with the class labels similar with the previous predictors, which indicate the success of the entire process, i.e. production of the diffraction-quality crystals. The DB_CRYS dataset includes two labels, non-crystallizable chains (MF, PF and CF) versus crystallizable chains (CRYS). The chains in the DB_CRYS dataset were annotated with the four class labels, i.e. the non-crystallizable chains were annotated as MF, PF or CF, to create the DB_4CL dataset. The number of trials in each dataset is shown in Supplementary Table 1.
We randomly divided the MF, PF, CF and CRYS sets into two equal sized subsets, the training and the test sets. We used the training subsets to create the corresponding training DB_MF, DB_PF, DB_CF and DB_CRYS datasets, and the tests subsets to create the independent test datasets. We designed our predictor based on the training datasets (using 5-fold cross-validation protocol) and then we performed evaluation and comparison with the existing methods on the independent test datasets. We note that the sequence identity between chains from the same class in the training and test sets is <25%.
2.3 Architecture of the proposed predictor
The prediction is performed in two steps: (i) the input sequences are converted into a set of numerical features that describe certain, relevant characteristics of the protein chain; and (ii) the feature values are fed into four predictive models that output the predicted propensity for material production, purification, crystallization and diffraction quality crystallization, respectively; we use a support vector machine (SVM), which was previously shown to provide high-quality predictions in this area (Kandaswamy et al., 2010; Smialowski et al. 2006), to implement these four models and their outputs are aggregated together to provide a four-class prediction. The architecture of the PPCpred method is shown in Figure 1.
Fig. 1.

The overall architecture of the proposed PPCpred method.
2.3.1 Quality measures and evaluation protocols
The assessment of the predictions uses the same criteria that were used to evaluate prior work in this area (Chen et al., 2007; Kandaswamy et al., 2010; Kurgan et al., 2009; Mizianty and Kurgan, 2009; Overton and Barton, 2006; Overton et al., 2008; Slabinski et al., 2007b; Smialowski et al., 2006). The evaluation was performed per-protein at two levels: (i) the binary values that define whether a given trial/sequence is positive (passes a given step) or negative (fails to pass the step) are evaluated using the Matthews correlation coefficient (MCC), accuracy (ACC), Sensitivity and Specificity measures; and (ii) the real values that quantify the probability of prediction which corresponds to the propensity of the chain to pass the test, i.e. to produce the protein material, to purify, and to produce crystals, are evaluated with the receiver operating characteristic (ROC) and area under the ROC curve (AUC) measures. Detailed explanation of the above-mentioned quality measures is given in the Supplementary Material. The aggregated predictions of four classes are assessed based on the overall accuracy (number of correctly predicted trials over all trials) and mean MCC (over the four MCC values computed for each class label).
We analyze statistical significance of the differences between PPCpred and the other methods. For each test set, we compared 100 paired results for MCC and ACC obtained using the bootstrapping with 25% of randomly selected trials. Since the measurements follow normal distribution, as tested using Shapiro–Wilk test at the 0.05 significance, we use paired t-test and we measure significance of the differences at the 0.01 level.
We designed the proposed predictor, which includes filtration of the considered features, parameterization of the SVM-based classifiers, feature selection and selection of the thresholds for the aggregation of the four predictors (which are described below), using 5-fold cross-validation on the training datasets. In this test, we randomly divide the training dataset into five equal-sized subsets of protein chains. We use four of these subsets to form a training fold that is utilized to compute the model and the fifth subset constitutes the testing fold that is used to perform the evaluation. This is repeated five times, each time choosing a different subset to be the test fold. The tests on the independent test datasets was performed using the model trained on the corresponding training dataset using the parameters and feature sets that were established based on the cross-validation.
2.3.2 Features
Our method considers a comprehensive set of features generated using several information sources including the sequence and the sequence-derived isoelectric point, the encoding of amino acids in the sequence with several property-based indices (e.g. hydrophobicity and energy) from the AAIndex database (Kawashima et al., 2008), solvent accessibility predicted using Real-SPINE3 (Faraggi et al., 2009), disorder predicted using DISOPRED2 (Ward et al., 2004), and secondary structure predicted with PSIPRED 3.2 (Jones, 1999). The importance of the information derived directly from the protein chain, including the composition of certain amino acids, the isoelectric point, etc., for prediction of the crystallization success was demonstrated in numerous studies (Chandonia et al., 2006; Chen et al., 2007; Goh et al., 2004; Kandaswamy et al., 2010; Kantardjieff and Rupp, 2004; Kantardjieff et al., 2004; Kurgan et al., 2009; Overton and Barton, 2006; Overton et al., 2008; Price et al., 2009; Slabinski et al., 2007b; Smialowski et al., 2006). The usage of the energy and hydrophobicity of the constituent residues is motivated by the work in (Babnigg and Joachimiak, 2010; Chen et al., 2007; Goh et al., 2004; Kandaswamy et al., 2010; Kurgan et al., 2009; Overton and Barton, 2006; Overton et al., 2008; Price et al., 2009). The predicted secondary structure, disorder and solvent accessibility were found to be useful to predict propensity of the crystallization in (Chandonia et al., 2006; Kandaswamy et al., 2010; Mizianty and Kurgan, 2009; Price et al., 2009; Slabinski et al., 2007a, b). We note that we also use the above information to predict the propensity of the material production and purification, which is one of the novel aspects of this study. We considered 64 hydrophobicity- and energy-based indices from the AAIndex1 database, see Supplementary Table 2, and the side-chain entropy (Creamer, 2000) that was found useful in (Price et al., 2009); we disregarded amino acid indices related to the solvent accessibility and secondary structure, as we already include these predictions.
We combine information based on the amino acids indices, predicted secondary structure and disorder with the predicted solvent accessibility by computing the values separately for the exposed and buried residues; we define buried residues as the residues for which the predicted relative solvent accessibility is <25%; otherwise a given residue is assumed to be solvent exposed. In total, we generated 828 features, which include:
AA_{AAi} Composition of the 20 standard amino acid (AA) types, i.e. the count divided by the sequence length, where AAi stands for one of 20 AAs (20 features).
AA_{exp,bur}_{AAi} Composition of the exposed/buried AAs (count of the exposed/buried AAi divided by the number of all exposed/buried residues in a given chain) (40 features).
pI The isoelectric point (1 feature).
{AAIndex} The average value of a given amino acids index AAIndex over the whole sequence (65 features).
{AAIndex}_{min,max}_{5,10,15,20} The minimal/maximal average value of the amino acid index AAIndex among all sliding windows of sizes 5, 10, 15 and 20. For chains shorter than a given window size, we use the window size equal the length of the sequence. These features are motivated by the work in (Babnigg and Joachimiak, 2010) (65×4×2=520 features).
{AAIndex}_{exp,bur} The summed value of the amino acid index AAIndex for exposed/buried residues, divided by the number of exposed/buried residues in the sequence These features are motivated by the work in (Price et al., 2009) (65×2=130 features).
DIS_AVG_VAL The average value of the predicted disorder probabilities (1 feature).
DIS_SEG Number of the predicted disorder segments (1 feature).
DIS_RES_seg{1,5,10,15,20} Number of the predicted disorder residues in the disorder segments which are at least 1, 5, 10, 15 and 20 residues long, divided by the sequence length. For segments with at least one residue, this feature represents content of the predicted disorder. (5 features).
DIS_avg The average length of the predicted disorder segments divided by the sequence length (1 feature).
DIS_max The maximal length of the predicted disorder segment divided by the sequence length (1 feature).
DIS_{exp,bur} Number of the predicted exposed/buried disordered residues divided by the number of exposed/buried residues (2 features).
DIS_{exp,bur}_AVG_VAL The summed value of the predicted disorder probability for the predicted exposed/buried residues divided by the number of predicted exposed/buried residues (2 features).
SS_{SSi}_RES_seg{1,5,10,15,20} Number of residues in the predicted coil/helix/strand segments, SSi∈{C,H,E}, which are at least 1, 5, 10, 15 and 20 residues long, divided by the sequence length. For segments with at least one residue, these feature represents content of the predicted coils, helices and strands (15 features).
SS_{SSi}_avg The average length of the predicted SSi segments divided by the sequence length (3 features).
SS_{SSi}_max The maximal length of the predicted SSi segments divided by the sequence length (3 features).
SS_{SSi_AVG_VAL The average predicted probability be in the secondary structure state SSi (3 features).
SS_{exp,burr}_{SSi} Number of the predicted exposed/buried residues in the secondary structure state SSi divided by the number of exposed/buried residues (6 features).
RSA_AVG_VAL The average value of predicted relative solvent accessibility (1 feature).
{EXP,BUR}_RES_seg{1,5,10,15,20} Number of the predicted exposed/buried residues in the exposed/buried segments which are at least 1, 5, 10, 15 and 20 residues long divided by the sequence length. For segments with at least 1 residue, these features represent content of the exposed/buried residues. We note that there were no predicted exposed segments with over 15 residues, and thus the corresponding two features were removed (8 features).
2.3.3 Filtration of the considered features
We note that some of the considered features may be correlated with each other and may not be useful to differentiate between the considered class labels (i.e. the annotation of the protein production, purification, crystallization and diffraction-quality crystallization, respectively). We performed filtration to remove the highly correlated and low-quality features. The training dataset was divided at random into five training and test fold (as in the 5-fold cross-validation) and we ranked the features according to their average, over the five training folds, biserial correlation with the class labels. We selected the feature with the highest average biserial correlation, and we added the next ranked feature into the set of the selected features only if the Pearson correlation coefficient of this feature with every feature in the selected feature set was <0.7. This step removed the highly correlated features. Next, we computed the average value of the average (over the 5-folds) absolute biserial correlations (with the class labels) for the selected features, and we removed the features with the correlations below the average. The latter step removes the low-quality features. At the end, we selected 86, 100, 115 and 95 features for the DB_MF (material production), DB_PF (purification), DB_CF (crystallization) and DB_CRYS (diffraction-quality crystallization) training datasets, respectively.
2.3.4 Parameterization and feature selection
For each dataset we built three SVM models, implemented with the LibSVM package (Chang and Lin, 2001), that are based on the three available kernels including the radial basis function (RBF), polynomial (POLY), which also includes the linear kernel, and sigmoid (SIG). We computed total of 12 models (three different kernels for four datasets: DB_MF, DB_PF, DB_CF and DB_CRYS). Each model was built using the same procedure. First, the model for a given dataset was parameterized using 10 features with the highest average biserial correlation from the features selected in Section 2.3.3. Using 5-fold cross-validation on the training set, we performed grid search to find parameters that maximize the MCC. For each parameter, except the degree of the polynomial, we considered consecutive powers of 2. In case when the selected parameter values were at the border of the search grid, we extended the search to the next consecutive power of 2. The selected parameters were used to perform the wrapper-based feature selection (Hall and Smith, 1999) using the best first search strategy. Supplementary Material provides detailed description of the considered parameters and feature search procedure. After the feature selection is completed, we parameterized the SVMs using the selected feature set and the same grid search as above. Finally, we select the SVM that provides the highest MCC (among the three kernel types) for each dataset, see Supplementary Table 3, i.e. we use the POLY kernels for the prediction of diffraction-quality crystallization, purification and crystallization, and RBF kernel for the prediction of the material production.
2.3.5 The 4-class prediction
We aggregate outputs from the best four SVM models developed in Section 2.3.4 to perform the 4-class prediction. We investigated two approaches to aggregate predictions: (i) by selecting the class with the maximal predicted probability (max-based); and (ii) by choosing the class based on the order of the steps in the crystallization protocol (order-based). In the latter case, we select the class by checking the outputs of the SVMs in the order defined in Table 1, from MF, to PF, to CF and to CRYS. We predict the corresponding outcome if the predicted probability is above a cut-off threshold, e.g. we predict that the material production fails if the output from the corresponding SVM, which quantifies the probability of the material production failure, is greater that the threshold. If none of the probabilities are above the threshold then we select the outcome with the highest probability. We tried the cut-off values from between 0.01 and 1 with step 0.01 and selected the value = 0.43 that maximizes the mean MCC (over the four classes) on the DB_4CL training dataset. The max-based aggregation obtains the mean MCC and accuracy equal 0.293 and 49.0%, respectively, while the order-based method obtains 0.345 and 54.8%, respectively. Supplementary Table 3 also shows that the order-based aggregation also performs better, when compared with the max-based aggregation, for the prediction of the individual outcomes for the material production, purification and crystallization. Consequently, we use the order-based aggregation to implement the proposed PPCpred method.
2.3.6 BLAST-based predictor
The existing predictors address only the prediction of the diffraction-quality crystallization; they do not predict the other three classes considered in this work. Therefore, we consider a baseline score implemented using sequence alignment to comparatively evaluate the predictive quality of our method. Each test trial/chain was aligned against the sequences in the corresponding training dataset using PSI-BLAST (Altschul et al., 1997) and we use the class label of the most similar chain as the prediction. In the case when no alignments are found, the test chain is predicted with the label of the most populated, i.e. more probable, class. This is repeated for each of the five training and test dataset pairs, DB_MF, DB_PF, DB_CF, DB_CRYS and DB_4CL.
3 RESULTS
The proposed PPCpred method, which utilizes the order-based aggregation of the propensities predicted by the four SVM models, was evaluated on the test datasets for the predictions of each of the four outcomes, i.e. prediction of material production, purification, crystallization and diffraction-quality crystallization, as well as for the 4-class prediction. We compare the results generated by the PPCpred with the existing methods for the prediction of the crystallization propensity, with the BLAST-based solution, and with the maximum-based aggregation scheme.
3.1 Comparison of the diffraction-quality crystallization propensity predictions
The PPCpred is compared with the recent predictors of the crystallization propensity including OBScore (Overton and Barton, 2006), XtalPred (Slabinski et al., 2007b), ParCrys (Overton et al., 2008), CRYSTALP2 (Kurgan et al., 2009), MetaPPCP (Mizianty and Kurgan, 2009) and SVMCrys (Kandaswamy et al., 2010), with the BLAST-based method, and with our SVM predictor of the diffraction-quality crystallization (SVM_POLY), see the results on the DB_CRYS dataset in Table 2. The PPCpred outperforms the existing solutions in both the binary prediction (based on the MCC and ACC scores) and the real-valued propensities (based on the AUC values). The best, existing predictor is XtalPred, which is likely due to the usage of the sequence alignment against the PDB and nr databases, followed by SMVCrys and MetaPPCP. The PPCpred improves over the SVM_POLY method, which demonstrates that aggregation of the results from the four SVMs is helpful. Also, the maximum-based aggregation is shown to be inferior to the order-based aggregation used in the PPCpred for the binary predictions, but the magnitude of this difference is relatively small. Table 2 shows that the improvements in MCC and ACC offered by PPCpred are statistically significant. The binary predictions from PPCpred are characterized by high specificity (high success rate among the native non-crystallizable proteins) at about 85%. This means that we relatively rarely mispredict these chains to be crystallizable, which would save resources to solve other chains.
Table 2.
Summary of results for the prediction of the propensity of the diffraction-quality crystallization success (based on the DB_CRYS test dataset), the prediction of the propensity of the material production failure (DB_MF test set), the prediction of the propensity of the purification failure (DB_PF test set) and the prediction of the propensity of the crystallization failure (DB_CF test set)
| Test dataset (prediction target) | Method | MCC |
ACC |
SPEC | SENS | AUC | ||
|---|---|---|---|---|---|---|---|---|
| value | sig | value | sig | |||||
| DB_CRYS (propensity of the diffraction-quality crystallization success) | ParCrys | 0.108 | + | 47.5 | + | 31.8 | 78.6 | 0.561 |
| OBScore | 0.124 | + | 47.8 | + | 31.4 | 80.3 | 0.572 | |
| BLAST-based | 0.188 | + | 65.6 | + | 79.5 | 38.0 | N/A | |
| CRYSTALP2 | 0.195 | + | 55.3 | + | 45.7 | 74.4 | 0.648 | |
| MetaPPCP | 0.195 | + | 59.9 | + | 59.0 | 61.7 | 0.620 | |
| SVMCrys | 0.213 | + | 56.3 | + | 46.7 | 75.2 | N/A | |
| XtalPred | 0.278 | + | 63.9 | + | 62.3 | 67.0 | 0.683 | |
| SVM_POLY | 0.398 | + | 74.6 | + | 88.1 | 47.9 | 0.779 | |
| max-based | 0.467 | + | 76.1 | + | 81.6 | 65.3 | 0.793 | |
| PPCpred | 0.471 | 76.8 | 84.8 | 61.2 | 0.789 | |||
| DB_MF (propensity of the material production failure) | BLAST-based | 0.014 | + | 55.4 | + | 35.3 | 66.0 | N/A |
| max-based | 0.339 | + | 71.6 | + | 45.4 | 85.5 | 0.621 | |
| SVM_RBF | 0.423 | + | 74.6 | + | 56.1 | 84.5 | 0.791 | |
| PPCpred | 0.462 | 75.0 | 69.2 | 78.0 | 0.755 | |||
| DB_PF (propensity of the purification failure) | BLAST-based | 0.102 | + | 60.0 | + | 43.2 | 67.4 | N/A |
| max-based | 0.246 | + | 70.8 | + | 34.4 | 86.9 | 0.609 | |
| SVM_POLY | 0.290 | + | 73.2 | – | 30.8 | 91.8 | 0.741 | |
| PPCpred | 0.324 | 72.0 | 50.1 | 81.6 | 0.697 | |||
| DB_CF (propensity of the crystallization failure) | BLAST-based | 0.060 | + | 60.9 | + | 37.0 | 69.4 | N/A |
| SVM_POLY | 0.346 | + | 77.0 | = | 40.1 | 90.0 | 0.814 | |
| PPCpred | 0.457 | 76.6 | 70.8 | 78.7 | 0.811 | |||
| max-based | 0.461 | – | 76.9 | – | 70.5 | 79.2 | 0.813 | |
The proposed PPCpred is compared against results on the OBScore, XtalPred, ParCrys, CRYSTALP2, MetaPPCP and SVMCrys on the DB_CRYS dataset, and against the maximum-based aggregation method (max-based), the best performing SVM classifier (SVM_POLY or SVM_RBF), and the BLAST-based predictor on the four datasets. The methods are sorted in the ascending order based on their MCC scores, and the highest values for each quality index and dataset are shown in bold. The BLAST and SVMCrys provide only binary prediction and thus we could not compute their AUC. Results of tests of significance of the differences in MCC and ACC between PPCpred and the other methods are given in the ‘sig’ columns. The tests compare values over 100 bootstrapping repetitions. The ‘+’ and ‘−’ mean that PPCpred is statistically significantly better/worse with P<0.01, and ‘=’ means that results are not significantly different.
The ROC curves of the considered predictors, except for the BLAST and SVMCrys that provide only the binary predictions, are shown in Supplementary Figure 1. PPCpred outperforms the other solutions for true positive (TP) rates >0.85 and false positive (FP) rates >0.38, while the maximum-based aggregation works better for smaller TP and FP rates. This demonstrates that PPCpred is particularly useful when the user requires high TP rates, i.e. the number of false negatives (crystallizable chains predicted are non-crystallizable) is low. In this case, PPCpred would relatively rarely mispredict chains that can be successfully solved, which would protect against abandoning solvable targets. The high TP rate comes as a trade-off for the higher FP rate (higher rate of predicting the non-crystalizable chains as crystallizable), which means that PPCpred would more often mistakenly advise to crystallize a difficult target, which consequently would waste resources.
We note that although the existing predictors achieve positive MCC values, they are generally lower than the values reported in the original publications on their test datasets. A possible explanation for that is that our annotation is somehow different and that the existing models were trained on relatively old, from before 2006, trials. To test the latter hypothesis, we sorted the trials based on their date of the last activity to investigate whether the predictive quality varies with this timestamp, see Figure 2. The values of both MCC and AUC are lower for the more recent trials for majority of the methods, except for the PPCpred, XtalPred and the BLAST-based predictor. This confirms that the likely reason for the overall relatively low performance of the ParCrys, OBScore, CRYSTALP2, MetaPPCP and SVMCrys is the fact that they utilize older training data. We note that XtalPred was updated in mid-2007 and it uses sequence alignment against recent contents of the PDB and nr databases, which helps to keep its predictions more up-to-date. This finding suggests that the advances in the crystallization protocols may render older predictors absolute, which motivates development of new up-to-date methods.
3.2 Comparison of the prediction of the propensity of material production, purification, crystallization and diffraction-quality crystallization
The results for each individual target outcome of the PPCpred, BLAST-based predictors, our four SVM-based predictors of the material production, purification, crystallization and diffraction-quality crystallization (SVM_POLY and SVM_RBF), and the maximum-based method for combining the four SVMs predictors are summarized in Table 2. Using the MCC measure, PPCpred significantly outperforms the other methods for the binary prediction of the material production, purification and diffraction-quality crystallization, and it provides comparable predictive quality with the maximum-based aggregator for the prediction of the crystallization, i.e. the maximum-based aggregator provides an improvement with small magnitude that is statistically significant. PPCpred also provides well-balanced values of the sensitivity and specificity. Our method provides reasonably high values of MCC, between 0.32 and 0.47, which indicate that it provides useful outputs.
The evaluation for the 4-class predictions on the DB_4CL test dataset is shown in Table 3. The output of the predictor indicates whether a given chain will provide high-quality crystal, will fail to crystallize, or whether the purification or material production will fail. The methods are evaluated using the overall accuracy (fraction of the correctly predicted chains) and mean MCC (over the four MCC values computed for each class/outcome). Only the PPCpred, the alignment based predictor, and the maximum-based aggregation method can be compared—the other methods predict only one of the outcomes. The overall accuracy of PPCpred equals 55.6%, which is higher by 5 and 21% than the accuracy of the other two solutions. The improvements are statistically significant. We believe that this level of predictive quality should be acceptable for the potential users given the current crystallization success rates, which are at about 4.6% (Kurgan and Mizianty, 2009).
Table 3.
Results for the 4-class prediction (failure in material production, failure in purification, failure in crystallization and success in the generation of the diffraction-quality crystals) on the DB_4CL test dataset
| Method | Mean MCC |
ACC |
||
|---|---|---|---|---|
| Value | sig | Value | sig | |
| BLAST-based | 0.041 | + | 31.1 | + |
| max-based | 0.294 | + | 49.0 | + |
| PPCpred | 0.353 | 55.6 | ||
The proposed PPCpred is compared against the maximum-based aggregation method (max-based), and the BLAST-based predictor. The methods are sorted in the ascending order based on their MCC scores, and the highest values for each quality index and dataset are shown in bold. Results of tests of significance of the differences in mean MCC and ACC between PPCpred and the other methods are given in the ‘sig’ columns. The tests compare values over 100 bootstrapping repetitions. The ‘+’ and ‘−’ mean that PPCpred is statistically significantly better/worse with P<0.01, and ‘=’ means that results are not significantly different.
3.3 Factors related to prediction of crystallization, purification and material production propensity
Table 4 summarizes the features that were used in PPCpred. These features utilize all considered information sources, including the energy and hydrophobicity-based indices, composition of certain amino acid types, the predicted disorder, secondary structure and solvent accessibility, and content of certain buried and exposed residues; Supplementary Table 4 lists the selected features. This shows that the success/failure in the considered steps of the crystallization process depends on a combination of multiple factors. We observe the strong presence of information derived from the hydrophobicity indices, which agrees with the observations in Goh et al. (2004), Overton and Barton (2006), Chen et al. (2007), Overton et al. (2008), Kurgan et al. (2009) and Babnigg and Joachimiak (2010). Importantly, our features demonstrate the importance of the influence of the hydrophobic or hydrophilic segments in the protein chain on the success/failure on all considered steps in the crystallization protocol, i.e. several selected features for prediction of each of the four considered steps are based on the minimal or maximal hydrophobicity in a sliding window. Our features also suggest the importance of Cys residues for the prediction of the material production and diffraction-quality crystallization, and buried Cys for the prediction of purification. This agrees with the observations in (Overton et al., 2008; Slabinski et al., 2007b), but these studies investigated only the propensity for the diffraction-quality crystallization and did not consider the influence of the solvent accessibility. Another factor related to the crystallization success is the content of the buried His. This agrees with results in Overton et al. (2008) and Kurgan et al. (2009), but these studies again considered the overall content of His, without the influence of the solvent accessibility.
Table 4.
Summary of the features types selected for the prediction of the material production, purification and crystallization
| Features types | Number of features selected for the prediction of |
|||
|---|---|---|---|---|
| Material production | Purification | Crystallization | Diffraction-quality crystallization | |
| Hydrophobicity index | 2 | 2 | 5 | 5 |
| Energy-based index | 4 | 0 | 2 | 3 |
| Composition of AAs | 1 | 3 | 1 | 1 |
| Isoelectric point | 0 | 1 | 0 | 0 |
| Solvent accessibility | 3 | 4 | 1 | 3 |
| Disorder | 1 | 0 | 1 | 1 |
| Secondary structure | 0 | 0 | 0 | 1 |
| Considered AA types | Arg, Cys, Glu | Asn, Cys, Ser, Met | His | Cys, His, Ser |
Figure 3 shows scatter plots of three pairs of features that were selected for the prediction of the crystallization, diffraction-quality crystallization and purification, respectively. The two features used to predict crystallization, GOLD730101_min_10 and WERD780103_min_5 (Fig. 3A), and based on the minimal average values of the hydrophobicity (Goldsack and Chalifoux, 1973) and energy (specifically the energy of transfer in water of an isolated residue from a non-regular structure to the helical conformation) (Wertz and Scheraga, 1978) indices in the sliding windows of sizes 10 and 5, respectively. This means that the sequence segments with low hydrophobicity and transfer energy values are characteristic to chains that are difficult to crystallize. Importantly, combining these two features allows for improved separation between the successful and unsuccessful crystallization trials, i.e. trials for a given range of values of one index are further separated by the values of the other index. The diffraction-quality crystallization is impacted by the DIS_SEG and AA_bur_S features (Fig. 3B), which quantify the number of the predicted disorder segments and the content of the predicted buried Ser, respectively. The content of Ser was shown to be important for the prediction of crystallization propensity in Overton et al. (2008) and Kurgan et al. (2009), but these studies investigated the overall Ser content, while we show that the (predicted) buried Ser provides strong discriminatory power. Similarly, while the content of the predicted disordered residues was used in several related studies (Slabinski et al., 2007b; Price et al., 2009), our analysis reveals the strong influence of the number of disordered segments. The plot shows that chains with larger number of disordered segments and larger number of buried Ser are more difficult to crystallize. Finally, Figure 3C shows that chains with larger amount of buried Ser (AA_bur_S feature) and high hydrophobicity in a long-sliding window (GOLD730101_max_20 feature) are more challenging to purify.
Fig. 3.

Scatter plots of three pairs of features used by the PPCpred: features used for the prediction of crystallization (A); for the diffraction-quality crystallization (B) and for the purification (C). Size of the markers denotes the number of trials and color denotes their membership, green for the successful and black for the failed trials.
Overall, the factors that we identified are intuitive, physically reasonable and they are well aligned with the existing ‘rules of thumb’. Our main contributions are in providing additional details (e.g. related to solvent accessibility of selected residues types) and the fact that our model provides a novel way of balancing these factors to obtain good predictive performance.
4 CONCLUSIONS
We developed a first-of-its-kind in silico method, PPCpred, which predicts the success/failure for four main steps in the protein crystallization protocols, including the material production, purification, crystallization and diffraction-quality crystallization. PPCpred significantly outperforms the alignment-based predictor as well as the several modern crystallization propensity predictors. Our method provides the overall accuracy at 56% and average MCC at 0.35, which given current low success rates in the experimental protocols should provide useful input for the SG centers and crystallographers/biologists who are interested in participation in the PSI:Biology phase. We also developed an improved protocol to annotate progress of protein chains along the crystallization process using the PepcDB, and we shows/confirm several interesting markers (based on the features included in our predictors) that influence the success/failure of the above-mentioned steps.
The predictions generated by PPCpred could be used to guide crystallographers to select more feasible alternative targets or, in case when the target is already selected, to rank different constructs of the same target. The former application is evaluated in our work, while the evaluation of the latter one will be performed in a feature study when large enough amount of suitable data becomes available. Users of PPCpred could also find out which of the crystallization steps is the most likely obstacle in the crystallization process, and try to modify the target to increase chances to pass that step, e.g. the user may introduce tags at sequence termini to ease purification when the purification failure is predicted. The success of crystallization also depends on the crystallization protocols. Our method was designed using data from several SG centers, which allows us to generalize over multiple protocols. At the same time, our model takes into account only the intra-molecular factors that are encoded in the protein chain. Therefore, the PPCpred as well as the other crystallization propensity predictors may not provide reliable predictions when the inter-molecular factors such as the specific characteristics of the expression systems, protein–protein and/or protein–precipitant interactions, buffer composition, precipitant diffusion method, gravity, etc., must be considered.
Supplementary Material
ACKNOWLEDGEMENTS
We gratefully acknowledge the help from Drs Helen Berman and John Westbrook from Rutgers University in handling the PepcDB and TragetDB, design of the annotation protocol and for constructive comments on the draft of this article.
Funding: Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant (to L.K.); Izaak Walton Killam Memorial scholarship (to M.J.M.).
Conflict of Interest: none declared.
REFERENCES
- Altschul S.F., et al. Gapped BLASTand PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babnigg G., Joachimiak A. Predicting protein crystallization propensity from protein sequence. J Struct. Funct. Genet. 2010;11:71–80. doi: 10.1007/s10969-010-9080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biertumpfel C., et al. Practical implementations for improving the throughput in a manual crystallization setup. J Appl. Crystal. 2005;38:568–570. [Google Scholar]
- Brenner S.E. Target selection for structural genomics. Nat. Struct. Biol. 2000;7:967–969. doi: 10.1038/80747. [DOI] [PubMed] [Google Scholar]
- Brenner S.E., et al. The PRESAGE database for structural genomics. Nucleic Acids Res. 1999;27:251–253. doi: 10.1093/nar/27.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandonia J.M., Brenner S.E. Implications of structural genomics target selection strategies: Pfam5000, whole genome, random approaches. Proteins. 2005;58:166–179. doi: 10.1002/prot.20298. [DOI] [PubMed] [Google Scholar]
- Chandonia J.M., Brenner S.E. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- Chandonia J.M., et al. Target selection and deselection at the Berkeley Structural Genomics Center. Proteins. 2006;62:356–370. doi: 10.1002/prot.20674. [DOI] [PubMed] [Google Scholar]
- Chang C.-C., Lin C.-J. LIBSVM: a library for support vector machines. 2001 Software available at http://www.csie.ntu.edu.tw/~cjlin/libsvm(version 3.0, last accessed December 3, 2010) [Google Scholar]
- Chayen N.E. Turning protein crystallisation from an art into a science. Curr. Opin. Struct. Biol. 2004;14:577–583. doi: 10.1016/j.sbi.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Chen K., et al. Prediction of protein crystallization using collocation of amino acid pairs. Biochem. Biophys. Res. Comm. 2007;355:764–769. doi: 10.1016/j.bbrc.2007.02.040. [DOI] [PubMed] [Google Scholar]
- Chen L., et al. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20:2860–2862. doi: 10.1093/bioinformatics/bth300. [DOI] [PubMed] [Google Scholar]
- Creamer T.P. Side-chain conformational entropy in protein unfolded states. Proteins. 2000;40:443–445. doi: 10.1002/1097-0134(20000815)40:3<443::aid-prot100>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Dessailly B.H., et al. PSI-2: structural genomics to cover protein domain family space. Structure. 2009;17:869–881. doi: 10.1016/j.str.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraggi E., et al. Improving the prediction accuracy of residue solvent accessibility and real-value backbone torsion angles of proteins by fast guided-learning through a two-layer neural network. Proteins. 2009;74:857–871. doi: 10.1002/prot.22193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh C.S., et al. Mining structural genomics pipeline: identification of protein properties that affect high-throughput experimental analysis. J Mol. Biol. 2004;336:115–130. doi: 10.1016/j.jmb.2003.11.053. [DOI] [PubMed] [Google Scholar]
- Goldsack D., Chalifoux R. Contribution of free energy of mixing of hydrophobic side chains to the stability of the tertiary structure. J. Theor. Biol. 1973;39:645–651. doi: 10.1016/0022-5193(73)90075-1. [DOI] [PubMed] [Google Scholar]
- Hall M., Smith L. Proc. FLAIRS. Orlando, Florida, USA: AAAI Press; 1999. Feature selection for machine learning: comparing a correlation-based filter approach to the wrapper; pp. 235–239. May 1–5, 1999. [Google Scholar]
- Hui R., Edward A. High-throughput protein crystallization. J. Struct. Biol. 2003;142:154–161. doi: 10.1016/s1047-8477(03)00046-7. [DOI] [PubMed] [Google Scholar]
- Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- Kantardjieff K.A., Rupp B. Protein isoelectric point as a predictor for increased crystallization screening efficiency. Bioinformatics. 2004;20:2162–2168. doi: 10.1093/bioinformatics/bth066. [DOI] [PubMed] [Google Scholar]
- Kantardjieff K.A., et al. Distributions of pI vs pH provide strong prior information for the design of crystallization screening experiments. Bioinformatics. 2004;20:2171–2174. [Google Scholar]
- Kandaswamy K., et al. SVMCRYS: an SVM approach for the prediction of protein crystallization propensity from protein sequence. Prot. Pept. Lett. 2010;17:423–430. doi: 10.2174/092986610790963726. [DOI] [PubMed] [Google Scholar]
- Kawashima S., et al. AAindex: amino acid index database, progress report. Nucleic Acids Res. 2008;36:D202–D205. doi: 10.1093/nar/gkm998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouranov A., et al. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006;4:D302–D305. doi: 10.1093/nar/gkj120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgan L., Mizianty M.J. Sequence-based protein crystallization propensity prediction for structural genomics: review and comparative analysis. Nat. Sci. 2009;1:93–106. [Google Scholar]
- Kurgan L, et al. CRYSTALP2: sequence-based protein crystallization propensity prediction. BMC Struct. Biol. 2009;9:50. doi: 10.1186/1472-6807-9-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherson A. Protein crystallization in the structural genomics era. J. Struct. Funct. Genome. 2004;5:3–12. doi: 10.1023/B:JSFG.0000029199.43875.92. [DOI] [PubMed] [Google Scholar]
- Mizianty M.J., Kurgan L. Meta prediction of protein crystallization propensity. Biochem. Bioph. Res. Comm. 2009;390:10–15. doi: 10.1016/j.bbrc.2009.09.036. [DOI] [PubMed] [Google Scholar]
- Overton I.M., Barton G.J. A normalised scale for structural genomics target ranking: the OB-Score. FEBS Lett. 2006;580:4005–4009. doi: 10.1016/j.febslet.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Overton I.M., et al. ParCrys: a Parzen window density estimation approach to protein crystallization propensity prediction. Bioinformatics. 2008;24:901–907. doi: 10.1093/bioinformatics/btn055. [DOI] [PubMed] [Google Scholar]
- Price W.N., et al. Understanding the physical properties that control protein crystallization by analysis of large-scale experimental data. Nat. Biotechnol. 2009;27:51–57. doi: 10.1038/nbt.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PSI-Nature Structural Biology Knowledgebase (2011) http://sbkb.org/update/2011/01/full/sbkb.2010.62.html(last accessed date January 13, 2011)
- Puesy M., et al. Life in the fast lane for protein crystallization and X-ray crystallography. Progr. Biophys. Mol. Biol. 2005;88:359–386. doi: 10.1016/j.pbiomolbio.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Rodrigues A., Hubbard R.E. Making decisions for structural genomics. Brief. Bioinformatics. 2003;4:150–167. doi: 10.1093/bib/4.2.150. [DOI] [PubMed] [Google Scholar]
- Rupp B., Wang J.W. Predictive models for protein crystallization. Methods. 2004;34:391–408. doi: 10.1016/j.ymeth.2004.03.031. [DOI] [PubMed] [Google Scholar]
- Service R. Structural genomics, round 2. Science. 2005;307:1554–1558. doi: 10.1126/science.307.5715.1554. [DOI] [PubMed] [Google Scholar]
- Slabinski L., et al. The challenge of protein structure determination–lessons from structural genomics. Prot. Sci. 2007a;16:2472–2482. doi: 10.1110/ps.073037907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabinski L., et al. XtalPred: a web server for prediction of protein crystallizability. Bioinformatics. 2007b;23:3403–3405. doi: 10.1093/bioinformatics/btm477. [DOI] [PubMed] [Google Scholar]
- Smialowski P., et al. Will my protein crystallize? A sequence-based predictor. Proteins. 2006;62:343–355. doi: 10.1002/prot.20789. [DOI] [PubMed] [Google Scholar]
- Terwilliger T.C., et al. Class-directed structure determination: Foundation for a protein structure initiative. Prot. Sci. 1998;7:1851–1856. doi: 10.1002/pro.5560070901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.J., et al. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20:2138–2139. doi: 10.1093/bioinformatics/bth195. [DOI] [PubMed] [Google Scholar]
- Wertz D.H., Scheraga H.A. Influence of water on protein structure. An analysis of the preferences of amino acid residues for the inside or outside and for specific conformations in a protein molecule. Macromolecules. 1978;11:9–15. doi: 10.1021/ma60061a002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.