

Abstract
Amyloid-beta (Aβ) deposition is a defining feature of Alzheimer’s disease (AD). The toxicity of Aβ aggregation is thought to contribute to clinical deficits including progressive memory loss and cognitive dysfunction. Therefore, Aβ peptide has become the focus of many therapeutic approaches for the treatment of AD due to its central role in the development of neuropathology of AD. In the past decade, taking the advantage of multiphoton microscopy and molecular probes for amyloid peptide labeling, the dynamic progression of Aβ aggregation in amyloid plaques and cerebral amyloid angiopathy has been monitored in real time in transgenic mouse models of AD. Moreover, amyloid plaque-associated alterations in the brain including dendritic and synaptic abnormalities, changes of neuronal and astrocytic calcium homeostasis, microglial activation and recruitment in the plaque location have been extensively studied. These studies provide remarkable insight to understand the pathogenesis and pathogenicity of amyloid plaques in the context of AD. The ability to longitudinally image plaques and related structures facilitates the evaluation of therapeutic approaches targeting toward the clearance of plaques.
Keywords: Multiphoton imaging, Amyloid, Alzheimer’s disease, Dendritic spine, Calcium homeostasis, Microglial cells
1. Introduction
The definitive diagnosis of Alzheimer’s disease (AD) largely depends on neuropsychological assessments, which indicate the patients’ clinical symptoms, such as impairments in memory and cognition as well as post-mortem examination of brain tissue. Postmortem tissue from afflicted patients exhibits extensive neurofibrillary tangles and Aβ plaques. Therefore, plaque pathology remains of intrinsic diagnostic importance. Previously, identification of Aβ, the major component of senile plaques, in brain tissue was limited to static histological approaches. In the past decade, great efforts have being made to develop new imaging technologies that are sensitive for in vivo detection of amyloid plaques in the intact brain. These techniques have revolutionized our understanding of the structure and function of the living brain and will be extremely valuable for minimally invasive early diagnosis of AD. The imaging techniques developed to identify AD-associated pathological structures in the brain include positron emission tomography (PET) (Nordberg, 2008; Noble and Scarmeas, 2009; Ono, 2009), single photon emission computed tomography (SPECT) (Friedland et al., 1997; Kung et al., 2002; Ono, 2009), magnetic resonance imaging (MRI) (Killiany et al., 2000; Benveniste and Blackband, 2002; Poduslo et al., 2002; Jack et al., 2005, 2007; Higuchi et al., 2005; Benveniste et al., 2007; Dimou et al., 2009), near-infrared fluorescence imaging (NIR) (Hintersteiner et al., 2005; Ran et al., 2009; Raymond et al., 2008), and multiphoton microscopy (Klunk et al., 2002; Bacskai and Hyman, 2002; Bacskai et al., 2003; Skoch et al., 2005, 2006). The comparison of above techniques for in vivo imaging of AD-related pathology has been discussed in previous reviews (Bacskai et al., 2002b; Garcia-Alloza and Bacskai, 2004) and summarized in Table 1.
Table 1.
Comparison of in vivo imaging techniques for amyloid protein.
| Imaging Technique | Spatial Resolution (mm) | Sensitivity to Aβ | Time resolution | Imaging region | Invasive | Agents for imaging |
|---|---|---|---|---|---|---|
| Multiphoton Microscopy | 0.001 | High sensitivity | Fast (Second) | Barrel cortex | Yes | Methoxy-XO4 (Klunk et al., 2002); Thioflavin-S (Bacskai et al., 2001); PIB (Bacskai et al., 2003); fluorescent antibodies 10D5 or 3D6 (Bacskai et al., 2002a) |
| Positron Emission Tomography (PET) | 2–10 | Micromolar | Slow | Whole brain | No | Nuclear medicine probes (Nordberg, 2008; Ono, 2009) including [11C]PIB, [11C]SB-13, [18F]FDDNP, [123I]IMPY, [18F]BF-227, and [18F]BAY94-9172 |
| Single Photon Emission Computed Tomography (SPECT) | 1–10 | Lower than PET (non-quantifiable) | Slow | Whole brain | No | [123I]IMPY(Kung et al., 2002), [99Tc]-10H3 (Friedland et al., 1997) |
| Magnetic Resonance Imaging (MRI) | 0.04–1(Benveniste and Blackband, 2002; Jack et al., 2007) | Millimolar | Slow (2–10 h at mice models) (Jack et al., 2005) | Whole brain | No | Magnetic contrast agents 19F-containing Congo-red based compound (Higuchi et al., 2005), Put–Gd–Aβ (Poduslo et al., 2002) |
| NIR spectroscopy | 1–3 | High sensitivity | Fast | Whole brain | No | CRANAD-2 (Ran et al., 2009); AOI-987 (Hintersteiner et al., 2005; Raymond et al., 2008) |
Among these imaging techniques, multiphoton microscopy has been widely used in pre-clinical studies in animal models of AD, because it is minimally invasive, provides high resolution and can be performed repeatedly in the same animal. Multiphoton imaging is especially suitable for chronic observation of Aβ deposits and surrounding brain structures in the intact brain of living animals. In this review, we will focus on the use of multiphoton microscopy for in vivo imaging of mouse models of AD, and the most recent progress made by using this technique in the field. We will discuss the events correlated with amyloid deposition including the dendritic and spine abnormalities, alterations of neuronal and astrocytic calcium homeostasis, microglial recruitment in transgenic models of AD, and how these studies facilitate the evaluation of efficacy of potential therapeutics.
1.1. Longitudinal imaging of amyloid plaques in intact brain with multiphoton microscopy
Multiphoton microscopy has become a powerful tool for imaging the structure and function of brain cells in living animals. Imaging studies over intervals ranging from seconds to months provide significant insights into the structural plasticity of synapses in the intact brain (Pan and Gan, 2008) and reveal how neuronal connections are altered in animal models of neurodegeneration, acute brain injury, and cerebrovascular disease.
Multiphoton microscopy, a descendent of confocal microscopy, uses the raster scanning of a point source of laser excitation to excite fluorescent molecules. In contrast to confocal microscopy, which uses continuous wave laser excitation, multiphoton microscopy uses pulsed laser excitation to deliver high photon fluxes, which enable the simultaneous absorption of two long wavelength low energy photons. With this strategy many advantages are achieved over confocal microscopy and single photon excitation. Perhaps most important is that only fluorochromes at the focal point receive sufficient energy for two photon excitation. In this manner fluorochromes outside of the focal plane are not excited and therefore are not subject to photobleaching and associated cellular photodamage. Because low energy photons are delivered multiphoton microscopy is associated with less tissue damage, and because near-infrared excitation is delivered it is possible to image relatively deeper in the brain in comparison with single photon excitation. Of course in contrast to PET or MRI imaging, multiphoton microscopy still detects fluorescence relatively superficially within about 800 μm of the cortical surface. However, associated with this imaging is extremely high resolution of ~ 1 μm. Consequently, multiphoton microscopy is able to reliably image dendritic spines, the postsynaptic apparatus of excitatory synapses, dynamic processes such as Ca2+ transients as well as the appearance and growth of amyloid plaques.
Although amyloid deposits can exhibit intrinsic auto-fluorescence, high-resolution imaging is facilitated by the addition of amyloid specific fluorescent labels. Compounds that bind to amyloid deposits with high specificity have been modified to be suitable for in vivo imaging. The properties of such agents include high specificity for amyloid and their ability to cross the blood brain barrier (BBB). Comparison of agents used for in vivo imaging is described in Table 2 (Vassar and Culling, 1959; Elghetany and Saleem, 1988; Duyckaerts et al., 2009). Among these compounds, methoxy-X04, which is a Congo-red derivative, has been widely used for in vivo imaging of amyloid plaques due to its high affinity and specificity for the amyloid protein and ability to cross the BBB (Klunk et al., 2002). The specificity of methoxy-X04 labeling of dense-core plaques has been evaluated using counterstaining of thioflavine-S and immunohistochemistry with an anti-Aβ antibody (Klunk et al., 2002). Post-mortem histochemistry has shown a one-to-one correspondence between fluorescent structures stained by systemically administered methoxy-X04 and structures labeled with thioflavine-S. Immunohisochemistry with an anti-Aβ antibody (BAM10) has also confirmed that methoxy-X04 stained immunoreactive plaques and amyloid angiopathy throughout the brain. The methoxy-X04 fluorescence is detectable for up to 3 days after a single administration (Bacskai et al., 2002b), and can be reapplied for serial imaging studies. Using multiphoton microscopy, methoxy-X04-labeled plaques can be visualized in four dimensions, by volume and over time in transgenic mouse models of AD. One of the strengths of multiphoton microscopy is the ability to repeatedly image the same brain region in the same animal. To facilitate the ability to identify the same three-dimensional voxel (volumetric pixel), one can use a second label which identifies the vasculature thereby providing landmark for reference. To achieve this objective a fluorescent dye conjugated to dextran is injected into the tail vein. Fig. 1 presents a representative image of methoxy-X04-labeled plaques and rhodamine-dextran labeled vasculature.
Table 2.
Agents for multiphoton in vivo imaging of amyloid.
| Probes | Binding site | Cross BBB | Administration | Labeling |
|---|---|---|---|---|
| Thioflavin–S | Pleated beta-sheet protein structure | No | Topically | Dense core and CAA (Vassar and Culling, 1959; Elghetany and Saleem, 1988) |
| Thiazine red | Pleated beta-sheet protein structure | No | Topically | Dense core and CAA (Bacskai et al., 2003) |
| Methoxy-XO4 | Pleated beta-sheet protein structure | Yes | i.p. and i.v. | Dense core and CAA (Klunk et al., 2002) |
| Pittsburg Compound B (PIB) | Pleated beta-sheet protein structure | Yes | i.p. and i.v. | Dense core and CAA (Bacskai et al., 2003) |
| Antibodies | Epitope | No | Topically | Dense core, CAA and diffuse plaques (Elghetany and Saleem, 1988; Duyckaerts et al., 2009) |
Fig. 1.
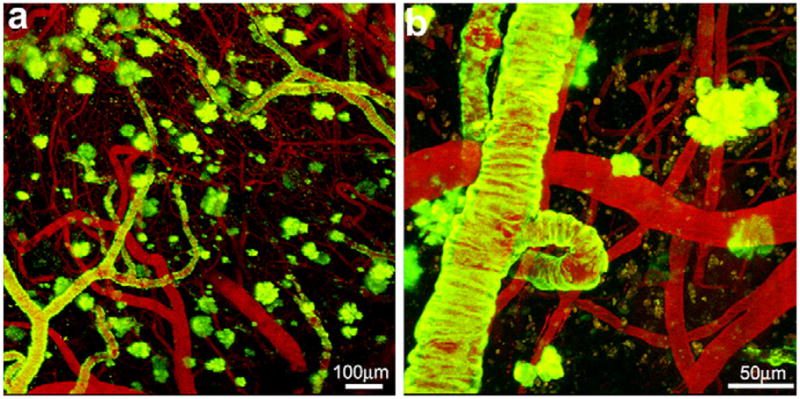
Methoxy-X04 labeled amyloid plaques and cerebral angiopathy in the barrel cortex of a PS1/APP mouse. Representative Z-series maximum intensity projection of methoxy-X04 labeled amyloid plaques and cerebral amyloid angiopathy in low (a) and high (b) magnification images. Amyloid deposits are labeled with methoxy-X04 (green) and cerebral vasculatures are labeled with rhodamine-dextran (red).
Because multiphoton microscopy can be used to simultaneously detect multiple fluorescent probes, it is feasible to track neuronal and glial structures together with plaques. For example, by using viral-mediated green fluorescent protein (GFP) expression in cortical neurons in a transgenic model of AD (Spires et al., 2005) or by crossing a transgenic model of AD with another mouse lines containing fluorescent reporter (Tsai et al., 2004; Meyer-Luehmann et al., 2008; Bolmont et al., 2008), it is possible to image dendrites and spines as well as microglia and plaques. The thy1-YFP trans-genic mouse expresses yellow fluorescent protein (YFP) at high levels in the dendritic and axonal structures in motor and sensory neurons, as well as in subsets of central neurons, for instance, layer 5 pyramidal neurons in the cortex. Crossing thy1-YFP mice with transgenic model of AD enables monitoring morphological alternations of axon and dendrite in the context of AD (Tsai et al., 2004). Transgenic mice with fluorescently labeled microglia have been generated by expressing GFP in microglia under the control of different genetic loci specific to microglia such as Iba-1 and Cx3cr1 (encoding the chemokine-fractalkine receptor). Crossing transgenic Iba1-GFP mouse (Bolmont et al., 2008) or Cx3cr1GFP+/− mouse (Davalos et al., 2005; Koenigsknecht-Talboo et al., 2008) with mouse model of AD enables high-resolution visualization and longitudinal assessment of dynamic interaction of microglia and amyloid plaques.
1.2. Using multiphoton microscopy to characterize kinetics of cerebral amyloid angiopathy (CAA) and senile plaque progression in transgenic mouse model of AD
Extracellular aggregation of Aβ peptide results in widespread individual amyloid plaques and cerebral amyloid angiopathy (CAA). Amyloid plaques have been reported to be toxic to dendrites and disrupt synaptic transmission underlying neuronal dysfunction in the brain of AD (Knowles et al., 1999; Lacor et al., 2007; Meyer-Luehmann et al., 2008). CAA is defined as the aggregation of Aβ peptide in cerebral vessel walls and has deleterious effects causing loss of smooth muscle cells (Mandybur, 1975; Vinters, 1987), disruption of vessels (Greenberg, 2002), and eventually parenchymal hemorrhage (Kalyan-Raman and Kalyan-Raman, 1984; Gilles et al., 1984; Mott and Hulette, 2005). Transgenic mouse models of AD develop both plaques and CAA in a progressive and age-dependent manner Fig. 2. Traditional studies that examine the amyloid deposition have focused on the examination of histological sections from the animals at a single age. This approach has been limited by an inability to monitor the sequential events during the development of amyloid plaques and CAA, therefore little is known about when and how aggregation initiates and the factors that influence the dynamics of their formation and growth in vivo. Multiphoton imaging provides the opportunity to define the subtleties of the initiation and progression of amyloid accumulation in the intact brain in living animals.
Fig. 2.

Age-dependent aggregation of CAA in a vessel segment. Repeated In vivo imaging of a PS1/APP mouse at two weeks intervals from 2 to 6-months of age. Serial imaging of the same volume in barrel cortex exhibited a progressive increase in cerebral amyloid angiopathy (labeled with methoxy-X04, green) in a typical vessel segment which was identified with rhodamine-dextran (red).
A recent study using serial in vivo imaging through a thinned-skull cranial window has characterized the appearance and growth of amyloid plaques in APP/PS1 transgenic mice over intervals ranging from one week to 3 months (Yan et al., 2009). Although repeated identification of the same three-dimensional location in the brain can be difficult, this approach has shown that APP/PS1 strain exhibits faster growth of plaques in 6-month-old compared with 10-month-old mice. The growth rate of existing plaques is size-related; smaller plaques exhibit a more rapid growth relative to larger plaques. In a separate study Meyer-Luehmann et al. (2008) have reported the rapid formation of new amyloid plaques within 24 h. Once a plaque was formed they then observed limited further growth. It is important to note that different procedures were used in these two studies. Because of the limited depth resolution of multiphoton microscopy it is necessary to reduce the pathlength between the microscope objective and the cortical region that is to be imaged. Two approaches are used: either a region of skull is removed and replaced with a glass coverslip window, or the skull is thinned to about 25 μm at which point it acts as a window into the cortex. Clearly there are concerns with each approach: in one the skull is removed but in the other great care has to be taken to prevent heating of the cortex during the process of skull thinning. This issue has been recently addressed by assessing the cellular ultrastructure using electron micrographs and quantifying the expression levels of markers of injury/inflammation between operated and non-operated hemispheres in perfusion-fixed brains (Holtmaat et al., 2009). Although Xu et al. reported that open-skull window preparations led to extensive glial activation, significantly higher turnover of dendritic spines and substantial loss of dendritic spines (Xu et al., 2007), more evidences have shown indistinguishable structural plasticity with the two types of preparation and that upregulation of glial cell protein markers under cranial windows is mild and transient.
Using fixed intact whole brains, the spatial and temporal distribution of CAA in leptomeningeal vessels has been studied in multiple transgenic models including Tg2576, PS1/Tg2576, PDAPP and TgCRND8, suggesting a consistent and stereotyped age-dependent spatial distribution of CAA (Domnitz et al., 2005). With multiphoton imaging via cranial windows, this study has been extended through serial observations over several weeks of individual leptomeningeal arterial segments in living Tg2576 mice (Robbins et al., 2006). The earliest appearance of CAA in leptomeningeal arteries typically begins by 9 month of age and process eventually saturated in mice older than 16 months, in which the CAA burden reached upwards of 75% of vasculature. The initial CAA exhibits band-like amyloid deposits and then it starts propagating primarily from existing deposits, rather than through initiation of new deposits on the vessel, which is a distinct pattern of kinetics from that of accumulation of amyloid plaques. CAA progression in APPswe/PS1dE9 mice has the same propagation model as described in Tg2576 but the overall rate of progression is slower (Garcia-Alloza et al., 2006).
1.3. Using multiphoton microscopy to study the local toxicity of amyloid plaques
1.3.1. Dendritic and spine abnormalities nearby amyloid plaques
Dendritic spines are essential for excitatory synaptic transmission and spine remodeling is thought to contribute to synaptic plasticity and represent the sub-cellular basis for learning and memory (Bhatt et al., 2009). Thus a disturbance of synaptic signaling and loss or alteration of dendritic spines may contribute to significant disruption of neuronal circuits, which eventually results in severe impairment in cognition and memory. In fact, a loss of spines has been described in patients with AD (Scheff and Price, 1993) and post-mortem examination also suggests that the extent of spine loss is strongly correlated with the degree of clinical impairment in the brain of AD (Dekosky and Scheff, 1990; Terry et al., 1991; Selkoe, 2002).
In transgenic animal models of AD, similar dendritic abnormalities including loss of spines, branch disruption, dendritic curvature, and varicosity formation, have also been observed (Grutzendler et al., 2007; Spires et al., 2005). It has been suggested that amyloid deposits and their surrounding microenviroment are toxic to dendrites and lead to the loss and alteration of spines and deficits in synaptic function. Such alteration is thought to be responsible for cognitive deficits in AD pathogenesis before or even in the absence of neuronal loss (Tsai et al., 2004).
Gan and coworkers, using time-lapse in vivo imaging in PSAPP/YFP mice, have revealed extensive formation and elimination of dendritic spines in the vicinity of amyloid deposits over days to weeks, suggesting amyloid deposits induce progressive remodeling of dendritic structures (Tsai et al., 2004). Dendrites passing through or nearby amyloid deposits develop prominent varicosities and eventually exhibit a net loss of spines due to the imbalance rate of spine formation and elimination (Fig. 3). Similar spine dynamics has also been observed in Tg2576 mice, in which neuronal dendrites are visualized with virus derived GFP and exhibit increased elimination but unchanged spine formation after extensive plaque deposition (Spires-Jones et al., 2007). In addition, it has been suggested that distance from the plaque correlates with increasing spine density (Spires et al., 2005). Dendrites within 15 μm of the plaque edge had significantly lower spine density than those farther away and a profound decrement in spine density extends about 20 μm from plaques edges, indicating a local synaptotoxic effect of amyloid plaques (Fig. 4). Dendrites surrounding amyloid deposits also exhibit curved trajectories and a smaller dendritic diameter (Spires-Jones et al., 2007; Tsai et al., 2004).
Fig. 3.
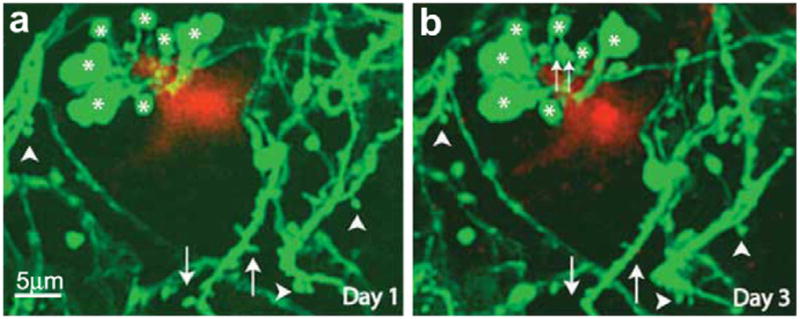
Axon and dendritic abnormalities near amyloid plaques. In vivo time-lapse imaging showed GFP-labeled cortical dendrites and axons near an amyloid plaque in the cortex of a PSAPP/YFP mouse at 6 month of age. Although the majority of spines (arrowheads) and varicosities (asterisks) were stable over 3 days, some structural changes (such as spine loss (arrows) and varicosity formation (double arrows)) did occur. (This figure is reproduced with permission from Tsai et al., 2004).
Fig. 4.
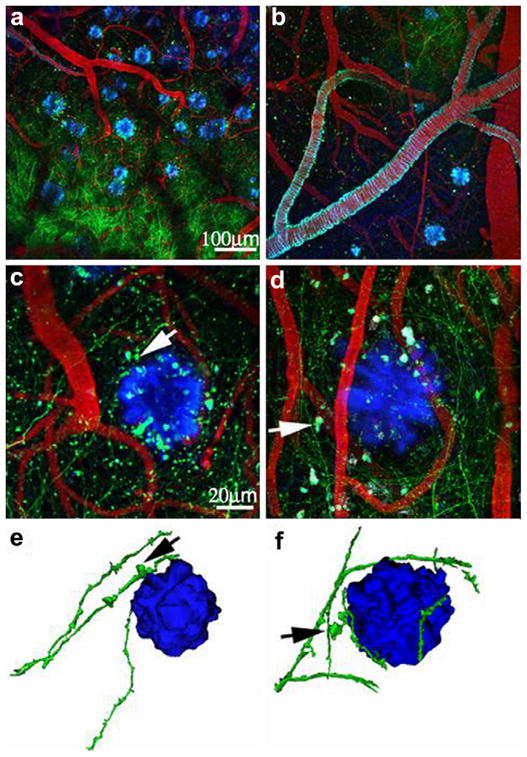
Amyloid plaques alter the morphology and trajectory of neurites in vivo. (a and b) Low magnification images exhibit an overview GFP-AAV injection site containing GFP-filled neurites (green), Texas red-labeled vasculature (red), and methoxy-X04 labeled amyloid deposition (blue). (c and d) In higher magnification images, arrows indicate amyloid-associated dystrophic neurites, showing trajectory curves around plaques. (e and f) Three-dimensional reconstructions of plaques and neurites clearly show this curvature around plaques and highlight dystrophies near plaques (arrows). (This figure is reproduced with permission from Spires et al., 2005).
The above in vivo observations of plaque-related spine loss have also been verified recently using an advanced histological preparation called array tomography, a technique that combines ultrathin sectioning of tissue with immunofluorescence (Koffie et al., 2009). Array tomography circumvents the limit of the z-resolution of multiphoton microscopy and allows the detection of synaptic elements and quantitative assessment of plaques-associated excitatory synapse loss. This study has shown that dense-core plaques are surrounded by a halo of oliogmeric Aβ which interacts with a subset of postsynaptic densities. Moreover, it has been found that the density of postsynaptic densities (PSDs) decreases significantly in the halo of oligomeric abeta surrounding plaques and correlates negatively with the proportion of oliogmeric Aβ-positive PSDs, suggesting that oligomeric Aβ surrounding plaques contributes to synapse loss in a mouse model of AD.
Recently, sequentially in vivo imaging on APPswe/PS1d9YFP transgenic mice provides insights into the temporal relation between plaque formation and the changes in local neuritic morphology and determines the effects of newly formed dense-core plaques on the microarchitecture of the brain (Meyer-Luehmann et al., 2008). This study, which shows that plaques can form extremely rapidly, also showed that dendrites that were morphologically normal the day prior to amyloid plaque formation exhibited rapid alterations on the day of appearance of a new plaque indicating that dendritic deformation is a secondaryeffect of plaque development. This supports the hypothesis that amyloid deposits precede and leads to neuronal dysfunction causing cognitive and memory deficits in AD.
1.3.2. In vivo calcium imaging in mouse models of AD
Calcium signaling is known to be important for multiple neuronal functions including neuronal excitability, synaptic plasticity and cell survival (Berridge et al., 2003; Mattson and Chan, 2003a). Alterations in calcium homeostasis have been reported to be one of the earliest molecular changes that occur in AD patients (Khachaturian, 1994). Animal studies have also shown that Aβ can disrupt neuronal Ca2+ homeostasis (Mattson et al., 1992, 1993) and affect astrocytic calcium signaling (Mattson and Chan, 2003b; Takano et al., 2007), for instance, Aβ-associated excitotoxicity by inducing influx of extracellular Ca2+ into the neuronal cytoplasm (Mattson et al., 1992). This view has been further supported by the observations that Aβ peptides impair membrane ATPase activities leading to Ca2+ destabilization (Mark et al., 1995) and that Aβ-mediated disruption of lipid membranes might increase membrane Ca2+ permeability (Kayed et al., 2004; Sokolov et al., 2006).
Two recent studies using multiphoton imaging to monitor neuronal calcium homeostasis in AD transgenic models have also supported the view that Aβ-induced calcium dysfunction may contribute to neuropathologic features of AD. By combining genetically-encoded calcium indicator and multiphoton imaging, one study measured neuronal calcium in individual neurites and spines in APP mice. This study shows overloaded calcium in neurites in the presence of amyloid plaques and suggests that amyloid plaques impair neuritic calcium homeostasis in vivo, leading to structural and functional disruption of the neuronal network (Kuchibhotla et al., 2008). Another recent study, using in vivo calcium imaging in APP23 × PS45 mice, has shown a redistribution of silent and hyperactive neurons in the cortex of AD brain, in which hyperactive neurons are exclusively near the amyloid plaques and display enhanced frequency of spontaneous calcium transient (Busche et al., 2008). Authors suggested that this hyperactivity is caused by impaired synaptic inhibition, rather than intracellular Ca2+ release from store signaling or intrinsic firing.
Recent studies have provided compelling evidence that astrocytes are vital for brain functions. In AD astrocytes become reactive and surround plaques and might play a role in Aβ deposition and clearance (Wegiel et al., 2001; Nagele et al., 2004). Given the significant impact of Aβ deposits on nearby neuronal calcium homeostasis, it is possible that astrocytic calcium signaling would also be perturbed and might contribute to neuronal dysfunction. In the context of AD, there are indications that astrocytic Ca2+ homeostasis is disturbed by the presence of Aβ peptide. For example, the administration of Aβ peptides to co-culture of hippocampal neurons and astrocytes causes abnormal Ca2+ transients and mitochondrial depolarization in astrocytes before any impairment is visible in neurons (Abramov et al., 2004a, 2004b).
Astrocytic calcium signaling which plays an important role in regulating vascular tone (Takano et al., 2006; Zonta et al., 2003; Mulligan and MacVicar, 2004) is perturbed in mouse models of AD. Multiphoton imaging has demonstrated an increased frequency of spontaneous astrocytic Ca2+ associated with oscillatory changes in arteriole diameter and limited vasodilatation in early stages of AD (Takano et al., 2007), suggesting that abnormal astrocytic calcium signaling may contribute to vascular instability in AD. A recent study in APPswe/PS1E9 mice has shown synchronized astrocytic intracellular calcium waves which initiate from astrocytes near amyloid plaque and spread radially (Kuchibhotla et al., 2009). Sustained astrocytic Ca2+ oscillations following status epilepticus lead to neuronal death (Ding et al., 2007) raising the possibility that synchronous astrocytic calcium signals triggered near amyloid plaques may be deleterious to neuronal function.
1.3.3. In vivo imaging of microglia surrounding amyloid plaque
Microglial activation in the vicinity of the amyloid plaques is an important pathogenic component in AD and may exhibit both neurotoxic and neuroprotective actions including the phagocytosis and clearance of amyloid deposits (D’Andrea et al., 2004). Microglial activation is dynamic and context-dependent. Time-lapse multiphoton imaging provides a minimally invasive tool to study the biology of this important glial cell in vivo. In the brain of normal mouse, resting microglial processes can extend and retract rapidly (Nimmerjahn et al., 2005) and exhibit directed extension to regions of cellular damage and in response to gradients of ATP, which is presumably released from damaged cells (Davalos et al., 2005). Using AD transgenic mice with GFP-labeled microglia enables longitudinal assessment of microglial recruitment to regions of amyloid plaques and investigation of microglia–amyloid interactions in vivo. Longitudinal imaging has shown that amyloid plaques develop surprisingly rapidly and microglia become reactive and are attracted to the sites of plaque formation within 24 h (Meyer-Luehmann et al., 2008).
Similar experiments were conducted by Bolmont et al. using APP/PS1 mice expressing GFP at Iba-1 locus (a calcium binding protein expressed in microglia). In vivo imaging confirmed that microglia responded rapidly to plaque formation by extending processes and migrating toward plaques (Bolmont et al., 2008). The number of microglia associated with each plaque increased at a rate of three per month independent of the size of the plaques. Interestingly, the size of the plaque influenced the volume of each microglial cell, with larger plaques associated with larger microglial cells. Further, the authors observed that the amyloid binding dye often exhibited a punctuate pattern within plaque-associated microglia in vivo that co-localized in post-mortem tissues with Aβ and microglial lysozymes, suggesting an ability of microglial cells to phagocytose Aβ in vivo (Bolmont et al., 2008).
1.4. Facilitate evaluation of the efficacy of therapeutics aimed at removing amyloid plaque
One therapeutic approach that has attracted significant attention is anti-Aβ immunotherapy. Immunization with Aβ (active immunization) or treatment with anti-Aβ antibodies (passive immunization) have been demonstrated to effectively reduce amyloid burden in transgenic AD mouse models (Schenk et al., 1999) and improve leaning and memory (Morgan et al., 2000; Janus et al., 2000). In contrast with active immunization, passive immunization by direct injection of anti-Aβ antibodies bypasses the requirement for an immune response to generate the antibodies (Bard et al., 2000). In the past ten years, the therapeutic effect of direct application of anti-Aβ antibodies to the brain in plaque clearance and other aspects of neuropathology such as neuritic abnormality and spine plasticity have been extensively studied, especially using multiphoton microscopy on transgenic animal models.
The first effective passive immunization in mouse model of AD was reported in 2000, in which peripheral administrations of 3D6 (IgG2bAβ1–15) or 10D5 (IgG1Aβ3–7) antibodies significantly reduce amyloid deposits in PDAPP mice (Bard et al., 2000). Following this study, taking advantage of multiphoton in vivo imaging, a remarkable clearance of thioflavine-S stained amyloid deposits were visualized within 3 days after direct application of antibodies through the craniotomy window (Bacskai et al., 2001). Furthermore, using the same technique it has been reported that topical applications of 10D5 or 3D6 to the brain of aged Tg2576 or PDAPP clear half of the diffuse amyloid deposits and reduce the size of dense-core deposits by 30% within 3 days (Bacskai et al., 2002a). In addition to amyloid plaques, cerebral amyloid angiopathy (CAA), an accumulation of Aβ peptide in the vessel wall of arteries, has also been shown to be effectively cleared with chronic administration of antibodies over two weeks using quantitative in vivo imaging (Prada et al., 2007).
It has been suggested that one of the beneficial actions of antibody administration is that neutralizing synaptotoxic soluble Aβ can prevent Aβ-related inhibition of LTP, which is directly associated with learning and memory (Klyubin et al., 2005). Therefore it is extremely intriguing to know the effect of antibody treatment on the dendritic morphology and plasticity in the AD brain. In vivo imaging of PDAPP/Thy1-YFP mice has shown the morphological recovery of amyloid-associated neuritic dystrophy 3 days after anti-Aβ antibody (10D5) treatment (Brendza et al., 2005). More recently, the acute effects of immunotherapy on neurite morphology have been also studied with in vivo imaging. This study suggests a rapid therapeutic effect on dendritic spine plasticity within 1 h of 3D6 antibody treatment, which is characterized by a small but significant increase in dendritic spine formation distant from plaques (Spires-Jones et al., 2009).
2. Summary
The use of multiphoton in vivo imaging has become an invaluable tool not only to investigate amyloid dynamics in AD but also to study cellular interactions with amyloid aggregation, which may help us understand this disease and facilitate the development of new therapeutic approaches.
Acknowledgments
This work was supported by grant A2010612 to J. Dong from American Health Assistance Foundation.
References
- Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004a;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Canevari L, Duchen MR. Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta. 2004b;1742:81–87. doi: 10.1016/j.bbamcr.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hyman BT. Alzheimer’s disease: what multiphoton microscopy teaches us. Neuroscientist. 2002;8:386–390. doi: 10.1177/107385802236963. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002a;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Klunk WE, Mathis CA, Hyman BT. Imaging amyloid-beta deposits in vivo. J Cereb Blood Flow Metab. 2002b;22:1035–1041. doi: 10.1097/00004647-200209000-00001. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hickey GA, Skoch J, Kajdasz ST, Wang Y, Huang GF, Mathis CA, Klunk WE, Hyman BT. Four-dimensional multiphoton imaging of brain entry, amyloid binding, and clearance of an amyloid-beta ligand in transgenic mice. Proc Natl Acad Sci USA. 2003;100:12462–12467. doi: 10.1073/pnas.2034101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–991. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Blackband S. MR microscopy and high resolution small animal MRI: applications in neuroscience research. Prog Neurobiol. 2002;67:393–420. doi: 10.1016/s0301-0082(02)00020-5. [DOI] [PubMed] [Google Scholar]
- Benveniste H, Ma Y, Dhawan J, Gifford A, Smith SD, Feinstein I, Du C, Grant SC, Hof PR. Anatomical and functional phenotyping of mice models of Alzheimer’s disease by MR microscopy. Ann N Y Acad Sci. 2007;1097:12–29. doi: 10.1196/annals.1379.006. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM. Anti-Abeta antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest. 2005;115:428–433. doi: 10.1172/JCI23269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Dekosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alz-heimer’s disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- Dimou E, Booij J, Rodrigues M, Prosch H, Attems J, Knoll P, Zajicek B, Dudczak R, Mostbeck G, Kuntner C, Langer O, Bruecke T, Mirzaei S. Amyloid PET and MRI in Alzheimer’s disease and mild cognitive impairment. Curr Alzheimer Res. 2009;6:312–319. doi: 10.2174/156720509788486563. [DOI] [PubMed] [Google Scholar]
- Ding S, Fellin T, Zhu Y, Lee SY, Auberson YP, Meaney DF, Coulter DA, Carmignoto G, Haydon PG. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J Neurosci. 2007;27 (40):10674–10684. doi: 10.1523/JNEUROSCI.2001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domnitz SB, Robbins EM, Hoang AW, Garcia-Alloza M, Hyman BT, Rebeck GW, Greenberg SM, Bacskai BJ, Frosch MP. Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J Neuropathol Exp Neurol. 2005;64:588–594. doi: 10.1097/01.jnen.0000171644.00180.fc. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- Elghetany MT, Saleem A. Methods for staining amyloid in tissues: a review. Stain Technol. 1988;63:201–212. doi: 10.3109/10520298809107185. [DOI] [PubMed] [Google Scholar]
- Friedland RP, Kalaria R, Berridge M, Miraldi F, Hedera P, Reno J, Lyle L, Marotta CA. Neuroimaging of vessel amyloid in Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:242–247. doi: 10.1111/j.1749-6632.1997.tb48475.x. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Bacskai BJ. Techniques for brain imaging in vivo. Neuro-molecular Med. 2004;6:65–78. doi: 10.1385/NMM:6:1:065. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Gilles C, Brucher JM, Khoubesserian P, Vanderhaeghen JJ. Cerebral amyloid angiopathy as a cause of multiple intracerebral hemorrhages. Neurology. 1984;34:730–735. doi: 10.1212/wnl.34.6.730. [DOI] [PubMed] [Google Scholar]
- Greenberg SM. Cerebral amyloid angiopathy and vessel dysfunction. Cere-brovasc Dis. 2002;13 (Suppl 2):42–47. doi: 10.1159/000049149. [DOI] [PubMed] [Google Scholar]
- Grutzendler J, Helmin K, Tsai J, Gan WB. Various dendritic abnormalities are associated with fibrillar amyloid deposits in Alzheimer’s disease. Ann N Y Acad Sci. 2007;1097:30–39. doi: 10.1196/annals.1379.003. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Iwata N, Matsuba Y, Sato K, Sasamoto K, Saido TC. 19F and 1H MRI detection of amyloid beta plaques in vivo. Nat Neurosci. 2005;8:527–533. doi: 10.1038/nn1422. [DOI] [PubMed] [Google Scholar]
- Hintersteiner M, Enz A, Frey P, Jaton AL, Kinzy W, Kneuer R, Neumann U, Rudin M, Staufenbiel M, Stoeckli M, Wiederhold KH, Gremlich HU. In vivo detection of amyloid-beta deposits by near-infrared imaging using an oxazine-derivative probe. Nat Biotechnol. 2005;23:577–583. doi: 10.1038/nbt1085. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4 (8):1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wengenack TM, Reyes DA, Garwood M, Curran GL, Borowski BJ, Lin J, Preboske GM, Holasek SS, Adriany G, Poduslo JF. In vivo magnetic resonance microimaging of individual amyloid plaques in Alzheimer’s transgenic mice. J Neurosci. 2005;25:10041–10048. doi: 10.1523/JNEUROSCI.2588-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Marjanska M, Wengenack TM, Reyes DA, Curran GL, Lin J, Preboske GM, Poduslo JF, Garwood M. Magnetic resonance imaging of Alzheimer’s pathology in the brains of living transgenic mice: a new tool in Alzheimer’s disease research. Neuroscientist. 2007;13:38–48. doi: 10.1177/1073858406295610. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Kalyan-Raman UP, Kalyan-Raman K. Cerebral amyloid angiopathy causing intracranial hemorrhage. Ann Neurol. 1984;16:321–329. doi: 10.1002/ana.410160308. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Calcium hypothesis of Alzheimer’s disease and brain aging. Ann N Y Acad Sci. 1994;747:1–11. doi: 10.1111/j.1749-6632.1994.tb44398.x. [DOI] [PubMed] [Google Scholar]
- Killiany RJ, Gomez-Isla T, Moss M, Kikinis R, Sandor T, Jolesz F, Tanzi R, Jones K, Hyman BT, Albert MS. Use of structural magnetic resonance imaging to predict who will get Alzheimer’s disease. Ann Neurol. 2000;47:430–439. [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nat Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer’s disease. Proc Natl Acad Sci USA. 1999;96:5274–5279. doi: 10.1073/pnas.96.9.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenigsknecht-Talboo J, Meyer-Luehmann M, Parsadanian M, Garcia-Alloza M, Finn MB, Hyman BT, Bacskai BJ, Holtzman DM. Rapid microglial response around amyloid pathology after systemic anti-Abeta antibody administration in PDAPP mice. J Neurosci. 2008;28:14156–14164. doi: 10.1523/JNEUROSCI.4147-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci USA. 2009;106 (10):4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008;59:214–225. doi: 10.1016/j.neuron.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ. Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009;323:1211–1215. doi: 10.1126/science.1169096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung MP, Hou C, Zhuang ZP, Skovronsky DM, Zhang B, Gur TL, Trojanowski JQ, Lee VM, Kung HF. Radioiodinated styrylbenzene derivatives as potential SPECT imaging agents for amyloid plaque detection in Alzheimer’s disease. J Mol Neurosci. 2002;19:7–10. doi: 10.1007/s12031-002-0003-9. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandybur TI. The incidence of cerebral amyloid angiopathy in Alzheimer’s disease. Neurology. 1975;25:120–126. doi: 10.1212/wnl.25.2.120. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003a;5:1041–1043. doi: 10.1038/ncb1203-1041. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer’s disease. Cell Calcium. 2003b;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. beta-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer’s disease. Trends Neurosci. 1993;16:409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Mott RT, Hulette CM. Neuropathology of Alzheimer’s disease. Neuro-imaging Clin N Am. 2005;15:755–765. ix. doi: 10.1016/j.nic.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging. 2004;25:663–674. doi: 10.1016/j.neurobiolaging.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Noble JM, Scarmeas N. Application of pet imaging to diagnosis of Alzheimer’s disease and mild cognitive impairment. Int Rev Neurobiol. 2009;84:133–149. doi: 10.1016/S0074-7742(09)00407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A. Amyloid imaging in Alzheimer’s disease. Neuropsychologia. 2008;46:1636–1641. doi: 10.1016/j.neuropsychologia.2008.03.020. [DOI] [PubMed] [Google Scholar]
- Ono M. Development of positron-emission tomography/single-photon emission computed tomography imaging probes for in vivo detection of beta-amyloid plaques in Alzheimer’s brains. Chem Pharm Bull (Tokyo) 2009;57:1029–1039. doi: 10.1248/cpb.57.1029. [DOI] [PubMed] [Google Scholar]
- Pan F, Gan WB. Two-photon imaging of dendritic spine development in the mouse cortex. Dev Neurobiol. 2008;68:771–778. doi: 10.1002/dneu.20630. [DOI] [PubMed] [Google Scholar]
- Poduslo JF, Wengenack TM, Curran GL, Wisniewski T, Sigurdsson EM, Macura SI, Borowski BJ, Jack CR., Jr Molecular targeting of Alzheimer’s amyloid plaques for contrast-enhanced magnetic resonance imaging. Neurobiol Dis. 2002;11:315–329. doi: 10.1006/nbdi.2002.0550. [DOI] [PubMed] [Google Scholar]
- Prada CM, Garcia-Alloza M, Betensky RA, Zhang-Nunes SX, Greenberg SM, Bacskai BJ, Frosch MP. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J Neurosci. 2007;27:1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran C, Xu X, Raymond SB, Ferrara BJ, Neal K, Bacskai BJ, Medarova Z, Moore A. Design, synthesis, and testing of difluoroboron-derivatized curcumins as near-infrared probes for in vivo detection of amyloid-beta deposits. J Am Chem Soc. 2009;131:15257–15261. doi: 10.1021/ja9047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond SB, Skoch J, Hills ID, Nesterov EE, Swager TM, Bacskai BJ. Smart optical probes for near-infrared fluorescence imaging of Alzheimer’s disease pathology. Eur J Nucl Med Mol Imaging. 2008;35 (Suppl 1):S93–S98. doi: 10.1007/s00259-007-0708-7. [DOI] [PubMed] [Google Scholar]
- Robbins EM, Betensky RA, Domnitz SB, Purcell SM, Garcia-Alloza M, Greenberg C, Rebeck GW, Hyman BT, Greenberg SM, Frosch MP, Bacskai BJ. Kinetics of cerebral amyloid angiopathy progression in a transgenic mouse model of Alzheimer disease. J Neurosci. 2006;26:365–371. doi: 10.1523/JNEUROSCI.3854-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synapse loss in the temporal lobe in Alzheimer’s disease. Ann Neurol. 1993;33:190–199. doi: 10.1002/ana.410330209. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Skoch J, Hickey GA, Kajdasz ST, Hyman BT, Bacskai BJ. In vivo imaging of amyloid-beta deposits in mouse brain with multiphoton microscopy. Methods Mol Biol. 2005;299:349–363. doi: 10.1385/1-59259-874-9:349. [DOI] [PubMed] [Google Scholar]
- Skoch J, Hyman BT, Bacskai BJ. Preclinical characterization of amyloid imaging probes with multiphoton microscopy. J Alzheimers Dis. 2006;9:401–407. doi: 10.3233/jad-2006-9s345. [DOI] [PubMed] [Google Scholar]
- Sokolov Y, Kozak JA, Kayed R, Chanturiya A, Glabe C, Hall JE. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J Gen Physiol. 2006;128:637–647. doi: 10.1085/jgp.200609533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Meyer-Luehmann M, Osetek JD, Jones PB, Stern EA, Bacskai BJ, Hyman BT. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am J Pathol. 2007;171:1304–1311. doi: 10.2353/ajpath.2007.070055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Mielke ML, Rozkalne A, Meyer-Luehmann M, de Calignon A, Bacskai BJ, Schenk D, Hyman BT. Passive immunotherapy rapidly increases structural plasticity in a mouse model of Alzheimer disease. Neuro-biol Dis. 2009;33:213–220. doi: 10.1016/j.nbd.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Takano T, Han X, Deane R, Zlokovic B, Nedergaard M. Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann NY Acad Sci. 2007;1097:40–50. doi: 10.1196/annals.1379.004. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004;7:1181–1183. doi: 10.1038/nn1335. [DOI] [PubMed] [Google Scholar]
- Vassar PS, Culling CF. Fluorescent stains, with special reference to amyloid and connective tissues. Arch Pathol. 1959;68:487–498. [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- Wegiel J, Wang KC, Imaki H, Rubenstein R, Wronska A, Osuchowski M, Lipinski WJ, Walker LC, LeVine H. The role of microglial cells and astrocytes in fibrillar plaque evolution in transgenic APP(SW) mice. Neurobiol Aging. 2001;22:49–61. doi: 10.1016/s0197-4580(00)00181-0. [DOI] [PubMed] [Google Scholar]
- Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
- Yan P, Bero AW, Cirrito JR, Xiao Q, Hu X, Wang Y, Gonzales E, Holtzman DM, Lee JM. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 2009;29:10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]