

Summary
Atopic dermatitis (AD) is an important chronic or relapsing inflammatory skin disease that often precedes asthma and allergic disorders. New insights into the genetics and pathophysiology of AD point to an important role of structural abnormalities in the epidermis as well as immune dysregulation not only for this skin disease but also for the development of asthma and allergies. Patients with AD have a unique predisposition to colonization or infection by microbial organisms, most notably Staphylococcus aureus and herpes simplex virus. Measures directed at healing and protecting the skin barrier and addressing the immune dysregulation are essential in the treatment of patients with AD and early intervention may improve outcomes for both the skin disease as well as other target organs.
Keywords: atopic dermatitis, antimicrobial peptides, eczema herpeticum, epidermal barrier, filaggrin, Staphylococcus aureus
Introduction
Atopic dermatitis (AD) is an important common chronic or relapsing inflammatory disease of the skin that often precedes asthma and allergic disorders (1, 2). Lifetime prevalence in school-aged children in the United States has been reported to be up to 17% (3). The wide range of prevalence (8.7–18%) reported in the most recent US data derived from the 2003 National Survey of Children’s Health (4) suggests that environmental or other unidentified factors may influence disease expression. High prevalence rates have also been observed in a number of other countries, with data from over a million children in 97 countries showing that AD is a major problem in developing as well as developed countries (5, 6). Atopy, the genetic predisposition to make IgE antibody responses to common environmental or food protein antigens, has been shown to be strongly associated with AD with an incidence of approximately 80% in infants with AD (7). Of note, an ‘atopic march’ occurs early, as > 50% of children with AD will develop asthma and/or allergies, typically by their third birthday (8). Patients with severe or persistent disease and their families experience significant impairment in their quality of life (9), and in addition, AD places a heavy economic burden on society as a whole (10, 11). This review highlights recent insights into the genetics and pathophysiology of AD focusing on the relationship between skin barrier abnormalities and immune dysregulation.
Genetics of AD: an evolving story
AD is a highly heritable disease with phenotype-specific genes likely playing an important role along with ‘generic’ atopy genes. In a large birth cohort study, the risk of a child having AD if one or both parents had AD was found to be higher [odds ratio (OR), 3.4; 95% CI, 2.6–4.4] compared with the risk if one or both parents had asthma (OR, 1.5; 95% CI, 1.0–2.2) or allergic rhinitis (OR, 1.4; 95% CI, 1.1–1.8) (12). However, the relatively low concordance observed in some studies, even for identical twins, points to the role of environmental factors influencing disease risk and manifestation (13). The approach to identifying genes associated with AD has included genome-wide linkage and candidate gene association studies (reviewed in 14). Five genome-wide linkage studies performed on AD plus a genome-wide linkage screen originally designed for asthma with analyses repeated for the AD outcome were all performed on families of European ancestry, except for one that was performed on Japanese families. Linkage to AD was found on chromosomes 1, 3, 4, 5, 11, 13, 15, 17, 18 19, 20 with only the 3p24 locus showing significant replication. While the genome-wide linkage approach has identified a number of candidate genes associated with AD, none has conclusively pinpointed a specific locus or gene. It is also worth noting that no candidate gene was identified with positional cloning in the AD genome-wide linkage studies (14). In contrast, a candidate gene approach focusing on a specific gene or set of genes believed to be causally involved in the underlying pathology of a certain disease is not limited to families and can be applied to case-control study designs, which possess certain advantages over family-based studies and are generally considered to be more powerful in detecting true associations once the gene has been identified (15). While > 100 studies have reported an association of AD and a candidate gene, most of these were insufficiently powered, and heterogeneity of the AD phenotype in the studies makes replication of the observed associations difficult. Nevertheless, with this approach, of 81 genes examined, 46 had at least one positive association study reported. Of these 46 genes, 13 (FLG, IL4, IL4RA, SPINK5, CMA1, IL13, RANTES, CD14, DEFB1, GSTP1, IL18, NOD1, TIM1) were positively associated in at least one other independent study with the gene encoding filament-aggregating protein, FLG, associated with AD the highest number of studies (14). The genes identified encode for proteins predominantly involved in skin barrier function as well as innate and adaptive immune responses.
In a recent study looking at the association between prenatal farm-related exposures and AD in a birth cohort, the authors also analyzed the association between the expression of innate immune genes at birth and AD (16). In this prospective study, gene expression of TLRs and CD14 was assessed in cord blood leukocytes by quantitative polymerase chain reaction (PCR). Maternal contact with farm animals and cats during pregnancy had a significantly protective effect on AD in the first two years of life. The risk of AD was reduced by more than half among children with mothers having contact with three or more farm animal species during pregnancy compared with children with mothers without such contacts. Elevated expression of TLR5 and TLR9 in cord blood was associated with a decreased diagnosis of AD. In addition, a significant interaction between polymorphism in TLR2 and prenatal cat exposure was observed in AD.
In a novel approach, Barnes (14) evaluated 81 genes reported to have association with AD using the Ingenuity Pathway Analysis (www.analysis.ingenuity.com). More than half of the genes studied were clustered in two major networks associated with immune dysregulation: the pathway associated with antigen presentation, cell-mediated and humoral immune responses, and the pathway associated with cell signaling and interaction, cellular movement, and hematologic system development and function (Table 1). CD14 (monocyte differentiation antigen), GATA3 (GATA-binding protein 3), IL4, IL18, NOD1 (nucleotide-binding oligomerization domain 1), and TLR2 (Toll-like receptor 2) genes previously shown to be significantly associated with AD, were clustered in the antigen presentation and immune response pathway, while BCL2A1 (BCL2-related protein A1), BDNF (brain-derived neutrophilic factor), RANTES (regulated upon activation, normally T-expressed, and presumably secreted), CSF2 (colony-stimulating factor 2), GSTP1 (glutathione S-transferase 1), IL5, IL12B, IL12RB1, and SOCS3 (suppressor of cytokine signaling 3) genes were clustered in the cell-signaling or movement pathway. This approach may prove especially useful in selecting optimal candidates for further genetic association studies.
Table 1.
Genes involved in immune dysregulation associated with atopic dermatitis (from ref 14)
| Antigen presentation, cell-mediated and humoral immune response pathway | Cell signaling and interaction, cellular movement and hematologic system development and function pathway |
|---|---|
| CD14 (monocyte differentiation antigen) | BCL2A1 (BCL2-related protein A1) |
| GATA3 (GATA-binding protein 3) | BDNF (brain-derived neutrophilic factor) |
| IL4 | RANTES (Regulated upon Activation, Normally T-Expressed, and presumably Secreted) |
| IL18 | CSF2 (colony-stimulating factor 2) |
| NOD1 (nucleotide-binding oligomerization domain 1) | GSTP1 (glutathione S-transferase 1) |
| TLR2 (Toll-like receptor 2) | IL5 |
| IL12B | |
| IL12RB1 | |
| SOCS3 (suppressor of cytokine signaling 3) |
Patients with AD have an increased susceptibility to infections and cutaneous colonization (2) and several immune function genes have been shown to be associated with this predisposition. Gene association studies on AD implicate TLR2, NOD1, NOD2, CD14 and DEFB1 (β-defensin 1). It is important to recognize that these and other AD-associated genes discussed above are not necessarily unique to AD. A number of AD candidate genes are associated with diseases of immune dysregulation including asthma and allergies, consistent with the hypothesis that certain disease genes are not disease specific, but contribute to related clinical phenotypes (17).
Linkage studies performed in AD have implicated loci containing clusters of genes associated with skin barrier dysfunction including those found in the epidermal differentiation complex (EDC) on chromosome 1q21 encoding epidermal cornification and S100 proteins (18). Of these, FLG has been the most widely studied and as discussed previously, to date FLG is the gene with the highest association with AD. In a landmark paper published in 2006, Palmer et al. (19) showed that in a European Caucasian population, homozygous nonsense mutations in FLG (R501X and 2282del4) which stop protein translation were strongly associated with AD. A growing number of additional mutations have been reported with varying frequency in various ethnic groups including in Asian and African-American patients (20–23). A meta-analysis of FLG R501X and 2282del4 mutations in 6,448 cases, 26,787 control subjects and 1,993 families all selected for AD confirmed that the two null mutations represent the strongest and most compelling genetic risk factors for AD (24). Beside mutations in FLG, genomic profiling of mRNA in AD has identified defects in expression of multiple genes encoding the cornified envelope, including loricrin (25) (Fig. 1).
Fig. 1. Complex pathophysiology of atopic dermatitis including barrier and immune abnormalities.
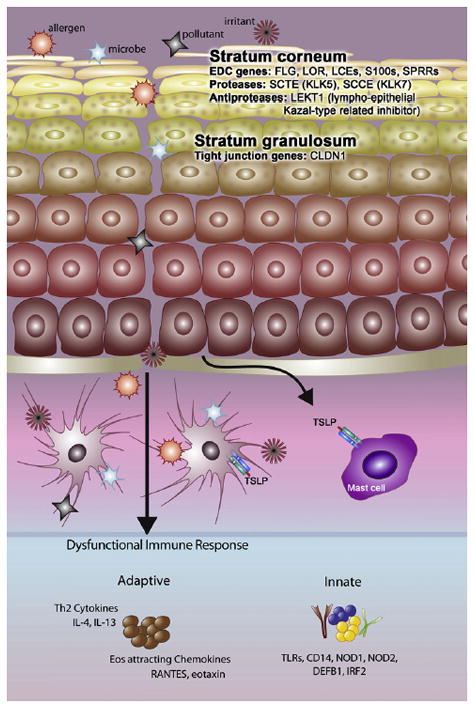
Reproduced with permission from: Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. J Allergy Clin Immunol 2010;125:16–29 [Figure 5A].
The study by Gao et al. (22) showed that the frequency of the FLG R501X mutation was three times higher and the relative risk for disease nearly double for AD patients with eczema herpeticum (EH), a serious viral complication, compared to AD without EH (OR, 11.8 vs 6.2; P = .0008). This is the first study providing insight into the genetic associations of AD and complicating factors, suggesting that skin barrier dysfunction associated with FLG mutations contributes to the increased propensity for disseminated viral skin infections seen in a subset of AD patients. More recently, a multicenter case-control study demonstrated an association between markers in thymic stromal lymphopoietin (TSLP) and its receptors, IL7R and TSLPR, and risk of AD and EH (26). The findings point to TSLP as an important candidate for AD, AD severity and ADEH and are the first to implicate TSLP as a potential casual gene for ADEH. Patients with AD are also at high risk to develop serious, potentially life-threatening reactions to vaccinia, the attenuated virus used for smallpox vaccination (27, 28). In a gene-profiling study of skin biopsy specimens to vaccinia virus, Grigoryev et al. (29) found differences in the transcriptional response to vaccinia virus in AD patients compared with patients with psoriasis or healthy subjects. The unique molecular signature in AD skin included genes involved in the innate immune response and wound healing.
More recently, genome-wide association studies (GWASs) have been introduced as an unbiased approach to search for genes controlling the risk for complex diseases (30). Results from the first GWAS in AD identified two novel genetic variants in association with eczema susceptibility: a single nucleotide polymorphism on chromosome 11q13.5 (rs7927894) and a single nucleotide polymorphism (rs877776) within the gene encoding hornerin on chromosome 1q21 (31). Subsequently, a case-control study investigated the association of rs7927894, rs877776, and the four most prevalent FLG null mutations with moderate-severe eczema in 511 Irish pediatric cases and 1000 Irish controls (32). Comprehensive testing for interaction between each of the loci was also performed. The association between rs7927894 and AD was replicated in this population (P = .0025; odds ratio, 1.27; 95% CI, 1.09–1.49), but no significant association between rs877776 AD was found. The four most common FLG null variants were strongly associated with AD (P = 1.26 × 10(−50); combined odds ratio, 5.81; 95% CI, 4.51–7.49). The rs7927894 association was found to be independent of the FLG risk alleles and may be multiplicative in its effect. Thus, the single nucleotide polymorphism rs7927894 appears to identify a genuine eczema susceptibility locus that will require further elucidation through fine mapping and functional analysis.
Complex interactions have been described between immune abnormalities characterizing AD such as elevated Th2 cytokine levels (1) and expression of genes involved in epidermal barrier function (33, 34). In a murine model, IL-4 was shown to decrease the expression of multiple genes associated with innate defense, including genes in the EDC that regulate epidermal barrier function. Mice that express a constitutively active Stat6 (Stat6VT) are prone to the development of allergic skin inflammation and have decreased expression of EDC genes. IL-4 deficiency was shown to protect Stat6VT transgenic mice from developing allergic skin inflammation and to decrease the time to recover barrier function with a concomitant increase in EDC gene expression (35). Insights from genetic studies suggest that immunologic and skin barrier related genetic variations may work synergistically to increase susceptibility to AD. In addition to susceptibility, FLG null alleles have also been shown to predispose to early onset AD that persists into adulthood, thus defining a possible unique phenotype of AD (36). Furthermore, patients 50 years of age or older with AD are at increased risk for having FLG mutations (37).
FLG mutations and risk of asthma and allergic disorders
Studies of FLG mutations have pointed to an important association of skin barrier abnormalities in the development of asthma and allergies. In the initial study that showed an association between AD and FLG mutations, Palmer et al. (19) also showed that these mutations were strongly associated with asthma, but only in those individuals who had eczema. Subsequent studies have confirmed this important association (24, 38–40). Other studies have shown the association of FLG mutations with allergic sensitization and allergic disorders (38, 39, 41) and support the concept of an atopic march with AD followed closely by the development of allergies and asthma (42). In a prospective clinical study from birth to school-age, a high-risk cohort of European children was assessed for development of AD, asthma, and allergies (43). Two common FLG variants were evaluated in Caucasian children and these were found to significantly increase risk of eczema, as well as developing recurrent wheeze, asthma, and asthma exacerbations (hazard ratio 1.82 [1.06–3.12], p = 0.03) expressed within the first 1.5 yr of life. The children with FLG mutations had a marked and persistent increase in acute severe asthma exacerbations from one yr of age [incidence ratio 2.40 (1.19–4.81), p = 0.01] and increased risk of asthma by age five [odds ratio 2.62 (1.12–6.11), p = 0.03]. FLG variants also increased risk of specific sensitization by age four [odds ratio 3.52 (1.72–7.25), p = 0.0007] but not age 1.5. While these associations have been described in predominantly European Caucasian populations, Imoto et al. (44) recently reported allergic sensitization associated with a FLG gene mutation in an Asian population. Of interest, in infants with eczema and sensitization to food allergens, FLG mutations were shown to predict childhood asthma with a positive predictive value of 100% (95% CI, 65.5% to 100%) (45). These findings point to a strong synergistic interaction between FLG-null alleles and early food sensitization in the disease transition from eczema to asthma (relative excess risk due to interaction, 2.64; 95% CI, 1.70–3.98; P = .00040) and suggest that FLG mutations and food sensitization represent distinct but closely interacting mechanisms in the development of asthma. Genotyping infants with eczema and food sensitization for FLG mutations could allow for the prediction of asthma before the onset of symptoms and facilitate the development of early subgroup-specific interventions to prevent progression in the atopic march.
Since filaggrin was not detected in bronchial biopsies from asthmatic and normal volunteers (46), this finding suggests that this skin barrier protein is not directly involved in lower airway permeability to environmental allergens. In addition, filaggrin was not expressed in nasal or esophageal epithelium (47) and is therefore unlikely to play a role in barrier function at these mucosal surfaces. The hypothesis that allergic sensitization occurs through a damaged skin barrier and leads to local and systemic immune responses (48) is supported by studies with a FLG-deficient mouse (49), as discussed below. Of interest, development of peanut allergy has been linked to transcutaneous exposure to peanut allergens in topical preparations (50) and in the environment (51), and early onset AD is a risk factor for peanut allergy (50). Whether FLG or other barrier-specific abnormalities will be linked to increased risk for peanut or other food allergy remains to be seen.
As noted by Barnes (14), very few of the genetic association studies in AD have looked at associations between genetic polymorphisms and environmental factors. Data from two birth cohorts demonstrated a significant interaction between FLG loss-of-function mutations and cat ownership at birth on the development of early-life eczema (52). These findings suggest that cat but not dog ownership substantially increases the risk of eczema in the first year of life in children with FLG mutations but not in those without such mutations and points to the need for early identification of at-risk individuals with appropriate environmental control measures. These findings are in contrast to the study of animal exposures and innate immune genes in AD discussed earlier (16).
Moving beyond genomics
After genomics, proteomics is considered the next step in the study of biological systems (53). Using a proteomic approach, Howell et al. (54) found the S100 calcium-binding protein A11 (S100/A11) to be significantly downregulated in the presence of IL-4 and IL-13 in AD. S100/A11, an important component of the cornified envelope encoded within the EDC on chromosome 1q21 (55) was also shown to be decreased in the skin of AD patients. In addition, the authors also showed an immunomodulatory effect of S100/A11 on FLG and HBD-3 gene expression pointing to immune dysregulation effecting both epidermal barrier integrity and innate immune response. Using a novel mass spectrometry-based proteomics approach on skin tape strippings, Broccardo et al. (56, 57) found that patients with AD have unique epidermal protein profiles. Proteins related to the skin barrier and generation of natural moisturizing factor (NMF) were expressed at significantly lower levels in lesional versus nonlesional sites of patients with AD with and without history of EH while epidermal fatty acid-binding protein was expressed at significantly higher levels in patients with methicillin-resistant S. aureus (MRSA). Lower expression of skin barrier proteins and enzymes involved in the generation of NMF could exacerbate barrier defects and perpetuate water loss from the epidermis, while greater expression of epidermal fatty acid-binding protein, especially in patients colonized with MRSA could perpetuate the inflammatory response through eicosanoid signaling.
Consequences of epidermal barrier abnormalities in AD
An intact, healthy skin barrier is a critical first line of defense against various microbes, irritants, and allergens. As discussed above, the epidermis of AD patients is characterized by significant barrier disruption, and AD patients have an increased susceptibility to allergic sensitization as well as microbial colonization and infections (2, 58). In normal subjects, formation of the cornified cell envelope involves dephosphorylation and cleavage of the large profilaggrin protein by serine proteases ending in the release of functional filaggrin (59). Filaggrin aggregates the keratin cytoskeleton to facilitate the collapse and flattening of keratinocytes in the outermost skin layer. Additionally, other proteins encoded by genes in the EDC discussed above including loricrin and involucrin are essential components of the epidermal barrier (60). As the water content of the stratum corneum drops, filaggrin is proteolyzed into pyrrolidine carboxylic acid and trans-urocanic acid which contribute to the composition of NMF (61). These breakdown products in turn act as osmolytes, accounting in large part for corneocyte hydration. The cellular basis for a permeability barrier abnormality remains to be fully elucidated (62). However, the most immediate result of FLG deficiency in AD is decreased stratum corneum hydration, resulting in a steeper water gradient across the stratum corneum and driving increased transepidermal water loss. In addition, filaggrin breakdown products play an important role in acidifying the stratum corneum and decreased generation of filaggrin metabolites could result in an increase in the stratum corneum pH of the stratum corneum with activation of a number of serine proteases (58). A pH-induced increase in serine protease activity could lead to both barrier breakdown and precipitate Th2 inflammation in AD (62). A recent in vitro study demonstrated that S. aureus growth rate and cell density were affected by the acidic filaggrin breakdown products urocanic acid and pyrrolidone carboxylic acid (63). Lower pH was associated with reduced expression of secreted and cell wall-associated proteins, including proteins involved in colonization such as clumping factor B and fibronectin binding protein A, as well as in immune evasion such as protein A. Importantly, exogenous proteases from S. aureus and dust mites could further perpetuate barrier and immune abnormalities (58).
Whether skin barrier dysfunction precedes skin inflammation and epidermal barrier defects initiate development of AD remains an unresolved issue. In a recent study, infants were evaluated for evidence of skin inflammation and barrier dysfunction at the age of three months (64). Those with FLG mutations were more likely to have AD than infants without defined FLG mutations. Importantly, infants with FLG mutations had abnormal epidermal barrier function compared to infants without defined FLG mutations, even in the absence of clinically evident eczema. While these results suggest that FLG mutations are associated with abnormal skin barrier function prior to the development of clinically overt inflammation, definitive proof of whether skin barrier dysfunction precedes skin inflammation and whether epidermal barrier defects are the key initiating factors in the development of AD awaits more sensitive analyses performed at multiple time points.
Skin barrier function as assessed by TEWL is intrinsically compromised in children with AD but not in children with other allergic conditions and also differs by race (65). The magnitude of skin barrier dysfunction correlates with AD disease severity. Skin barrier abnormalities may occur in AD patients irrespective of FLG genotype, suggesting that other factors may be important in modulating skin barrier integrity (66). Enhanced TEWL and diffusivity in AD may reflect defects in the intercellular lipid bilayers of the stratum corneum. However, disturbance of lipid organization in the stratum corneum could be impacted differently in AD patients with or without FLG mutations. Investigation of intercellular lipids of the stratum corneum in AD patients characterized for FLG genotype could provide further insights.
Lessons from mouse models
A number of mouse models of AD have been developed (reviewed in 67). These include eczema induced by epicutaneous application of sensitizers, transgenic mice overexpressing or lacking selective molecules, as well as mice that spontaneously develop AD-like skin lesions. More recently, Fallon et al. described a 1-bp deletion mutation, 5303delA, analogous to common human FLG mutations within the murine Flg gene in the spontaneous mouse mutant flaky tail (ft) (49). Topical application of allergen to mice homozygous for this mutation resulted in cutaneous inflammatory infiltrates and enhanced cutaneous allergen priming with development of allergen-specific antibody responses. These results support the flaky tail mouse as an important model of filaggrin deficiency. In addition, they support the hypothesis that antigen transfer through a defective epidermal barrier is a key mechanism underlying elevated IgE sensitization and initiation of cutaneous inflammation in humans with filaggrin-related atopic disease. Filaggrin-deficient ft/ft mice were also shown to exhibit TH17-dominated skin inflammation and eczematous changes with age as well as epicutaneous sensitization with protein antigen (68). The ft/ft mouse should provide new insights into the relationship of human FLG mutations and immune abnormalities in AD.
Digging deeper: tight junction abnormalities in AD
In addition to the stratum corneum, tight junctions found on opposing membranes of stratum granulosum keratinocytes directly below the stratum corneum form a second barrier (Fig. 1). Tight junctions are made up of a complex of adhesive and scaffolding proteins that control the passage of water, ions, and solutes through the paracellular pathway (69). The nonlesional epithelium of AD subjects has been shown to have bioelectric abnormalities indicative of a tight junction defect which could be the consequence of reduced levels of claudin-1 (CLDN1), a key tight junction adhesive protein (70). This is consistent with earlier work in KO mice that established the importance of epidermal tight junctions and claudin-1. CLDN1 knockout mice died within 24 h of birth with wrinkled skin, severe dehydration, and increased epidermal permeability as measured by dye studies and transepidermal water loss (71). In human AD studies, CLDN1 expression was shown to be inversely correlated with total circulating eosinophil counts and serum IgE, markers of Th2 polarity (70). This suggests that TJ defects may promote Th2 polarity, a characteristic of EH subjects (72). More recent mechanistic and genetic studies confirmed the critical role of tight junctions in maintaining barrier integrity and implicated reductions in claudin-1 levels in the spread of epidermal viral infections including EH (73).
Local and systemic immune dysregulation characterizes AD
A number of well-characterized systemic and cutaneous immune abnormalities have previously been described in AD, including increased serum IgE and sensitization to allergens, elevated Th2-type cytokine expression in acute lesions, increased numbers of T cells expressing cutaneous lymphocyte-associated antigen (CLA) (the homing receptor for the skin), increased expression of FcεRI on both Langerhans cells and inflammatory dendritic epidermal cells, as well as decreased expression of antimicrobial peptides (reviewed in 2). Whether skin barrier abnormalities precede immune dysregulation (‘outside-in’ hypothesis) (74) or immune dysregulation precedes barrier changes (‘inside-out’ hypothesis) continues to be an unresolved argument with data supporting both hypotheses. It is important to recognize that not all patients with AD have FLG mutations, and conversely, individuals with FLG -null alleles may not develop AD (59). In addition, while the natural history of AD shows that adults with AD are more likely to have FLG mutations (37), other studies show that some of these patients do outgrow their disease or go into an extended remission, although they tend to have a more prolonged course than those patients without FLG mutations (38).
A critical link between the barrier defect in AD patients with FLG mutations and Th2 polarization could be explained in part by enhanced allergen penetration through the damaged epidermis accompanied by increased production of thymic stromal lymphopoietin (TSLP) by keratinocytes, leading to a Th2-type milieu. TSLP has been called a ‘master switch for allergic inflammation (75) and shown to exert effects on a number of key cells involved in cutaneous inflammation, including mast cells, basophils, and eosinophils (reviewed in 76). AD patients with more polarized Th2-type disease with associated asthma and allergies as well as increased biomarkers including TSLP, cutaneous T cell-attracting chemokine, and serum IgE were more likely to have severe skin disease complicated by bacterial and viral skin infections (77). TSLP was recently shown to be induced in the epidermis after mechanical stimulation (78) and plays a role in activation of Th2 cytokine-secreting invariant natural killer cells, which are increased in AD skin (79). Polymorphisms of the TSLP gene have been shown to contribute to Th2-polarized immunity through higher TSLP production (80). As discussed previously, genetic variants in TSLP were recently shown to be associated with AD and EH (26). Still, the relationship of skin barrier and immune abnormalities to the increased susceptibility to microbial colonization and infections remains to be fully elucidated (81). Observations that topical calcineurin inhibitors can partially correct the barrier defect in AD and that gentamicin can restore the production of functional filaggrin chains provides further evidence of the complex relationship of the epidermal barrier and the immune system (82).
New subsets of T cells in AD
Naturally occurring CD4+CD25+ FoxP3+ T-regulatory (Treg) cells with normal immunosuppressive activity appear to be expanded in the peripheral blood of patients with AD (83). However, after stimulation by the superantigen (SAg) staphylococcal enterotoxin B (SEB), Treg cells appear to lose their immunosuppressive activity. This observation suggests a novel mechanism by which SAgs could augment T-cell activation in patients with AD (84). While increased numbers of Treg cells in the peripheral blood of patients with AD have been confirmed (85) and related to inflammatory signals with Treg cell suppressive activity subverted by proinflammatory mediators, CD4+CD25+FoxP3+ Treg cells were not found in lesional AD skin or in atopy patch test sites of AD patients (86). These studies contribute to previous observations of a number of discordant systemic versus skin-specific immune abnormalities (1).
The role of IL-17-secreting Th17 cells in AD has been investigated in several studies. IL-17 was shown to be preferentially elevated in acute vs chronic AD lesions (87). In addition, IL-17 expression was induced in the skin as well as in the airways in a murine epicutaneous antigen challenge model analogous to human AD (88). As discussed above, this group also showed that filaggrin-deficient mice exhibited Th17-dominated skin inflammation and eczematous changes with increased expression of IL-17 in the epidermis and increased antigen-specific IgE in their serum (68). In an atopy patch test model, IL-17 secretion was shown to be enhanced by SEB (89). Induced IL-17 upregulated the antimicrobial peptide HBD-2 in human keratinocytes in vivo, although co-expressed IL-4/IL-13 partially inhibited this effect. Thus, although IL-17 secreting T cells have been shown to infiltrate acute AD lesions and IL-17 secretion can be triggered by SAgs, ineffective IL-17-dependent upregulation of HBD-2 in patients with AD may result from partial inhibition by the Th2 cytokine milieu. A study in IL-4 and IL-13 knockout mice supports a role for IL-4 as a Th2 cytokine that downregulates the IL-17 response in epicutaneously sensitized mice (90). This abnormality could in part be responsible for the persistent colonization by S. aureus characteristically observed in AD patients. In addition, compared to psoriasis, a chronic inflammatory skin disease with abnormal barrier but without the predisposition to skin infections or colonization, Th17 cells appear to have a diminished role in AD skin and the associated reduced expression of innate immune defense genes may contribute to increased skin infections in AD (91). Both IL-4 and IL-13 were recently found to significantly antagonize IL-17 induction of antimicrobial genes such as DEFB4 (92). These findings have possible therapeutic implications, as measures directed at increasing signaling through intact IL-17 receptors, despite low ligand expression, could potentially reverse the relative AMP deficiency seen in AD (93). Neutralizing Th2 cytokines as previously demonstrated in skin explants from AD patients (94), thus allowing existing IL-17 to function could potentially restore AMP expression and decrease microbial colonization, although such an approach could tip the balance towards AMP-driven inflammation. In addition, data from the same group showed that T cells could independently express IL-22 even with low expression of IL-17 (95). This observation suggests that T cells could have functional specialization with ‘T17’ T cells inducing AMP and ‘T22’ T cells driving epidermal hyperplasia. Further characterization of T22 T cells is warranted and may have clinical as well as therapeutic implications.
Immunologic basis for pruritus in AD
Pruritus is the major symptom of AD that impacts most significantly on the quality of life of patients. Antihistamine therapy is frequently ineffective, suggesting that mediators other than histamine such as cytokines and neuropeptides may be involved (96). IL-31 is increased in AD skin lesions (97) and has been implicated in the development of chronic dermatitis in transgenic mice that overexpress IL-31 (98). Sources of IL-31 include skin-infiltrating CLA+ T cells and peripheral blood CD45RO CLA+ T cells (99). Irrespective of the atopic phenotype, serum IL-31 levels have been shown to correlate with disease activity in AD (100). S. aureus-derived SAgs have been shown to rapidly induce IL-31 mRNA expression in the skin of atopic individuals in vivo and in PBMCs in vitro, suggesting that chronic colonization and superinfection by this microbial organism can contribute to pruritus and inflammatory changes in AD (97).
Role of microbial organisms in AD
Patients with AD have a unique propensity to be colonized or infected by a number of microbial organisms (2). Dysregulation of the adaptive immune response with elevated total and specific IgE levels has been associated with disease severity and infectious complications (101, 102). Innate immune system abnormalities, including reduction in antimicrobial peptides, diminished recruitment of cells such as neutrophils to the skin, and Toll-like receptor defects (103) as well as epidermal barrier abnormalities discussed previously (58) play an important role in microbial colonization or infection in AD. In addition, the relationship between innate and adaptive immune responses with infectious complications of AD continues to be elucidated. Mobilization of HBD-3 and killing of S. aureus by keratinocytes from AD patients were both shown to be significantly inhibited by IL-4 and IL-13, while neutralization of these cytokines significantly improved these activities (104). Serum IgE levels in AD patients with HSV infections were shown to correlate inversely with cathelicidin LL-37 expression (72). How aberrations in adaptive and innate immune responses and barrier abnormalities interact in AD remains to be fully investigated.
S. aureus can be cultured from 90% of skin lesions and importantly can colonize normal appearing skin in AD (105). S. aureus can exacerbate or contribute to persistent skin inflammation in AD by secreting toxins with superantigenic properties, resulting in marked activation of T cells and other immune cells (106). Application of SEB to the skin can induce eczematous changes accompanied by infiltration of T cells selectively expanded in response to the SAg (107). In addition, AD patients can make specific IgE antibodies directed against the toxins found on their skin with basophils from these patients releasing histamine on exposure to the relevant toxin (108, 109). This suggests that SAgs can induce mast cell degranulation after penetrating the epidermal barrier and contribute to pruritus and acute inflammatory events along with participating in chronic skin inflammation. S. aureus isolates from patients with steroid-resistant AD have been shown to produce increased numbers of SAgs compared with isolates from controls suggesting a possible selective advantage of SAgs for colonization of patients (110).
Other products of S. aureus can also contribute to clinical disease in AD. Patients with impetiginized AD were found to have elevated levels of lipoteichoic acid (LTA) that correlated with clinical scores and S. aureus CFU (111). The amounts of LTA in the skin lesions were sufficient to exert biological effects on various cell types in vitro, as well as epidermal cytokine gene expression when skin was exposed to LTA ex vivo providing a novel mechanism by which S. aureus can exacerbate AD.
MRSA has emerged as an important pathogen that has rapidly evolved from a cause of nosocomial to community-acquired infections (106). MRSA invariably produce SAgs and AD patients may be particularly susceptible to colonization and infection by MRSA as well as to the inflammatory consequences of these SAgs, as they are frequently treated, often with extended courses with anti-staphylococcal antibiotics (112). Phenol-soluble modulins (PSMs) represent a novel class of secreted S. aureus peptides that contribute to the enhanced virulence of community acquired (CA)-MRSA (113). These peptides appear to target neutrophils, recruiting, activating and then lysing these cells. PSMs are under the regulatory control of the accessory gene regulator (agr) quorum-sensing system, which controls the expression of a number of staphylococcal virulence factors. Understanding the underlying mechanisms for infection and colonization by S. aureus of the skin of patients with AD including differences between methicillin sensitive S. aureus versus MRSA is critical for developing more effective treatment strategies for this growing public health problem.
In addition to bacterial infections, a subset of AD patients is at risk for localized and disseminated cutaneous viral infections, most often caused by HSV (114). EH is a potentially life-threatening infection due to disseminated HSV that occurs in 10–20% of patients with AD. Risk factors for EH include early onset of AD, severe and untreated AD, head and neck dermatitis, previous EH or HSV infections, elevated total serum IgE with higher level of allergic sensitizations (115). As discussed previously, AD patients with EH have more severe, Th2-polarized disease with greater allergen sensitization compared to AD patients without a history of EH (77). Patients with ADEH also have a defect in the inducibility of the AMPs, HBD-2, HBD-3 and cathelicidin (116). AD patients of both European and African ancestry with FLG R501X mutations have been found to have an even greater risk for EH (22), and genetic variants in TSLP have also been shown to be associated with ADEH (26), implicating both skin barrier and immune abnormalities as contributors to this serious complication. AD patients are also at risk for potentially life-threatening complications from vaccinia virus (VV) used to vaccinate against smallpox (27, 28). Skin explants from AD subjects inoculated with VV showed increased viral replication compared to healthy controls (94) with low levels of LL-37 and increased expression of IL-4 and IL-13. Importantly, neutralizing antibodies against these Th2 cytokines inhibited VV growth and increased levels of LL-37. Further studies may provide insights into identifying which AD patients are at greatest risk for complications from immunization with vaccinia virus.
Implications of skin barrier and immune abnormalities for novel therapeutic strategies in AD patients
Jensen et al. (117) looked at TEWL, as well as several other parameters of epidermal barrier including stratum corneum hydration and dye penetration, and showed improvement in all parameters when AD patients were treated with both a topical steroid and a topical calcineurin inhibitor. Electron microscopic evaluation of barrier structure showed prevalently ordered stratum corneum lipid layers and regular lamellar body extrusion in the calcineurin inhibitor treated skin but inconsistent extracellular lipid bilayers and only partially filled lamellar bodies in the steroid treated skin. Both treatments normalized epidermal differentiation and reduced epidermal hyperproliferation. Of note, while expression of filaggrin was also reduced in patients with AD, it was completely restored on treatment with either anti-inflammatory therapy. This is consistent with studies showing that filaggrin and other barrier protein expression in AD were downregulated through Th2 cytokine activity (33, 34). Of interest, tumor necrosis factor-α (TNF-α) was recently shown to modulate filaggrin and loricrin protein expression via a c-Jun N-terminal kinase dependent pathway (118). Psoriasis patients treated with a TNF-α antagonist had significant enhancement of epidermal barrier protein expression, suggesting that TNF-α acts to inhibit key skin barrier proteins and use of specific antagonists may improve barrier protein expression. Thus, specific immunomodulatory therapy could be of benefit in AD and other diseases with epidermal barrier abnormalities. Vitamin D deficiency is being increasingly recognized as playing a role in allergic diseases (119).
Vitamin D appears to also be involved in regulation of AMPs in keratinocytes (120). The results of a trial with oral vitamin D in AD patients support this hypothesis (121). In addition, children with AD treated with oral vitamin D in a randomized, controlled trial showed clinical improvement versus placebo (122). Larger multi-center trials with oral vitamin D in AD are currently in progress. An analysis of the National Health and Nutrition Examination Survey 2001–2004 found that individuals with vitamin D deficiency had a statistically significant increased risk of MRSA carriage (123). Whether vitamin D supplementation can decrease MRSA colonization remains to be studied.
The emergence of CA-MRSA has created an urgency to develop novel strategies to combat this microbial organism. An important consideration in devising new therapeutic strategies against S. aureus is recognizing that protective immunity to staphylococcal infections does not appear to exist to any significant degree, due in part to the fact that our immune system is in constant contact with staphylococcal antigens and many strains are commensal organisms (106). In addition, S. aureus produces protein A to help it evade acquired host defense. While several attempts to develop protective vaccines have met with failure in clinical trials (e.g. StaphVax), promising results based on using a combination of systematically selected antigens have been reported (124). These combinatory vaccines target microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), a family of bacterial proteins that bind to human extracellular matrix components. A vaccine based on a combination of antigens was shown to provide complete protection from lethal doses of S. aureus in a murine challenge model (125). Importantly, MSCRAMM vaccines have been shown to interfere with S. aureus colonization (126). Whether this will result in decreased infection rates remains to be determined. Another potential antigenic target for vaccine development against S. aureus is teichoic acid, which has been implicated in nasal colonization and biofilm formation (127). Patients with impetiginized AD have been found to have significant levels of lipoteichoic acid in infected lesions that were able to induce epidermal cytokine gene expression ex vivo (111). Trials with a conjugated vaccine that includes both a-toxin and Panton-Valentine leukocidin are currently in progress. In addition, a phase I, multicenter, double-blind study of a novel vaccine containing the conserved S. aureus iron surface determinant B conducted to assess the immunogenicity and safety of an adjuvanted formulation of V710 showed lasting immune responses in adult subjects with no immediate or serious adverse events reported (128).
As discussed previously, S. aureus secretes exotoxins with properties of SAgs and a novel approach involves engineering binding affinity agents capable of neutralizing these SAgs. Soluble forms of the engineered Vβ proteins produced in E. coli were shown to be effective inhibitors of SEB-mediated T-cell activation and completely neutralized the lethal activity of SEB in animal models (129). Subsequently, the same group was able to express Vβ domains in tandem as a single-chain protein and neutralized SEB and TSST-1 with a single agent demonstrating the feasibility of engineering a broader spectrum antagonist capable of neutralizing multiple toxins (130). Such an approach may be especially relevant to AD, as S. aureus isolates from patients with steroid-resistant AD have been shown to produce increased numbers of SAgs compared with isolates from controls (110).
Although cathelicidins and HBD-3 exhibit potent antiviral activity against VV (131), their utility as clinically useful anti-VV agents is limited due to rapid degradation by endogenous tissue proteases. In contrast, ceragenins are synthetic antimicrobial compounds designed to mimic the structure and function of endogenous antimicrobial peptides (132) that can disrupt bacterial membranes without damaging mammalian cell membranes (133). Due to their synthetic nature, ceragenins are not subject to human protease degradation allowing for a longer tissue half-life. One candidate compound (CSA-13) was found to exhibit potent antiviral activity against VV via direct antiviral effects and by stimulating the expression of endogenous antimicrobial peptides with known antiviral activity (134). Further topical application of CSA-13 in mice reduced VV-induced satellite lesion formation, suggesting that treatment with CSA-13 may be an intervention for patients with disseminated VV skin infection.
Conclusions
Patients with AD have genetically determined risk factors that affect their skin barrier function and immune responses and interact with environmental triggers. Clinically, this results in intensely pruritic, inflamed skin that allows penetration of irritants and allergens and predisposes patients to colonization and infection by microbial organisms. New techniques such as GWAS and high-throughput expression profiling should greatly improve our ability to identify relevant gene polymorphisms, including rare variants in this complex disorder as well as other allergic diseases. Insights into the complex relationship between skin barrier and immune abnormalities should lead to more targeted therapy for AD and associated infectious complications. New methods to categorize unique phenotypes of AD may also lead to novel early interventions strategies that could also interrupt the development of asthma and allergic disorders.
Figure 2.
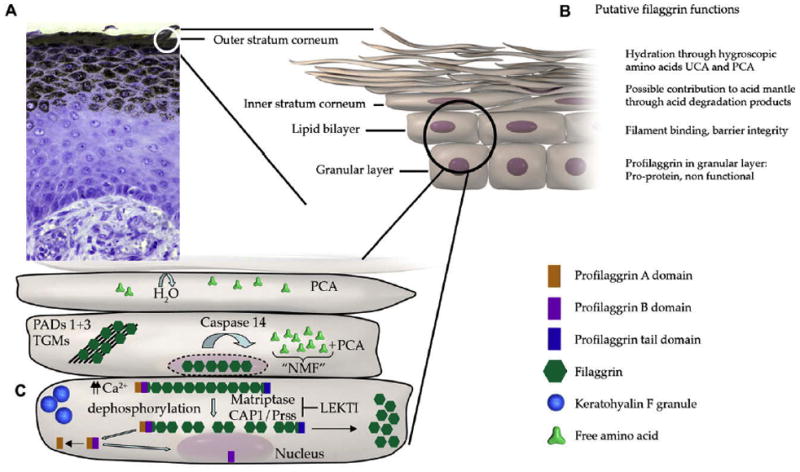
Filaggrin products, e.g. PCA, contribute to natural moisturizing factor and inhibit growth of Staphylococcus aureus. (Reproduced with permission from: O’Regan GM et al. Filaggrin in atopic dermatitis. J Allergy Clin Immunol 2008;122:689–93 [Figure 1]).
Acknowledgments
This work was supported by NIH/NIAID contracts N01 AI 40029 and AR41256.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Leung DY, Boguniewicz M, Howell MD, Nomura I, Hamid QA. New insights into atopic dermatitis. J Clin Invest. 2004;113:651–657. doi: 10.1172/JCI21060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boguniewicz M, Leung DY. Recent insights into atopic dermatitis and implications for management of infectious complications. J Allergy Clin Immunol. 2010;125:4–13. doi: 10.1016/j.jaci.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laughter D, Istvan JA, Tofte SJ, Hanifin JM. The prevalence of atopic dermatitis in Oregon schoolchildren. J Am Acad Dermatol. 2000;43:649–655. doi: 10.1067/mjd.2000.107773. [DOI] [PubMed] [Google Scholar]
- 4.Shaw TE, Currie GP, Koudelka CW, Simpson EL. Eczema prevalence in the United States: data from the 2003 National Survey of Children’s Health. J Invest Dermatol. 2011;131:67–73. doi: 10.1038/jid.2010.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Williams H, Stewart A, von Mutius E, Cookson W, Anderson HR. Is eczema really on the increase worldwide? J Allergy Clin Immunol. 2008;121:947–954. doi: 10.1016/j.jaci.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 6.Odhiambo JA, Williams HC, Clayton TO, Robertson CF, Asher MI. Global variations in prevalence of eczema symptoms in children from ISAAC Phase Three. J Allergy Clin Immunol. 2009;124:1251–1258. doi: 10.1016/j.jaci.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 7.de Benedictis FM, et al. The allergic sensitization in infants with atopic eczema from different countries. Allergy. 2009;64:295–303. doi: 10.1111/j.1398-9995.2008.01779.x. [DOI] [PubMed] [Google Scholar]
- 8.Kapoor R, Menon C, Hoffstad O, Bilker W, Leclerc P, Margolis DJ. The prevalence of atopic triad in children with physician-confirmed atopic dermatitis. J Am Acad Dermatol. 2008;58:68–73. doi: 10.1016/j.jaad.2007.06.041. [DOI] [PubMed] [Google Scholar]
- 9.Beattie PE, Lewis-Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol. 2006;155:145–151. doi: 10.1111/j.1365-2133.2006.07185.x. [DOI] [PubMed] [Google Scholar]
- 10.Boguniewicz M, et al. A multiple-domain framework of clinical, economic, and patient-reported outcomes for evaluating benefits of intervention in atopic dermatitis. J Drugs Dermatol. 2007;6:416–423. [PubMed] [Google Scholar]
- 11.Mancini AJ, Kaulback K, Chamlin SL. The socioeconomic impact of atopic dermatitis in the United States: a systematic review. Pediatr Dermatol. 2008;25:1–6. doi: 10.1111/j.1525-1470.2007.00572.x. [DOI] [PubMed] [Google Scholar]
- 12.Dold S, Wjst M, von Mutius E, Reitmeir P, Stiepel E. Genetic risk for asthma, allergic rhinitis, and atopic dermatitis. Arch Dis Child. 1992;67:1018–1022. doi: 10.1136/adc.67.8.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strachan DP, Wong HJ, Spector TD. Concordance and interrelationship of atopic diseases and markers of allergic sensitization among adult female twins. J Allergy Clin Immunol. 2001;108:901–907. doi: 10.1067/mai.2001.119408. [DOI] [PubMed] [Google Scholar]
- 14.Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. J Allergy Clin Immunol. 2010;125:16–29. doi: 10.1016/j.jaci.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 16.Roduit C, et al. Prenatal animal contact and gene expression of innate immunity receptors at birth are associated with atopic dermatitis. J Allergy Clin Immunol. 2011;127:179–185. doi: 10.1016/j.jaci.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 17.Gao L, et al. Polymorphisms in the myosin light chain kinase gene that confer risk of severe sepsis are associated with a lower risk of asthma. J Allergy Clin Immunol. 2007;119:1111–1118. doi: 10.1016/j.jaci.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Segre JA. Epidermal differentiation complex yields a secret: mutations in the cornification protein filaggrin underlie ichthyosis vulgaris. J Invest Dermatol. 2006;126:1202–1204. doi: 10.1038/sj.jid.5700367. [DOI] [PubMed] [Google Scholar]
- 19.Palmer CN, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 20.Nomura T, et al. Unique mutations in the filaggrin gene in Japanese patients with ichthyosis vulgaris and atopic dermatitis. J Allergy Clin Immunol. 2007;119:434–440. doi: 10.1016/j.jaci.2006.12.646. [DOI] [PubMed] [Google Scholar]
- 21.Sandilands A, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 22.Gao PS, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol. 2009;124:507–513. doi: 10.1016/j.jaci.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akiyama M. FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol. 2010;162:472–477. doi: 10.1111/j.1365-2133.2009.09582.x. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez E, et al. Meta-analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. J Allergy Clin Immunol. 2009;123:1361–1370. doi: 10.1016/j.jaci.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 25.Guttman-Yassky E, et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J Allergy Clin Immunol. 2009;124:1235–1244. doi: 10.1016/j.jaci.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 26.Gao PS, et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol. 2010;125:1403–1407. doi: 10.1016/j.jaci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Engler RJ, Kenner J, Leung DY. Smallpox vaccination: Risk considerations for patients with atopic dermatitis. J Allergy Clin Immunol. 2002;110:357–365. doi: 10.1067/mai.2002.128052. [DOI] [PubMed] [Google Scholar]
- 28.Vora S, et al. Severe eczema vaccinatum in a household contact of a smallpox vaccinee. Clin Infect Dis. 2008;46:1555–1561. doi: 10.1086/587668. [DOI] [PubMed] [Google Scholar]
- 29.Grigoryev DN, et al. Vaccinia virus-specific molecular signature in atopic dermatitis skin. J Allergy Clin Immunol. 2010;125:153–159. doi: 10.1016/j.jaci.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 31.Esparza-Gordillo J, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet. 2009;41:596–601. doi: 10.1038/ng.347. [DOI] [PubMed] [Google Scholar]
- 32.O’Regan GM, Campbell LE, Cordell HJ, Irvine AD, McLean WH, Brown SJ. Chromosome 11q13.5 variant associated with childhood eczema: an effect supplementary to filaggrin mutations. J Allergy Clin Immunol. 2010;125:170–174. doi: 10.1016/j.jaci.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Howell MD, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120:150–155. doi: 10.1016/j.jaci.2007.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126:332–337. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sehra S, et al. IL-4 regulates skin homeostasis and the predisposition toward allergic skin inflammation. J Immunol. 2010;184:3186–3190. doi: 10.4049/jimmunol.0901860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barker JN, et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J Invest Dermatol. 2007;127:564–567. doi: 10.1038/sj.jid.5700587. [DOI] [PubMed] [Google Scholar]
- 37.Rice NE, et al. Filaggrin gene mutations are associated with asthma and eczema in later life. J Allergy Clin Immunol. 2008;122:834–836. doi: 10.1016/j.jaci.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henderson J, et al. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. J Allergy Clin Immunol. 2008;121:872–877. doi: 10.1016/j.jaci.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 39.Weidinger S, et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol. 2008;121:1203–1209. doi: 10.1016/j.jaci.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 40.Brown SJ, et al. Filaggrin null mutations and childhood atopic eczema: a population-based case-control study. J Allergy Clin Immunol. 2008;121:940–946. doi: 10.1016/j.jaci.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Oord RA, Sheikh A. Filaggrin gene defects and risk of developing allergic sensitisation and allergic disorders: systematic review and meta-analysis. BMJ. 2009;339:b2433. doi: 10.1136/bmj.b2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marenholz I, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol. 2006;118:866–871. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 43.Bonnelykke K, Pipper CB, Tavendale R, Palmer CN, Bisgaard H. Filaggrin gene variants and atopic diseases in early childhood assessed longitudinally from birth. Pediatr Allergy Immunol. 2010;21:954–961. doi: 10.1111/j.1399-3038.2010.01073.x. [DOI] [PubMed] [Google Scholar]
- 44.Imoto Y, et al. S2554X mutation in the filaggrin gene is associated with allergen sensitization in the Japanese population. J Allergy Clin Immunol. 2010;125:498–500. doi: 10.1016/j.jaci.2009.10.062. [DOI] [PubMed] [Google Scholar]
- 45.Marenholz I, et al. An interaction between filaggrin mutations and early food sensitization improves the prediction of childhood asthma. J Allergy Clin Immunol. 2009;123:911–916. doi: 10.1016/j.jaci.2009.01.051. [DOI] [PubMed] [Google Scholar]
- 46.Ying S, Meng Q, Corrigan CJ, Lee TH. Lack of filaggrin expression in the human bronchial mucosa. J Allergy Clin Immunol. 2006;118:1386–1388. doi: 10.1016/j.jaci.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 47.De Benedetto A, Qualia CM, Baroody FM, Beck LA. Filaggrin expression in oral, nasal, and esophageal mucosa. J Invest Dermatol. 2008;128:1594–1597. doi: 10.1038/sj.jid.5701208. [DOI] [PubMed] [Google Scholar]
- 48.McLean WH, Palmer CN, Henderson J, Kabesch M, Weidinger S, Irvine AD. Filaggrin variants confer susceptibility to asthma. J Allergy Clin Immunol. 2008;121:1294–1295. doi: 10.1016/j.jaci.2008.02.039. [DOI] [PubMed] [Google Scholar]
- 49.Fallon PG, et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat Genet. 2009;41:602–608. doi: 10.1038/ng.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lack G, Fox D, Northstone K, Golding J. Factors associated with the development of peanut allergy in childhood. N Engl J Med. 2003;348:977–985. doi: 10.1056/NEJMoa013536. [DOI] [PubMed] [Google Scholar]
- 51.Fox AT, Sasieni P, du Toit G, Syed H, Lack G. Household peanut consumption as a risk factor for the development of peanut allergy. J Allergy Clin Immunol. 2009;123:417–423. doi: 10.1016/j.jaci.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 52.Bisgaard H, et al. Gene-environment interaction in the onset of eczema in infancy: filaggrin loss-of-function mutations enhanced by neonatal cat exposure. PLoS Med. 2008;5:e131. doi: 10.1371/journal.pmed.0050131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilkins MR, et al. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (NY) 1996;14:61–65. doi: 10.1038/nbt0196-61. [DOI] [PubMed] [Google Scholar]
- 54.Howell MD, et al. Th2 cytokines act on S100/A11 to downregulate keratinocyte differentiation. J Invest Dermatol. 2008;128:2248–2258. doi: 10.1038/jid.2008.74. [DOI] [PubMed] [Google Scholar]
- 55.Eckert RL, Broome AM, Ruse M, Robinson N, Ryan D, Lee K. S100 proteins in the epidermis. J Invest Dermatol. 2004;123:23–33. doi: 10.1111/j.0022-202X.2004.22719.x. [DOI] [PubMed] [Google Scholar]
- 56.Broccardo CJ, Mahaffey SB, Strand M, Reisdorph NA, Leung DY. Peeling off the layers: skin taping and a novel proteomics approach to study atopic dermatitis. J Allergy Clin Immunol. 2009;124:1113–1115. doi: 10.1016/j.jaci.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Broccardo CJ, et al. Comparative proteomic profiling of patients with atopic dermatitis based on history of eczema herpeticum infection and Staphylococcus aureus colonization. J Allergy Clin Immunol. 2011;127:186–193. doi: 10.1016/j.jaci.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cork MJ, et al. Epidermal barrier dysfunction in atopic dermatitis. J Invest Dermatol. 2009;129:1892–1908. doi: 10.1038/jid.2009.133. [DOI] [PubMed] [Google Scholar]
- 59.O’Regan GM, Sandilands A, McLean WH, Irvine AD. Filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2008;122:689–693. doi: 10.1016/j.jaci.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 61.Rawlings AV, Scott IR, Harding CR, Bowser PA. Stratum corneum moisturization at the molecular level. J Invest Dermatol. 1994;103:731–741. doi: 10.1111/1523-1747.ep12398620. [DOI] [PubMed] [Google Scholar]
- 62.Elias PM. Therapeutic implications of a barrier-based pathogenesis of atopic dermatitis. Ann Dermatol. 2010;22:245–254. doi: 10.5021/ad.2010.22.3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miajlovic H, Fallon PG, Irvine AD, Foster TJ. Effect of filaggrin breakdown products on growth of and protein expression by Staphylococcus aureus. J Allergy Clin Immunol. 2010;126:1184–1190. doi: 10.1016/j.jaci.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Flohr C, et al. Filaggrin loss-of-function mutations are associated with early-onset eczema, eczema severity and transepidermal water loss at 3 months of age. Br J Dermatol. 2010;163:1333–1336. doi: 10.1111/j.1365-2133.2010.10068.x. [DOI] [PubMed] [Google Scholar]
- 65.Gupta J, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity. J Allergy Clin Immunol. 2008;121:725–730. doi: 10.1016/j.jaci.2007.12.1161. [DOI] [PubMed] [Google Scholar]
- 66.Jakasa I, et al. Skin barrier function in healthy subjects and patients with atopic dermatitis in relation to filaggrin loss-of-function mutations. J Invest Dermatol. 2011;131:540–542. doi: 10.1038/jid.2010.307. [DOI] [PubMed] [Google Scholar]
- 67.Jin H, He R, Oyoshi M, Geha RS. Animal models of atopic dermatitis. J Invest Dermatol. 2009;129:31–40. doi: 10.1038/jid.2008.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol. 2009;124:485–493. doi: 10.1016/j.jaci.2009.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127:2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- 70.De Benedetto A, et al. Tight junctions defects in atopic dermatitis. J Allergy Clin Immunol. 2011 Dec 14; doi: 10.1016/j.jaci.2010.10.018. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Furuse M, et al. Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol. 2002;156:1099–1111. doi: 10.1083/jcb.200110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Howell MD, et al. Cathelicidin deficiency predisposes to eczema herpeticum. J Allergy Clin Immunol. 2006;117:836–841. doi: 10.1016/j.jaci.2005.12.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Benedetto A, et al. Reductions in Claudin-1 may enhance susceptibility to HSV-1infections in atopic dermatitis. J Allergy Clin Immunol. 2011 doi: 10.1016/j.jaci.2011.02.014. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms. J Allergy Clin Immunol. 2008;121:1337–1343. doi: 10.1016/j.jaci.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu YJ. Thymic stromal lymphopoietin: master switch for allergic inflammation. J Exp Med. 2006;203:269–273. doi: 10.1084/jem.20051745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ziegler SF, Artis D. Sensing the outside world: TSLP regulates barrier immunity. Nat Immunol. 2010;11:289–293. doi: 10.1038/ni.1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beck LA, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124:260–269. doi: 10.1016/j.jaci.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oyoshi MK, Larson RP, Ziegler SF, Geha RS. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J Allergy Clin Immunol. 2010;126:976–984. doi: 10.1016/j.jaci.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu WH, et al. Thymic stromal lymphopoietin-activated invariant natural killer T cells trigger an innate allergic immune response in atopic dermatitis. J Allergy Clin Immunol. 2010;126:290–299. doi: 10.1016/j.jaci.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 80.Harada M, et al. Functional analysis of the thymic stromal lymphopoietin variants in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 2009;40:368–374. doi: 10.1165/rcmb.2008-0041OC. [DOI] [PubMed] [Google Scholar]
- 81.Leung DY. Our evolving understanding of the functional role of filaggrin in atopic dermatitis. J Allergy Clin Immunol. 2009;124:494–495. doi: 10.1016/j.jaci.2009.07.041. [DOI] [PubMed] [Google Scholar]
- 82.Jung T, Stingl G. Atopic dermatitis: therapeutic concepts evolving from new pathophysiologic insights. J Allergy Clin Immunol. 2008;122:1074–1081. doi: 10.1016/j.jaci.2008.09.042. [DOI] [PubMed] [Google Scholar]
- 83.Ou LS, Goleva E, Hall C, Leung DY. T regulatory cells in atopic dermatitis and subversion of their activity by superantigens. J Allergy Clin Immunol. 2004;113:756–763. doi: 10.1016/j.jaci.2004.01.772. [DOI] [PubMed] [Google Scholar]
- 84.Cardona ID, Goleva E, Ou LS, Leung DY. Staphylococcal enterotoxin B inhibits regulatory T cells by inducing glucocorticoid-induced TNF receptor-related protein ligand on monocytes. J Allergy Clin Immunol. 2006;117:688–695. doi: 10.1016/j.jaci.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 85.Hijnen D, et al. Cyclosporin A reduces CD4(+)CD25(+) regulatory T-cell numbers in patients with atopic dermatitis. J Allergy Clin Immunol. 2009;124:856–858. doi: 10.1016/j.jaci.2009.07.056. [DOI] [PubMed] [Google Scholar]
- 86.Verhagen J, et al. Absence of T-regulatory cell expression and function in atopic dermatitis skin. J Allergy Clin Immunol. 2006;117:176–183. doi: 10.1016/j.jaci.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 87.Toda M, et al. Polarized in vivo expression of IL-11 and IL-17 between acute and chronic skin lesions. J Allergy Clin Immunol. 2003;111:875–881. doi: 10.1067/mai.2003.1414. [DOI] [PubMed] [Google Scholar]
- 88.He R, Oyoshi MK, Jin H, Geha RS. Epicutaneous antigen exposure induces a Th17 response that drives airway inflammation after inhalation challenge. Proc Natl Acad Sci USA. 2007;104:15817–15822. doi: 10.1073/pnas.0706942104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eyerich K, et al. IL-17 in atopic eczema: linking allergen-specific adaptive and microbial-triggered innate immune response. J Allergy Clin Immunol. 2009;123:59–66. doi: 10.1016/j.jaci.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 90.He R, et al. Exaggerated IL-17 response to epicutaneous sensitization mediates airway inflammation in the absence of IL-4 and IL-13. J Allergy Clin Immunol. 2009;124:761–770. doi: 10.1016/j.jaci.2009.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guttman-Yassky E, et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J Immunol. 2008;181:7420–7427. doi: 10.4049/jimmunol.181.10.7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nograles KE, et al. Atopic dermatitis keratinocytes exhibit normal T(H)17 cytokine responses. J Allergy Clin Immunol. 2010;125:744–746. doi: 10.1016/j.jaci.2009.12.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ong PY, et al. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N Engl J Med. 2002;347:1151–1160. doi: 10.1056/NEJMoa021481. [DOI] [PubMed] [Google Scholar]
- 94.Howell MD, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24:341–348. doi: 10.1016/j.immuni.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 95.Nograles KE, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–1252. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF. Cellular basis of itch sensation. Science. 2009;325:1531–1534. doi: 10.1126/science.1174868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sonkoly E, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117:411–417. doi: 10.1016/j.jaci.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 98.Dillon SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 99.Bilsborough J, et al. IL-31 is associated with cutaneous lymphocyte antigen-positive skin homing T cells in patients with atopic dermatitis. J Allergy Clin Immunol. 2006;117:418–425. doi: 10.1016/j.jaci.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 100.Raap U, et al. Correlation of IL-31 serum levels with severity of atopic dermatitis. J Allergy Clin Immunol. 2008;122:421–423. doi: 10.1016/j.jaci.2008.05.047. [DOI] [PubMed] [Google Scholar]
- 101.Salt BH, Boguniewicz M, Leung DY. Severe refractory atopic dermatitis in adults is highly atopic. J Allergy Clin Immunol. 2007;119:508–509. doi: 10.1016/j.jaci.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 102.Bunikowski R, et al. Prevalence and role of serum IgE antibodies to the Staphylococcus aureus-derived superantigens SEA and SEB in children with atopic dermatitis. J Allergy Clin Immunol. 1999;103:119–124. doi: 10.1016/s0091-6749(99)70535-x. [DOI] [PubMed] [Google Scholar]
- 103.De Benedetto A, Agnihothri R, McGirt LY, Bankova LG, Beck LA. Atopic dermatitis: a disease caused by innate immune defects? J Invest Dermatol. 2009;129:14–30. doi: 10.1038/jid.2008.259. [DOI] [PubMed] [Google Scholar]
- 104.Kisich KO, Carspecken CW, Fieve S, Boguniewicz M, Leung DY. Defective killing of Staphylococcus aureus in atopic dermatitis is associated with reduced mobilization of human beta-defensin-3. J Allergy Clin Immunol. 2008;122:62–68. doi: 10.1016/j.jaci.2008.04.022. [DOI] [PubMed] [Google Scholar]
- 105.Cho SH, Strickland I, Boguniewicz M, Leung DY. Fibronectin and fibrinogen contribute to the enhanced binding of Staphylococcus aureus to atopic skin. J Allergy Clin Immunol. 2001;108:269–274. doi: 10.1067/mai.2001.117455. [DOI] [PubMed] [Google Scholar]
- 106.Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DY. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol. 2010;125:39–49. doi: 10.1016/j.jaci.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Skov L, Olsen JV, Giorno R, Schlievert PM, Baadsgaard O, Leung DY. Application of Staphylococcal enterotoxin B on normal and atopic skin induces up-regulation of T cells by a superantigen-mediated mechanism. J Allergy Clin Immunol. 2000;105:820–826. doi: 10.1067/mai.2000.105524. [DOI] [PubMed] [Google Scholar]
- 108.Leung DY, et al. Presence of IgE antibodies to staphylococcal exotoxins on the skin of patients with atopic dermatitis. Evidence for a new group of allergens. J Clin Invest. 1993;92:1374–1380. doi: 10.1172/JCI116711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bunikowski R, et al. Evidence for a disease-promoting effect of Staphylococcus aureus-derived exotoxins in atopic dermatitis. J Allergy Clin Immunol. 2000;105:814–819. doi: 10.1067/mai.2000.105528. [DOI] [PubMed] [Google Scholar]
- 110.Schlievert PM, Case LC, Strandberg KL, Abrams BB, Leung DY. Superantigen profile of Staphylococcus aureus isolates from patients with steroid-resistant atopic dermatitis. Clin Infect Dis. 2008;46:1562–1567. doi: 10.1086/586746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Travers JB, et al. Infected atopic dermatitis lesions contain pharmacologic amounts of lipoteichoic acid. J Allergy Clin Immunol. 2010;125:146–152. doi: 10.1016/j.jaci.2009.09.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boguniewicz M, Sampson H, Leung SB, Harbeck R, Leung DY. Effects of cefuroxime axetil on Staphylococcus aureus colonization and superantigen production in atopic dermatitis. J Allergy Clin Immunol. 2001;108:651–652. doi: 10.1067/mai.2001.118598. [DOI] [PubMed] [Google Scholar]
- 113.Wang R, et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med. 2007;13:1510–1514. doi: 10.1038/nm1656. [DOI] [PubMed] [Google Scholar]
- 114.Wollenberg A, Wetzel S, Burgdorf WH, Haas J. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667–674. doi: 10.1016/j.jaci.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 115.Peng WM, et al. Risk factors of atopic dermatitis patients for eczema herpeticum. J Invest Dermatol. 2007;127:1261–1263. doi: 10.1038/sj.jid.5700657. [DOI] [PubMed] [Google Scholar]
- 116.Hata TR, et al. History of eczema herpeticum is associated with the inability to induce human beta-defensin (HBD)-2, HBD-3 and cathelicidin in the skin of patients with atopic dermatitis. Br J Dermatol. 2010;163:659–661. doi: 10.1111/j.1365-2133.2010.09892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jensen JM, et al. Different effects of pimecrolimus and betamethasone on the skin barrier in patients with atopic dermatitis. J Allergy Clin Immunol. 2009;123:1124–1133. doi: 10.1016/j.jaci.2009.03.032. [DOI] [PubMed] [Google Scholar]
- 118.Kim BE, et al. TNF-alpha down-regulates filaggrin and loricrin through c-Jun N-terminal kinase: Role for TNF-alpha antagonists to improve skin barrier. J Invest Dermatol. 2011 Feb 24; doi: 10.1038/jid.2011.24. [Epub ahead of print] PMID: 21346775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Searing DA, Leung DY. Vitamin D in atopic dermatitis, asthma and allergic diseases. Immunol Allergy Clin North Am. 2010;30:397–409. doi: 10.1016/j.iac.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol. 2008;122:261–266. doi: 10.1016/j.jaci.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hata TR, et al. Administration of oral vitamin D induces cathelicidin production in atopic individuals. J Allergy Clin Immunol. 2008;122:829–831. doi: 10.1016/j.jaci.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sidbury R, Sullivan AF, Thadhani RI, Camargo CA., Jr Randomized controlled trial of vitamin D supplementation for winter-related atopic dermatitis in Boston: a pilot study. Br J Dermatol. 2008;159:245–247. doi: 10.1111/j.1365-2133.2008.08601.x. [DOI] [PubMed] [Google Scholar]
- 123.Matheson EM, Mainous AG, 3rd, Hueston WJ, Diaz VA, Everett CJ. Vitamin D and methicillin-resistant Staphylococcus aureus nasal carriage. Scand J Infect Dis. 2010;42:455–460. doi: 10.3109/00365541003602049. [DOI] [PubMed] [Google Scholar]
- 124.Otto M. Targeted immunotherapy for staphylococcal infections: focus on anti-MSCRAMM antibodies. BioDrugs. 2008;22:27–36. doi: 10.2165/00063030-200822010-00003. [DOI] [PubMed] [Google Scholar]
- 125.Stranger-Jones YK, Bae T, Schneewind O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2006;103:16942–16947. doi: 10.1073/pnas.0606863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clarke SR, et al. Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J Infect Dis. 2006;193:1098–1108. doi: 10.1086/501471. [DOI] [PubMed] [Google Scholar]
- 127.Weidenmaier C, et al. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat Med. 2004;10:243–245. doi: 10.1038/nm991. [DOI] [PubMed] [Google Scholar]
- 128.Harro C, et al. Safety and immunogenicity of a novel Staphylococcus aureus vaccine: results from the first study of the vaccine dose range in humans. Clin Vaccine Immunol. 2010;17:1868–1874. doi: 10.1128/CVI.00356-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Buonpane RA, et al. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat Med. 2007;13:725–729. doi: 10.1038/nm1584. [DOI] [PubMed] [Google Scholar]
- 130.Yang X, et al. Neutralization of multiple staphylococcal superantigens by a single-chain protein consisting of affinity-matured, variable domain repeats. J Infect Dis. 2008;198:344–348. doi: 10.1086/589776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Howell MD, Jones JF, Kisich KO, Streib JE, Gallo RL, Leung DY. Selective killing of vaccinia virus by LL-37: implications for eczema vaccinatum. J Immunol. 2004;172:1763–1767. doi: 10.4049/jimmunol.172.3.1763. [DOI] [PubMed] [Google Scholar]
- 132.Savage PB, Li C, Taotafa U, Ding B, Guan Q. Antibacterial properties of cationic steroid antibiotics. FEMS Microbiol Lett. 2002;217:1–7. doi: 10.1111/j.1574-6968.2002.tb11448.x. [DOI] [PubMed] [Google Scholar]
- 133.Ding B, Taotofa U, Orsak T, Chadwell M, Savage PB. Synthesis and characterization of peptide-cationic steroid antibiotic conjugates. Org Lett. 2004;6:3433–3436. doi: 10.1021/ol048845t. [DOI] [PubMed] [Google Scholar]
- 134.Howell MD, et al. Ceragenins: a class of antiviral compounds to treat orthopox infections. J Invest Dermatol. 2009;129:2668–2675. doi: 10.1038/jid.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]