

Abstract
Th17 cells have recently emerged as a major player in inflammatory and autoimmune diseases via the production of pro-inflammatory cytokines IL-17, IL-17F, and IL-22. The differentiation of Th17 cells and the associated cytokine production is directly controlled by RORγt. Here we show that ursolic acid (UA), a small molecule present in herbal medicine, selectively and effectively inhibits the function of RORγt, resulting in greatly decreased IL-17 expression in both developing and differentiated Th17 cells. In addition, treatment with UA ameliorated experimental autoimmune encephalomyelitis. The results thus suggest UA as a valuable drug candidate or leading compound for developing treatments of Th17-mediated inflammatory diseases and cancer.
Keywords: Cytokine, Immunology, Inflammation, Nuclear Receptors, Transcription Regulation, RORg, Th17
Introduction
Th17 cells have been recently discovered as the third effector CD4+ T helper subset (1, 2). Th17 cells produce IL-17, IL-17F, and IL-22 (3, 4). Although Th17 cells play important roles in host defense against bacterial and fungal infections, they have been also linked to many immune-related diseases, including psoriasis, rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases, periodontal diseases, and asthma/airway inflammatory diseases (4, 5). Anti-IL-17 was recently shown to have good efficacy in treatment of multiple human diseases (6).
In Th17 cells, the transcription of IL-17 and IL-17F is mediated by Th17-specific transcriptional regulators RORγt6 and RORα, although the latter plays a less significant role in mice (7, 8). Mice deficient in RORγ and those deficient in both RORγt and RORα are defective in production of IL-17 and IL-17F and are resistant to experimental autoimmune encephalomyelitis (EAE), a mouse model for multiple sclerosis (7, 8). Therefore, developing ROR inhibitors represents a promising therapeutic strategy in treatment of Th17-mediated diseases.
In the current study, we screened a small chemical library and identified ursolic acid (UA), a natural carboxylic acid ubiquitously present in plants, as a strong and selective inhibitor for RORγt function. UA inhibited IL-17 production not only in developing Th17 cells but also in mature Th17 cells. Mice receiving UA were resistant to EAE, indicating that UA can be used for developing treatment of Th17-mediated diseases.
EXPERIMENTAL PROCEDURES
T Cell Analysis
Human and mouse T cell differentiation and retroviral transduction were performed and analyzed by intracellular staining or by quantitative real-time RT-PCR assays as described (8–10). UA (dissolved in DMSO) or DMSO was added into the culture medium for inhibition assays.
Luciferase Reporter Assays
The CNS2-Il17a and RORE reporter constructs were used for Dual-Luciferase reporter assays in EL4 and 293T cells, respectively, as reported (8, 11). The reporter activity was normalized against Renilla luciferase activity.
Co-activator Binding Assays
The effect of UA on the interaction of coactivator peptides with RORγ was determined by terbium-mediated time-resolved fluorescence energy transfer assays using the LanthaScreen TR-FRET from Invitrogen. The experiments were conducted with 50 nm human RORγ LBD-GST (amino acids 250–518) or RORα LBD-GST (amino acids 271–523), 50 nm terbium-anti-GST, and 1.5 μm fluorescein co-activator peptide (GPQTPQAQQKSLLQQLLTE) containing LXXLL motif derived from SRC-1 following the manufacturer's instructions. The binding signals were determined as the ratios of emission 520 nm and emission 495 nm, and the results from three repeats were normalized relative to the binding in the absence of UA. All the reagents including LBD-GST were from Invitrogen. The binding curve was generated with SigmaPlot and followed the equation: y = min + (max − min)/[1 + (1/EC50)hillscope].
EAE
EAE was induced by immunizing mice (five mice/group) twice with 300 μg of MOG35–55 peptide (amino acids 35–55; MEVGWYRSPFSROVHLYRNGK) emulsified in complete Freund's adjuvant followed by pertussis toxin injection and analyzed as described (8). The disease scores were assigned on a scale of 0–5 as follows: 0, none; 1, limp tail or waddling gait with tail tonicity; 2, wobbly gait; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, death. When indicated, DMSO or UA was given to mice at a dose of ∼150 mg/kg of body weight by intraperitoneal (i.p.) injection every other day after first MOG immunization.
Calculations and Statistic Analysis
All our in vitro data were repeated at least 2–5 times with consistent results. When indicated, the statistical significance was determined by Student's t test. (* represents p < 0.05; ** represents p < 0.03; *** represents p < 0.01).
RESULTS AND DISCUSSION
Ursolic Acid Inhibits Th17 Differentiation
In this study, the human Th17 differentiation system was used as the starting point to screen for Th17 inhibitors. We set up high-throughput 96-well plate Th17 cultures in the presence of various compounds. Consistent with previous reports (9, 10), TGF-β, together with IL-1β, IL-6, and IL-23, induced ∼6–12% IL-17+ cells from human cord blood CD4+ naive T cells after 7 days of culture (data not shown). After screening more than 2,000 known bioactive compounds, we identified UA as a Th17 inhibitor (Fig. 1A).
FIGURE 1.

UA dose-dependently inhibits Th17 differentiation. A, the effect of UA on human Th17 differentiation. Left, intracellular staining. Right, real-time RT-PCR (normalized to GAPDH). B, the effect of UA on mouse Th17 cell differentiation. Left, intracellular staining. Right, real-time RT-PCR (normalized to β-actin). The in vitro differentiation was repeated >4 times, and real-time RT-PCR was repeated 2 times with consistent results.
To confirm our above result, we performed in vitro T cell differentiation using naive CD4+ cells in the presence of different amounts of UA and found that UA dose-dependently inhibited both human and mouse Th17 cell development and 2 μm UA inhibited >80% IL-17 expression in Th17 cells (Fig. 1). This result was confirmed by real-time RT-PCR assays (Fig. 1). Interestingly, UA did not alter the mRNA level of RORα, RORγt, RUNX1, and IRF4 in Th17 cells (Fig. 1 and data not shown), which are known to be important transcription regulators in Th17 cytokine expression (12). In addition, UA did not cause noticeable changes of IFN-γ, IL-4, or Foxp3 gene expression in human and mouse Th1, Th2, or iTreg cells, respectively (supplemental Fig. S1).
UA Selectively Inhibits the Function of RORγ
UA has a similar structure to cholesterol and hydroxycholesterols, the putative ligands for ROR factors (13), suggesting that UA may target RORγt and RORα to inhibit Th17 cells. To assess this, RORα or RORγt was retrovirally overexpressed in T cells differentiated under neutral condition. As reported previously (8), both RORα and RORγt induced significant amounts of IL-17 and IL-17F in non-polarized cells (Fig. 2A). 2 μm UA strongly inhibited RORγt-mediated but not RORα-mediated IL-17 and IL-17F expression to almost background level (8).
FIGURE 2.

UA selectively blocks the function of RORγt but not RORα. A, mouse naive CD4+ T cells were differentiated under neutral condition and infected with RORα, RORγt, or control pMIG viruses on day 1. 2 μm UA or DMSO was added 6 h after viral infection. The cells were restimulated on day 4 for intracellular staining (gated on hCD2+ cells). B, 293T cells were transfected with the RORE reporter together with RORα, RORγt, or control plasmids. UA or DMSO was added after transfection, and the cells were harvested next day for Dual-Luciferase activity assays. The data were normalized to an internal control Renilla luciferase. C, the dose-dependent inhibitory results of UA on RORγt/RORα binding to its co-activator peptide or on Th17 differentiation were fitted to a sigmoidal dose-response curve to determine the corresponding IC50 values. x axis, log concentration (nm) of UA. y axis, relative binding of RORγt/RORα to its co-activator peptide or relative percentage of IL-17+ cells. All the assays were repeated at least 2 times with consistent results.
We have previously identified CNS2 as a cis-regulatory element that enhances the Il17 promoter activity in an ROR-dependent manner (8). RORγt-induced CNS2-Il17p reporter activity was abolished by 2 μm UA, whereas the RORα-dependent reporter activity was not affected (supplemental Fig. S2). In addition, we also examined the effect of UA in non-lymphoid cells by measuring the RORE reporter activity in 293T cells (11). The results again demonstrate that UA selectively suppressed the function of RORγt but not RORα (Fig. 2B).
Inhibitory Kinetics of UA on RORγt and Th17 Cells
The transcriptional activity of RORγt is regulated by its co-activators, such as SRC (steroid receptor co-activator), through binding by the LXXLL motif (14). To determine potency of UA, we examined the effect of UA on the interaction of RORγt ligand binding domain (LBD) with an LXXLL motif-containing peptide derived from SRC-1. The results showed that UA dose-dependently inhibited the binding of RORγt-LBD to the LXXLL co-activator peptide (Fig. 2C). Consistent with the results of retroviral overexpression and reporter gene assays, UA did not inhibit the binding of RORα-LBD to the coactivator peptide (Fig. 2C), suggesting UA as an RORγt-specific antagonist. By fitting the inhibitory binding data to a sigmoidal dose-response curve, the IC50 (half-maximal inhibitory concentration) of UA to RORγt was determined to be 0.68 ± 0.1 μm. Using the same method, the IC50 of UA on Th17 cells was determined to be 0.56 ± 0.1 μm (Fig. 2C). The similar IC50 values further suggest RORγt as the direct target of UA in Th17 cells.
UA Inhibits IL-17 Expression in Mature Th17 Cells
Our data thus far established UA as an RORγt-specific inhibitor in Th17 cell differentiation. To assess whether UA can inhibit the production of IL-17 in mature Th17 cells, which is more important in clinical settings, we first generated mature human and mouse Th17 cells from naive CD4+ T cells. After preincubation with UA, the mature Th17 cells were then restimulated with plate-bound anti-CD3 overnight for cytokine secretion measurements. The results demonstrated that UA indeed inhibited the secretion of IL-17 from differentiated Th17 cells of both human and mouse sources (Fig. 3).
FIGURE 3.
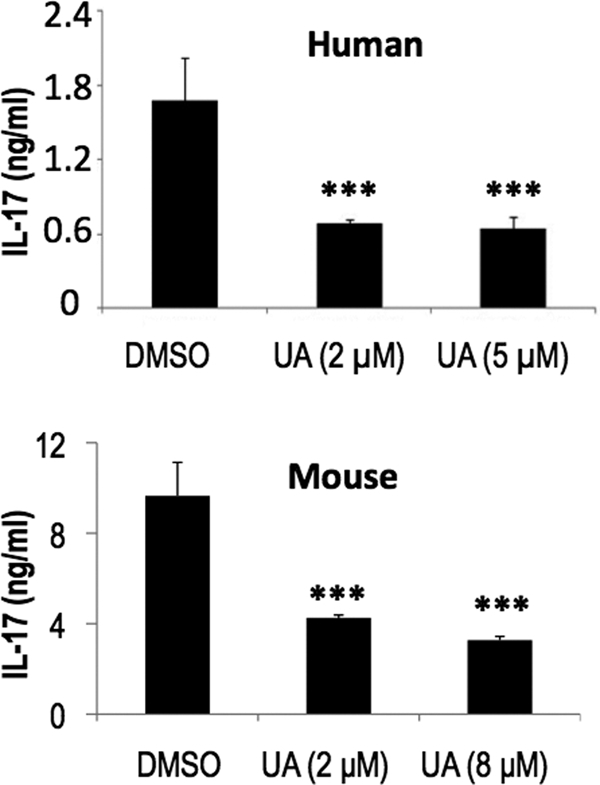
UA suppresses IL-17 production in mature Th17 cells. After being preincubated with UA, the mature human Th17 cells or mouse Th17 cells generated in vitro were then restimulated with anti-CD3 overnight for secreted cytokine analysis in the presence of the indicated amounts of UA. Both human and mouse experiments were repeated 2–3 times with consistent results.
UA Ameliorated MOG-induced EAE in Mice
To investigate the therapeutic potential of UA in Th17-mediated autoimmune diseases, we examined the effect of UA on MOG-induced EAE in mice and found that UA treatment not only delayed the onset of disease in mice (Fig. 4A) but also more significantly ameliorated disease symptom (Fig. 4B) in comparison with the control group. Consistently, the UA-treated mice contained significantly fewer IL-17+ cells as well as IFN-γ+ cells in their central nervous system (Fig. 4C). Furthermore, in both MOG-immunized mice (data not shown) and EAE mice (Fig. 4D), UA treatment also caused a reduction in IL-17 production in the spleen. These data suggest that UA may be used in treatment of Th17-mediated inflammatory diseases.
FIGURE 4.

UA treatment ameliorated EAE disease in mice. For EAE induction, mice were given either DMSO or UA by i.p. injection every other day after the first MOG immunization and monitored daily for clinical symptom development after the second MOG immunization. Results are the combination of two independent EAE experiments, and the disease onset date of DMSO-treated mice was set to day 1 for statistical analysis. A, the percentage of mice that developed EAE disease (n = 10 for DMSO group and n = 8 for UA group). B, the clinical scores of diseased mice (n = 7 for the DMSO group and n = 6 for the UA group). C, the total number of CD4+, IL-17+, and IFN-γ+ cells in the central nervous system of EAE mice. D, the effect of UA on MOG-specific IL-17 production in the spleens of EAE mice.
UA contains many pharmacological activities, including strong hepatoprotective, anti-tumor, and anti-inflammation effects partly through targeting NF-κB and STAT3 (15–19). In Th17 cells, we found that UA did not affect the expression of STAT3 downstream targets, such as IL-21 and RORγt, indicating that STAT3 is not the target in Th17 cells. To confirm this, we performed a Western blot to check the activation of STAT3 in Th17 cells and found that 2 μm UA did not have any effect on IL-6-induced STAT3 phosphorylation (data not shown). Moreover, UA has a relatively high IC50 value for STAT3 and NF-κB and inhibits these two molecules only when used at 25 μm or above (18–20), which is at least 30-fold higher than the IC50 value of UA for Th17 cells (0.56 ± 0.1 μm) and RORγt (0.68 ± 0.1 μm), further excluding them from the targets of UA in Th17 cells. In addition, we also exclude the possibility that UA may inhibit Th17 cells through inducing apoptosis (data not shown), as reported in other cells (16, 20).
As a natural small molecule ubiquitously present in plants and even human diets, UA is relatively non-toxic and is well tolerated orally and topically in both human and rodents. The acute toxicity (LD50) of UA in rodents was determined to be >637 mg/kg for intraperitoneal injection and 8,330 mg/kg for oral administration (21). In addition, UA has been identified as a major effective component in many medical herbs, which have a long history in clinical practice in ancient China and Asian countries (15). Due to its important pharmacological activities, UA has been used for treatment of liver diseases and skin cancer (15). These clinical practices and its relatively low toxicity provide UA a great advantage over other ROR inhibitors recently reported (22, 23) in developing therapeutics against Th17-mediated autoimmune diseases. Considering the broad function of Th17 cells in inflammatory diseases and cancer, it is of interest in assessing whether the therapeutic effects of UA are via inhibition of RORγt or Th17 cell function.
Supplementary Material
Acknowledgments
We thank Dr. Anton Jetten for RORE reporter vectors, Min Xie for assistance with chemical analysis, D. Watry for assistance with FACS analyses and co-activator binding assay, and Gustavo J. Martinez for helping in the RORE reporter gene assays.
A patent disclosure has been filed by M.D. Anderson Cancer Center based on the results of this paper.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- ROR
- RAR-related orphan receptor
- RAR
- retinoic acid receptor
- RORE
- ROR response element
- EAE
- experimental autoimmune encephalomyelitis
- UA
- ursolic acid
- LBD
- ligand binding domain
- SRC
- steroid receptor co-activator
- DMSO
- dimethyl sulfoxide
- MOG
- myelin oligodendrocyte glycoprotein.
REFERENCES
- 1. Park H., Li Z., Yang X. O., Chang S. H., Nurieva R., Wang Y. H., Wang Y., Hood L., Zhu Z., Tian Q., Dong C. (2005) Nat. Immunol. 6, 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harrington L. E., Hatton R. D., Mangan P. R., Turner H., Murphy T. L., Murphy K. M., Weaver C. T. (2005) Nat. Immunol. 6, 1123–1132 [DOI] [PubMed] [Google Scholar]
- 3. Dong C. (2008) Nat. Rev. Immunol. 8, 337–348 [DOI] [PubMed] [Google Scholar]
- 4. Korn T., Bettelli E., Oukka M., Kuchroo V. K. (2009) Annu. Rev. Immunol. 27, 485–517 [DOI] [PubMed] [Google Scholar]
- 5. Tesmer L. A., Lundy S. K., Sarkar S., Fox D. A. (2008) Immunol. Rev. 223, 87–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hueber W., Patel D. D., Dryja T., Wright A. M., Koroleva I., Bruin G., Antoni C., Draelos Z., Gold M. H., Durez P., Tak P. P., Gomez-Reino J. J., Foster C. S., Kim R. Y., Samson C. M., Falk N. S., Chu D. S., Callanan D., Nguyen Q. D., Rose K., Haider A., Di Padova F. (2010) Sci. Transl. Med. 2, 52ra72 [DOI] [PubMed] [Google Scholar]
- 7. Ivanov II, McKenzie B. S., Zhou L., Tadokoro C. E., Lepelley A., Lafaille J. J., Cua D. J., Littman D. R. (2006) Cell 126, 1121–1133 [DOI] [PubMed] [Google Scholar]
- 8. Yang X. O., Pappu B. P., Nurieva R., Akimzhanov A., Kang H. S., Chung Y., Ma L., Shah B., Panopoulos A. D., Schluns K. S., Watowich S. S., Tian Q., Jetten A. M., Dong C. (2008) Immunity 28, 29–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manel N., Unutmaz D., Littman D. R. (2008) Nat. Immunol. 9, 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Volpe E., Servant N., Zollinger R., Bogiatzi S. I., Hupé P., Barillot E., Soumelis V. (2008) Nat. Immunol. 9, 650–657 [DOI] [PubMed] [Google Scholar]
- 11. Yang X. O., Nurieva R., Martinez G. J., Kang H. S., Chung Y., Pappu B. P., Shah B., Chang S. H., Schluns K. S., Watowich S. S., Feng X. H., Jetten A. M., Dong C. (2008) Immunity 29, 44–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou L., Littman D. R. (2009) Curr. Opin. Immunol. 21, 146–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin L., Martynowski D., Zheng S., Wada T., Xie W., Li Y. (2010) Mol. Endocrinol. 24, 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xie H., Sadim M. S., Sun Z. (2005) J. Immunol. 175, 3800–3809 [DOI] [PubMed] [Google Scholar]
- 15. Liu J. (1995) J. Ethnopharmacol. 49, 57–68 [DOI] [PubMed] [Google Scholar]
- 16. Ikeda Y., Murakami A., Ohigashi H. (2008) Mol. Nutr. Food Res. 52, 26–42 [DOI] [PubMed] [Google Scholar]
- 17. Huang H. C., Huang C. Y., Lin-Shiau S. Y., Lin J. K. (2009) Mol. Carcinog. 48, 517–531 [DOI] [PubMed] [Google Scholar]
- 18. Shishodia S., Majumdar S., Banerjee S., Aggarwal B. B. (2003) Cancer Res. 63, 4375–4383 [PubMed] [Google Scholar]
- 19. Pathak A. K., Bhutani M., Nair A. S., Ahn K. S., Chakraborty A., Kadara H., Guha S., Sethi G., Aggarwal B. B. (2007) Mol. Cancer Res. 5, 943–955 [DOI] [PubMed] [Google Scholar]
- 20. Lauthier F., Taillet L., Trouillas P., Delage C., Simon A. (2000) Anticancer Drugs 11, 737–745 [DOI] [PubMed] [Google Scholar]
- 21. Lee A. W., Chen T. L., Shih C. M., Huang C. Y., Tsao N. W., Chang N. C., Chen Y. H., Fong T. H., Lin F. Y. (2010) J. Agric. Food Chem. 58, 12941–12949 [DOI] [PubMed] [Google Scholar]
- 22. Solt L. A., Kumar N., Nuhant P., Wang Y., Lauer J. L., Liu J., Istrate M. A., Kamenecka T. M., Roush W. R., Vidović D., Schürer S. C., Xu J., Wagoner G., Drew P. D., Griffin P. R., Burris T. P. (2011) Nature 472, 491–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huh J. R., Leung M. W., Huang P., Ryan D. A., Krout M. R., Malapaka R. R., Chow J., Manel N., Ciofani M., Kim S. V., Cuesta A., Santori F. R., Lafaille J. J., Xu H. E., Gin D. Y., Rastinejad F., Littman D. R. (2011) Nature 472, 486–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.