

Abstract
Genetic investigation of crescentic glomerulonephritis (Crgn) susceptibility in the Wistar Kyoto rat, a strain uniquely susceptible to nephrotoxic nephritis (NTN), allowed us to positionally clone the activator protein-1 transcription factor Jund as a susceptibility gene associated with Crgn. To study the influence of Jund deficiency (Jund-/-) on immune-mediated renal disease, susceptibility to accelerated NTN was examined in Jund-/- mice and C57BL/6 wild-type (WT) controls. Jund-/- mice showed exacerbated glomerular crescent formation and macrophage infiltration, 10 days after NTN induction. Serum urea levels were also significantly increased in the Jund-/- mice compared with the WT controls. There was no evidence of immune response differences between Jund-/- and WT animals because the quantitative immunofluorescence for sheep and mouse IgG deposition in glomeruli was similar. Because murine Jund was inactivated by replacement with a bacterial LacZ reporter gene, we then investigated its glomerular expression by IHC and found that the Jund promoter is mainly active in Jund-/- podocytes. Furthermore, cultured glomeruli from Jund-/- mice showed relatively increased expression of vascular endothelial growth factor A (Vegfa), Cxcr4, and Cxcl12, well-known HIF target genes. Accordingly, small-interfering RNA–mediated JUND knockdown in conditionally immortalized human podocyte cell lines led to increased VEGFA and HIF1A expression. Our findings suggest that deficiency of Jund may cause increased oxidative stress in podocytes, leading to altered VEGFA expression and subsequent glomerular injury in Crgn.
JunD is a ubiquitously expressed DNA-binding protein, one of the components of the activator protein-1 (AP-1) transcription factor. AP-1 members are dimeric complexes composed of Jun (c-Jun, JunB, v-Jun, and JunD), Fos (c-Fos, FosB, Fra-1, and Fra-2), and ATF (ATF1-4, ATF-6, β-ATF, and ATFx) proteins. AP-1 can either activate or repress transcription, depending on the specific components of the complex and the cellular environment.1 Jund expression and JunD protein-protein interactions modulate tumor angiogenesis, cellular differentiation, proliferation, and apoptosis.1, 2 An additional function of Jund in the control of oxidative stress and angiogenic switch was recently discovered.3, 4, 5 Moreover, Jund-mediated oxidative stress has played a pivotal role in systemic regulation of insulin that affects lifespan6 and in tumor development by altering the microenvironment.7
Glomerulonephritis is a major cause of kidney failure in humans. Crescentic glomerulonephritis (Crgn) is the most severe form and may be seen with immune complex deposition in glomeruli, with antibodies directed against the glomerular basement membrane, or in systemic vasculitis. Well-documented variation in susceptibility to Crgn between inbred strains of rodents has strongly suggested the influence of genetic predisposing factors in animal models.8, 9, 10, 11, 12 The rat nephrotoxic nephritis (NTN) model leads to severe Crgn in the Wistar Kyoto (WKY) rat, whereas the Lewis rat that shares the same major histocompatibility complex haplotype is NTN resistant.8, 13 This model is highly reproducible and heritable, and the increased genetic susceptibility of the WKY rat is explained by both circulating and renal intrinsic factors.10, 14 By using genomewide linkage and glomerular expression analyses in NTN-susceptible WKY and Lewis rats, Jund was previously positionally cloned as a susceptibility gene associated with Crgn.9 Jund is markedly overexpressed in the NTN-susceptible WKY glomeruli and bone marrow–derived macrophages (BMDMs) in basal conditions. Small-interfering RNA (siRNA)–mediated Jund knockdown in WKY BMDMs led to decreased macrophage activation, suggesting a novel role for Jund in macrophage activation,9 an important hallmark of the pathophysiological features of glomerulonephritis. Although Jund is not differentially expressed between WKY and Lewis mesangial cells, our microarray analysis showed that it was markedly overexpressed in the basal (without NTN induction) and nephritic WKY glomeruli 10 days after nephrotoxic serum (NTS) injection.9 This finding suggests that Jund expression by glomerular, other than mesangial, cells contributes to glomerular crescent formation. To elucidate the cellular mechanisms by which Jund contributes to glomerular inflammation, we studied the effect of targeted deletion of Jund in the accelerated NTN model in the mice.
Herein, we report that Jund-deficient (Jund-/-) mice show exacerbated glomerular crescent formation, 10 days after NTN induction, with significantly increased serum urea levels when compared with WT controls. We show that Jund promoter is mainly active in Jund-/- podocytes and that cultured glomeruli from Jund-/- mice showed increased expression of vascular endothelial growth factor A (Vegfa), Cxcr4, and Cxcl12. Consistent with these findings, siRNA-mediated JUND knockdown in conditionally immortalized human podocyte cell lines led to increased VEGFA and HIF1A expression, suggesting that deficiency of Jund may cause increased oxidative stress in podocytes, leading to glomerular injury in Crgn.
Materials and Methods
Mice
The Jund-/- colony was maintained through the breeding of heterozygous animals on a C57BL/6–129 mixed genetic background. Because male Jund-/- mice were previously reported to be sterile,15 6- to 10-week-old Jund-/- and wild-type (WT) female littermates were used in all procedures. Genotyping was performed from mice tail snips by using Jund- and LacZ-specific primers, as previously described.15 All mice were housed with free access to food and water, according to institutional guidelines.
Induction of Accelerated NTN
Nephrotoxic globulin was prepared as previously described.16 Briefly, a sheep was immunized with murine glomerular lysate initially in complete and then incomplete Freund's adjuvant (Sigma, Dorset, UK). After ammonium sulfate precipitation, a γ-globulin–enriched fraction of immune serum was heat inactivated. The precipitate was extensively dialyzed against sterile PBS (Invitrogen, Paisley, UK) and stored at -80°C. The endotoxin content of the nephrotoxic globulin was undetectable after using a Biowhittaker QCL-1000 LPS assay kit (Biowhittaker, Walkersville, MD). Mice were immunized i.p. with 0.2 mg of sheep IgG in a v/v mix with complete Freund's adjuvant (Sigma). Five days after the immunization, 5 mg of NTS was injected i.v. to WT and Jund-/- mice (n = 10 per group). The mice were monitored clinically; 10 days after NTS injection, the mice were anesthetized with i.p. midazolam and fentanyl and exsanguinated before harvesting the kidneys. Serum was also collected from all mice on day 10 for urea nitrogen determination as an indicator of renal function.
Renal Function Assessment
The serum urea concentration was measured using an assay based on the hydrolysis of urea to ammonium and subsequent oxidization of NADH, according to the manufacturer's instructions (R-biopharm, Glasgow, UK). Serum albumin levels were assessed by a mouse albumin enzyme-linked immunosorbent assay (Cambridge Bioscience, Cambridge, UK) that uses a competitive enzyme immunoassay with a polyclonal antibody specific for mouse albumin.
Histological Studies and Quantitative Immunofluorescence
Kidneys were fixed in 10% formal saline, processed, embedded in paraffin wax, and stained with Periodic acid-Schiff reagent. Glomeruli were assessed for histological abnormalities on a semiquantitative scale from 0 to 4, where 0 is normal; 1 and 2, glomerular tuft hypercellularity; and 3 and 4, glomerular tuft hypercellularity with crescents. Glomerular crescents were defined as glomeruli containing two or more layers of cells in the Bowman's space. One hundred glomeruli were counted per sample, and results are presented as percentage of glomerular crescents.
For immunofluorescence, kidneys were embedded in OCT (Cellpath, Newtown, UK), snap frozen in isopentane cooled with liquid nitrogen, and stored at −80°C. Sections (5-μm thick) were fixed in acetone-methanol for 10 minutes.
Glomerular mouse and sheep IgG was visualized by direct immunofluorescence using fluorescein isothiocyanate (FITC)–conjugated goat anti-mouse IgG (Fc specific) and FITC–conjugated monoclonal mouse anti-sheep IgG clone GT34 (Sigma).
Glomerular C3 staining was assessed by direct immunofluorescence using a FITC–conjugated goat anti-mouse C3 IgG (MP Biomedicals, Cambridge, UK), as previously described.17 To quantitate immunofluorescence, sections were examined at ×100 magnification using an Olympus BX4 fluorescence microscope (Olympus Optical, London, UK) and a Photonic Science Color Coolview camera (Photonic Science, Robertsbridge, UK). Samples from each experiment were stained on the same occasion and measured together. Images of the sections were captured by the image analysis equipment, and the total pixel intensity for each glomerulus was measured and averaged for 20 glomeruli per section. The arbitrary units of fluorescence correspond to the mean pixel intensity for 20 glomeruli for each mouse.
For immunoperoxidase staining of macrophages, snap-frozen 5-μm kidney sections were stained with FA11 (monoclonal rat anti-mouse CD68; AbD Serotec, Kidlington, UK). Polyclonal biotinylated rabbit anti-rat IgG was used as a secondary antibody (Stratech, Newmarket Suffolk, UK). Peroxidase-conjugated avidin-biotin complex (Vector Laboratories, Peterborough, UK) reagent was used to detect specific CD68 staining in the renal cortex.
For β-galactosidase IHC, paraffin sections were subjected to antigen retrieval by trypsin digestion for 10 minutes. The primary antibody was anti–β galactosidase (Promega Z378A) at 1:1000, and binding was detected using a Biogenex polymer horseradish peroxidase detection kit.
Splenocyte Proliferation
For splenocyte proliferation, spleens were harvested 7 days after immunization with sheep IgG. After the preparation of single-cell suspensions and red blood cell lysis, cells were cultured at 1 × 106 cells/mL for 72 hours in HL1 serum-free medium (Lonza, Slough, UK), in the presence or absence of 100 μg/mL heat-aggregated sheep IgG or 2 million CD3/CD28 Dynabeads (Invitrogen) per well as a positive control. At 72 hours, 1 μCi/well 3H thymidine was added for 24 hours before assessing splenocyte proliferation using a Wallac 1450 MicroBeta counter.
ELISA for IgG Subclasses
The level of serum IgG subclasses was determined by enzyme-linked immunosorbent assay using goat anti-mouse IgG1, IgG2a, IgG2b, and IgG3 (Southern Biotech, Birmingham, AL). Briefly, Nunc plates precoated with 100 μg/mL sheep IgG (Sigma) were blocked and then incubated with serum collected after NTN induction at different dilutions (1:1000 for IgG1 and 1:500 for IgG2a, IgG2b, and IgG3). After adding goat anti-mouse IgG conjugated to alkaline phosphatase (Southern Biotech), antibody binding was detected using p-nitrophenyl phosphate substrate (Sigma) at an OD of 405.
Glomerular Gene Expression Analysis
Glomeruli were sieved from the cortex of WT and Jund-/- kidneys, as previously described.18 Total RNA was extracted from sieved glomeruli of unmanipulated or NTN-induced Jund-/- mice and WT littermates by using the TRIzol method. Vegfa, Hif1a, Cxcr4, Cxcl12, fibronectin, c-Jun, and c-Fos expression levels were determined by quantitative RT-PCR (RT-qPCR) using forward and reverse primers, as follows: mouse Vegfa, 5′-TGTGATTCTGATAAAATAGACATTGC-3′ (forward) and 5′-TTTTTCCCCCACAATTATTACG-3′ (reverse); mouse Hif1a, 5′-CCCATACAAGGCAGCAGAAA-3′ (forward) and 5′-AGCCACCAGTGTCCAAAAAT-3′ (reverse); Cxcr4, 5′-CCTCAGCTGTTGCTGCATAA-3′ (forward) and 5′-CACCATTTCAGGCTTTGGTT-3′ (reverse); Cxcl12, 5′-CAGAGCCAACGTCAAGCA-3′ (forward) and 5′-AGGTACTCTTGGATCCAC-3′ (reverse); fibronectin, 5′-GCGACTCTGACTGGCCTTAC-3′ (forward) and 5′-CCGTGTAAGGGTCAAAGCAT-3′ (reverse); c-Jun, 5′-TGTCCTGCCCAGTGTTTGTA-3′ (forward) and 5′-GAGGTTGGGGGCTACTTTTC-3′ (reverse); and c-Fos, 5′- CCAGTCCTCACCTCTTCCAG-3′ (forward) and 5′- TCCAGCACCAGGTTAATTCC-3′ (reverse).
Human Podocyte Culture and siRNA Knockdown
Conditionally immortalized human podocytes were a gift from Moin Saleem (University of Bristol, Bristol, UK). These podocytes were derived by incorporating a temperature-sensitive SV40 gene that enables cells to proliferate at the permissive temperature (33°C) and to differentiate at the nonpermissive temperature (37°C). The characteristics of the WT human podocyte cell line were previously reported.19 Cells were cultured for 14 days at the nonpermissive temperature (37°C) in 6 well plates (Nunc) in RPMI 1640 medium with glutamine (Invitrogen, Paisley, UK), supplemented with 10% fetal calf serum (Biosera, East Sussex, UK) and 1% insulin transferrin sodium selenite (Sigma). siRNA knockdown experiments were performed as previously described.9 JUND knockdown and the effect of its knockdown on VEGFA and HIF1A expression were assessed by RT-qPCR by using forward and reverse primers, as follows: JUND, 5′-CTCAAGGACGAGCCACAGAC-3′ (forward) and 5′-GGGTCTTCACTTTCTCTTCCA-3′ (reverse); human VEGFA, 5′-TCCTCACACCATTGAAACCA-3′ (forward) and 5′-TTTTCTCTGCCTCCACAATG-3′ (reverse); and human HIF1A, 5′-GGGAGTTTATCCCTTTTTCG-3′ (forward) and 5′-TTGTGGCTACCACGTACTGC-3′ (reverse).
BMDM Culture, Fc OxyBURST, and Phagocytosis Assays
Femurs from adult Jund-/- and WT mice were isolated and flushed with HBSS. Total BM-derived cells were plated and cultured for 7 days, as previously described.10 For the Fc oxyBURST assay, 106 BMDMs (in triplicate) were suspended in Krebs' Ringer PBS with 1.0 mmol/L Ca2+, 1.5 mmol/L Mg2+, and 5.5 mmol/L glucose; warmed to 37°C; and stimulated with Fc oxyBURST reagent (240 μg/mL; Invitrogen). Individual data points consisting of 10,000 fluorescence events were collected at 0, 15, 45, 75, 90, 105, and 120 seconds in an FACSCalibur after a baseline fluorescence reading was taken to determine the intrinsic fluorescence of unstimulated cells. The percentage of fluorescence BMDMs corresponds to the percentage of activated gated cells after Fc receptor–mediated phagocytosis. Jund-/- and WT BMDMs were also assessed for phagocytosis, as previously described.9 Briefly, latex polystyrene 6.0-μm microspheres (50 beads per macrophage; Polysciences Inc., Warrington, PA) were opsonized with bovine serum albumin–anti–bovine serum albumin IgG (Sigma) and added to macrophages cultured in eight-well glass chamber slides. After staining with Diff-Quick fix (Dade Behring, Newark, DE), 100 macrophages were counted to determine the number of beads ingested per cell.
Statistics
Results describing glomerular injury and immune response are expressed as median (range). All RT-qPCR results and the assessment of glomerular CD68 and CD4+ cells are expressed as mean ± SE. Nonparametric tests of significance were applied throughout. For comparing two groups, the Mann-Whitney U-test was used and differences were considered significant when P < 0.05.
Results
Targeted Jund Deletion and Susceptibility to Crgn in Mice
Jund-/- mice showed marked susceptibility to Crgn on day 10 after the induction of accelerated NTN when compared with age- and sex-matched WT littermates (Figure 1, A–C). All of the WT mice showed glomerular abnormalities, ranging from mild segmental glomerular hypercellularity to occasional crescents. However, there was a marked increase in the glomerular hypercellularity and crescent formation in the Jund-/- mice when compared with WT controls (Figure 1, A and B). The kidney function assessed by measuring serum urea and urine protein levels confirmed the histological observations showing significantly increased serum urea and proteinuria in Jund-/- animals (Figure 1, D and E). We also assessed serum albumin levels and demonstrated reduced serum albumin in Jund-/- animals when compared with WT littermates, consistent with the increased albuminuria (Figure 1F). Glomerular CD68+ and CD4+ cell infiltration showed increased infiltration of positive cells for these markers in the Jund-/- mice when compared with WT littermates (Figure 1G).
Figure 1.
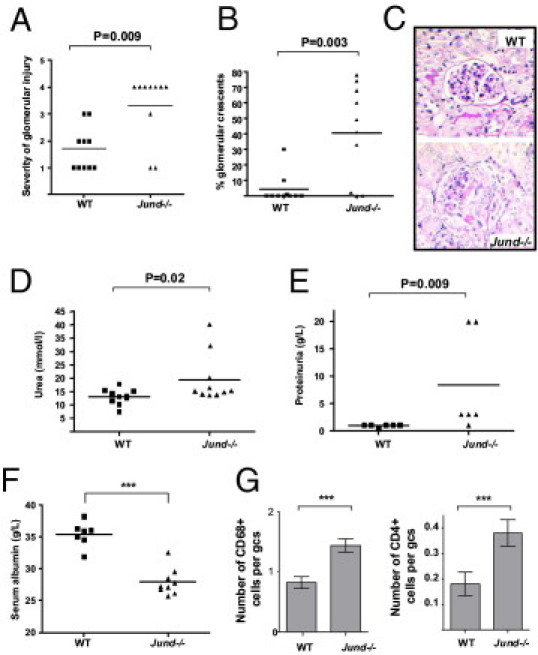
Targeted deletion of Jund leads to severe Crgn in mice. Severity of glomerular injury (scored by assessing glomerular hypercellularity and crescents on a semiquantitative scale, where 0 represents normal) (A), percentage of glomerular crescents (B), and representative H&E-stained glomeruli (C) showing crescent formation in the Jund-/- mice (day 10 after NTS injection). Serum urea (D) and proteinuria (E) levels are significantly increased in Jund-/- animals compared with WT controls 10 days after the injection of NTS. The kidney function assessed by measuring serum albumin also showed a significant reduction in serum albumin in Jund-/- animals when compared with WT controls (F). The numbers of CD68+ and CD4+ cells per glomerular cross section (gcs) were also significantly increased in the Jund-/- mice when compared with WT controls (G). Ten mice were used for all of the parameters, except for proteinuria (six mice used). ***P < 0.001.
Deposited Glomerular IgG and Humoral Immune Response Assessment
Quantitation of sheep IgG deposited on the glomerular basement membrane on day 10 showed no significant difference between Jund-/- and WT littermates (Figure 2, A and C). In addition, as a measure of humoral immune response, glomerular mouse IgG was also quantified on frozen kidney sections and no significant difference was found between the Jund-/- and WT mice (Figure 2, B and C). Serum levels of mouse total IgG specific for sheep IgG were also assessed (Figure 2D) and showed no significant differences between the two groups of mice. Serum levels of IgG subclasses (IgG1, IgG2a, IgG2b, and IgG3) were analyzed by enzyme-linked immunosorbent assay, and we did not find any significantly different levels of these antibodies in the serum samples of Jund-/- mice and WT littermates on day 10 after the induction of the accelerated NTNmice and WT littermates on day 10 after the induction of the accelerated NTN (see Supplemental Figure S1 at http://ajp.amjpathol.org). In addition, we assessed the C3 deposition in the glomeruli of Jund-/- and WT mice on day 10 after NTN induction and did not find any significant difference in the intensity of C3 staining in glomeruli between the two groups (data not shown). Splenocyte proliferation to sheep IgG was determined after preimmunization. There was no significant difference in splenocyte proliferation to antigen between the WT and Jund-/- mice (see Supplemental Figure S2 at http://ajp.amjpathol.org). Taken together, these results show clearly that the increased susceptibility of the Jund-/- mice to accelerated NTN is independent of the glomerular deposition of the nephrotoxic antibodies and the immune response against the implanted sheep IgG in the glomeruli.
Figure 2.
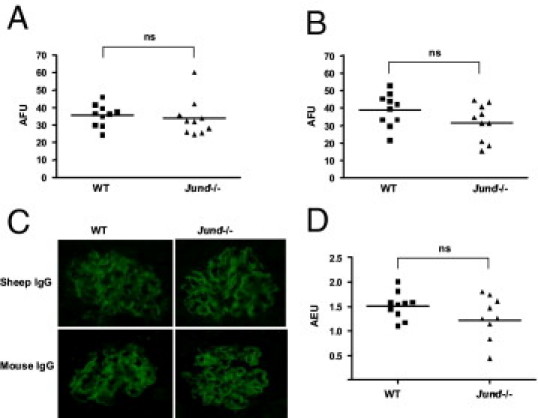
Deposited glomerular IgG and humoral immune response assessment in WT and Jund-/- mice 10 days after accelerated NTN induction. Quantitative immunofluorescence for sheep IgG (A) and mouse IgG (B). Representative glomeruli showing immunofluorescence for sheep and mouse IgG 10 days after the induction of accelerated NTN (C). Serum levels of mouse total IgG specific for sheep IgG (D). Ten mice were used in each group. AFU indicates arbitrary fluorescence unit; AEU, arbitrary enzyme-linked immunosorbent assay unit; ns, nonsignificant.
Jund Expression Is Restricted to Glomerular Podocytes
Given that Jund-targeted deletion was achieved by replacement of Jund with LacZ,15 the site Jund expression was assessed by IHC staining of β-galactosidase within the normal and nephritic kidney sections. Jund promoter is markedly active in glomerular podocytes in the basal and nephritic glomeruli 10 days after the induction of accelerated NTN (Figure 3). Given the previously established role of Jund in regulating oxidative stress and angiogenesis,3, 6 we hypothesized that the absence of Jund causes enhanced oxidative stress characterized by up-regulation of hypoxia-response downstream genes within the glomerulus.
Figure 3.
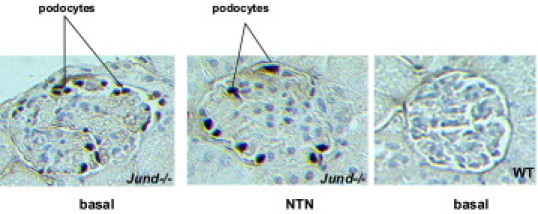
The Jund promoter is active in glomerular podocytes. Jund expression was assessed by IHC staining of β-galactosidase. The Jund promoter is markedly active in glomerular podocytes in the basal and nephritic glomeruli 10 days after the induction of accelerated NTN. WT mice glomeruli were used as a negative control of β-galactosidase staining.
Targeted Deletion of Jund and Glomerular Expression of Hypoxia-Induced Genes
Glomeruli from basal and NTN-induced Jund-/- and WT mice were analyzed for the expression of hypoxia-response downstream genes by RT-qPCR. We showed increased expression of Vegfa, Cxcr4, and Cxcl12, but not fibronectin, in basal Jund-/- glomeruli when compared with WT controls (Figure 4, A–D). NTN induction did not cause a significant difference in the expression of these genes, except for Cxcl12 and fibronectin mRNA levels, which decreased and increased, respectively, in the Jund-/- glomeruli when compared with WT controls (Figure 4, C and D). Furthermore, we assessed whether the targeted deletion of Jund had any effect on the expression of other AP-1 members, such as c-Jun and c-Fos (Figure 4, E and F). Both in basal and nephritic glomeruli, the expression of c-Jun and c-Fos was not different between the WT and Jund-/- individuals, suggesting that targeted deletion of Jund had a direct effect on the relative expression of hypoxia-induced genes.
Figure 4.
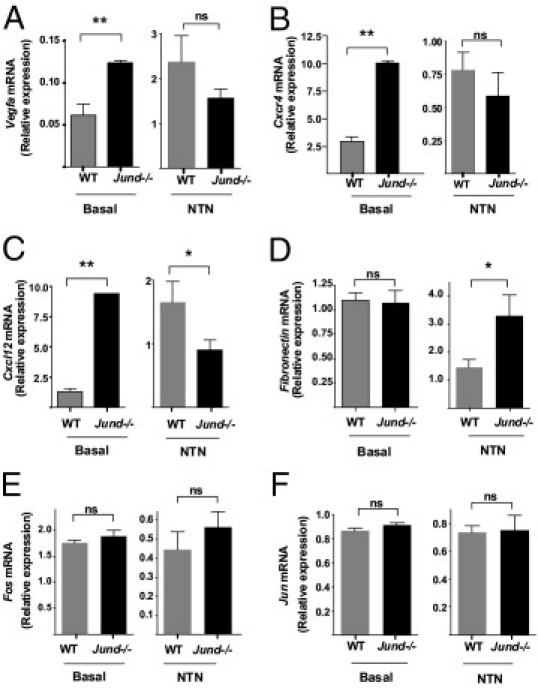
Targeted deletion of Jund and glomerular expression of hypoxia-induced genes. Glomerular expression of Vegfa (A), Cxcr4 (B), Cxcl12 (C), and fibronectin (D) in unmanipulated (basal) and NTN-induced (NTN) WT and Jund-/- mice assessed by RT-qPCR. Targeted deletion of Jund did not significantly affect the expression of other AP-1 members, such as c-Fos (E) and c-Jun (F). Six mice were used in each group. ns indicates nonsignificant. *P < 0.05, **P < 0.01.
Given the previously established role of Jund as a determinant of macrophage activation,9 we investigated whether targeted deletion of Jund had an effect on Fc receptor–dependant and Fc receptor–independent macrophage activation. We measured Fc receptor–mediated oxidative burst (Fc oxyBURST assay) and phagocytosis in WT and Jund-/- BMDMs (see Supplemental Figure S3, A and B, at http://ajp.amjpathol.org). In addition, we also measured Nos2, Tnfa, and Ccl2 expression in WT and Jund-/- BMDMs after stimulation with lipopolysaccharide (see Supplemental Figure S3C at http://ajp.amjpathol.org). The results show that the targeted deletion of Jund did not have an effect on mouse BMDM activation.
JUND Expression Controls VEGFA and HIF1A mRNA Levels in a Human Podocyte Cell Line
To examine the direct effect of JUND on the regulation of expression of VEGFA and HIF1A, we used a conditionally immortalized human podocyte cell line.19 These cells proliferate at 33°C; however, after transfer to the “nonpermissive” environment (37°C), they enter growth arrest and express markers of differentiated in vivo podocytes, mimicking the podocyte maturation in vivo19 (Figure 5A). We first showed that podocyte differentiation is associated with increased VEGFA expression. Interestingly, the increased VEGFA expression in differentiated podocytes was also associated with decreased JUND expression when compared with undifferentiated cells (Figure 5B). We then performed siRNA knockdown of JUND in the human podocyte cell line and showed that JUND mRNA knockdown is associated with increased VEGFA and HIF1A in the same cells (Figure 5C).
Figure 5.
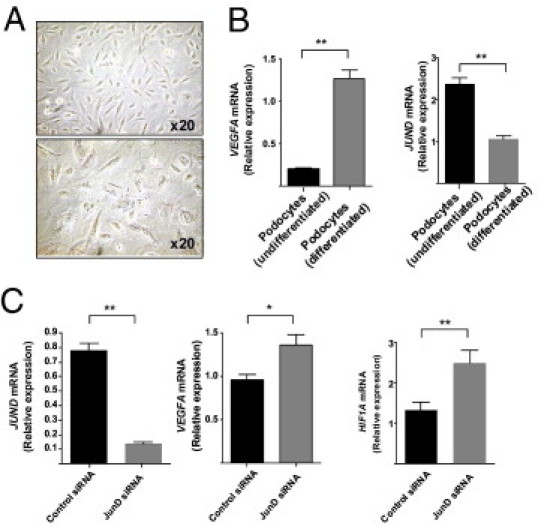
JUND knockdown in conditionally immortalized human podocyte cell lines. The human podocyte cell line was incubated for 14 days at 37°C to allow full differentiation and maturation. A: The difference in cell morphological features between the undifferentiated (33°C, top) and differentiated (37°C, bottom) podocytes is illustrated by light microscopy. B:VEGFA and JUND expression assessed by RT-qPCR in undifferentiated and differentiated podocytes. C: Knockdown with JUND siRNA (100 nmol/L) in differentiated podocytes showed 80% knockdown compared with scrambled siRNA (control siRNA, 100 nmol/L) by RT-qPCR. Both VEGFA and HIF1A expression levels were significantly increased when JUND expression was knockdown. *P < 0.05, **P < 0.01.
Discussion
JunD is a ubiquitously expressed member of the AP-1 transcription factor. Initial studies in fibroblasts showed its antiapoptotic and antimitogenic effects. In addition, JunD is not regulated by immediate-early gene transcriptional mechanisms. These previous studies2 suggest that JunD is an “atypical” member of AP-1 that can act as an activator or repressor of transcription of diverse cell type–specific genes involved in oxidative stress, cell proliferation, and differentiation.
The role of the AP-1 transcription factor in glomerulonephritis has been examined in several studies. In rats with immune complex nephritis, nuclear staining for AP-1 in several glomerular and tubulointerstitial cells was previously reported.20 In humans, tubular overactivation of AP-1 was described as a marker of progressive renal disease.21 Furthermore, c-Jun N-terminal kinase–mediated c-Jun/AP-1 activation was also detected in nephritic glomeruli of rats22; more recently, blockade of c-Jun N-terminal kinase signaling provided marked protection against the progression of crescentic anti–glomerular basement membrane glomerulonephritis in WKY rats.23 Interestingly, a recent study24 also showed that the epidermal loss of JunB leads to a systemic lupus erythematosus phenotype characterized by glomerular changes, such as mesangial hypercellularity with glomerular basement membrane thickening and luminal obstruction by hyaline immune complex deposits.
The role of JunD in immune response was first reported by Meixner and colleagues.25 They showed that JunD suppresses lymphocyte proliferation and has a negative effect on helper T-cell differentiation.25 Previously, JunD was identified as a determinant of macrophage activation in the WKY NTN model.9 In this model, Crgn is induced by injection of an NTS raised in rabbit without any preimmunization. On day 10 after the NTS injection, the inbred WKY rat gets severe glomerular crescent formation and macrophage infiltration in response to the planted antibodies on the glomerular basement membrane, whereas the Lewis rat is entirely resistant to any glomerular inflammation despite the similar deposition of nephrotoxic antibodies.13 Jund is markedly overexpressed in WKY BMDMs and nephritic glomeruli, and its cellular expression level control macrophage activation in rats and humans.9 Based on these results, we decided to investigate the role of targeted deletion of JunD in mouse NTN and showed herein that Jund-/- mice are markedly susceptible to accelerated NTN. In this model, we did not show any effect of the complete deletion of JunD on macrophage infiltration; however, we found that the absence of JunD in podocytes caused up-regulation of hypoxia-inducible gene targets. However, we cannot exclude a potential role of JunD in the regulation of oxidative stress in WKY rat podocytes. To answer these points, we are generating a Jund-knockout WKY rat to study the direct effect of the targeted deletion of JunD in the macrophage-dependant WKY NTN model.
Despite the marked susceptibility to accelerate NTN of the Jund-/- mice, the absence of any enhanced humoral immune response led us to question where the JunD promoter is active within the mice glomerulus. We clearly showed that, in the Jund-/- mice, the Jund promoter regulating LacZ expression is active in the glomerular podocytes. It is well documented that podocytes function as vascular-supporting cells producing Vegfa, a potent proangiogenic factor.26 Tight regulation of Vegfa signaling and dosage is required for development and maintenance of kidney function by regulating podocyte-endothelial cross talk in the glomerulus.26, 27 For instance, local reduction of VEGF within the kidney is sufficient to trigger the pathogenesis of thrombotic microangiopathy.28 In addition, a recent study29 identified VEGFA as a new susceptibility locus for chronic kidney disease and kidney function in a large genomewide association study. More importantly, a previous study30 also showed the importance of Hif1a stabilization due to podocyte-specific deletion of Vhlh and subsequent up-regulation of hypoxia-induced downstream genes, including Vegfa, causing rapidly progressive glomerulonephritis in the mice. Based on the results showing the accumulation of H2O2 in Jund-/- cells causing the Hif1a accumulation and enhanced transcription of Vegfa3, 6 and CXCL12,7 we hypothesized that deletion of JunD within the podocytes may cause oxidative stress characterized by increased expression of hypoxia-inducible genes, including Vegfa. Indeed, we showed in both the glomeruli isolated from Jund-/- and WT mice and an immortalized human podocyte cell line that JunD controls VEGFA expression in podocytes. Our results confirm a role for JunD in protecting the cells from oxidative stress and emphasize the importance of podocyte biology in the pathophysiological characteristics of Crgn.
In conclusion, the transcriptional mechanisms by which JunD regulate Vegfa expression within the podocytes require careful attention and JunD may be considered as a therapeutic target in glomerular diseases characterized by altered Vegfa production.
Acknowledgment
We thank Kylie McDonald for her excellent technical help.
Footnotes
Supported by the Wellcome Trust, intramural funding from the Medical Research Council Clinical Sciences Centre, INSERM, FP6 EURATools (European Union contract LSHG-CT-2005-019015), and an Imperial College Junior Research Fellowship (J.B.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.03.006.
Supplementary data
Serum levels of mouse IgG1, IgG2a, IgG2b, and IgG3 and anti-rabbit Ig antibodies were determined in serum samples of WT and Jund-/- mice (n = 8 mice, at least, in each group). There was no significant difference (ns) between the WT and Jund-/- for all of the antibody subclasses tested.
Splenocyte 3H thymidine incorporation 72 hours after exposure to sheep IgG (100 μg/mL) in vitro. Splenocytes were harvested 7 days after preimmunization. n = 6 mice, at least, in each group. ns indicates nonsignificant.
Macrophage activation in WT and Jund-/- macrophages. A: Macrophage activation, assessed by Fc receptor–mediated phagocytosis and oxidization, in WT and Jund-/- BMDMs. BMDMs were stimulated with Fc oxyBURST, and individual data points consisting of 10,000 fluorescence events were collected every 10 seconds in an FACSCalibur. B: Bead phagocytosis measured by the incubation of opsonized beads with WT and Jund-/- BMDMs. C: Relative expression of Nos2, Tnfa, and Ccl2 normalized to Gapdh measured by RT-qPCR in basal and lipopolysaccharide (LPS)–stimulated (100 ng/mL) WT and Jund-/- BMDMs. n = 4 mice, at least, in each group. ns indicates nonsignificant.
References
- 1.Hernandez J.M., Floyd D.H., Weilbaecher K.N., Green P.L., Boris-Lawrie K. Multiple facets of junD gene expression are atypical among AP-1 family members. Oncogene. 2008;27:4757–4767. doi: 10.1038/onc.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mechta-Grigoriou F., Gerald D., Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20:2378–2389. doi: 10.1038/sj.onc.1204381. [DOI] [PubMed] [Google Scholar]
- 3.Gerald D., Berra E., Frapart Y.M., Chan D.A., Giaccia A.J., Mansuy D., Pouyssegur J., Yaniv M., Mechta-Grigoriou F. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118:781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 4.Meixner A., Karreth F., Kenner L., Penninger J.M., Wagner E.F. Jun and JunD-dependent functions in cell proliferation and stress response. Cell Death Differ. 2010;17:1409–1419. doi: 10.1038/cdd.2010.22. [DOI] [PubMed] [Google Scholar]
- 5.Pouyssegur J., Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006;387:1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- 6.Laurent G., Solari F., Mateescu B., Karaca M., Castel J., Bourachot B., Magnan C., Billaud M., Mechta-Grigoriou F. Oxidative stress contributes to aging by enhancing pancreatic angiogenesis and insulin signaling. Cell Metab. 2008;7:113–124. doi: 10.1016/j.cmet.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 7.Toullec A., Gerald D., Despouy G., Bourachot B., Cardon M., Lefort S., Richardson M., Rigaill G., Parrini M.C., Lucchesi C., Bellanger D., Stern M.H., Dubois T., Sastre-Garau X., Delattre O., Vincent-Salomon A., Mechta-Grigoriou F. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. 2010;2:211–230. doi: 10.1002/emmm.201000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aitman T.J., Dong R., Vyse T.J., Norsworthy P.J., Johnson M.D., Smith J., Mangion J., Roberton-Lowe C., Marshall A.J., Petretto E., Hodges M.D., Bhangal G., Patel S.G., Sheehan-Rooney K., Duda M., Cook P.R., Evans D.J., Domin J., Flint J., Boyle J.J., Pusey C.D., Cook H.T. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–855. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 9.Behmoaras J., Bhangal G., Smith J., McDonald K., Mutch B., Lai P.C., Domin J., Game L., Salama A., Foxwell B.M., Pusey C.D., Cook H.T., Aitman T.J. Jund is a determinant of macrophage activation and is associated with glomerulonephritis susceptibility. Nat Genet. 2008;40:553–559. doi: 10.1038/ng.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behmoaras J., Smith J., D'Souza Z., Bhangal G., Chawanasuntoropoj R., Tam F.W., Pusey C.D., Aitman T.J., Cook H.T. Genetic loci modulate macrophage activity and glomerular damage in experimental glomerulonephritis. J Am Soc Nephrol. 2010;21:1136–1144. doi: 10.1681/ASN.2009090968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holdsworth S.R., Kitching A.R., Tipping P.G. Th1 and Th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999;55:1198–1216. doi: 10.1046/j.1523-1755.1999.00369.x. [DOI] [PubMed] [Google Scholar]
- 12.Liu K., Li Q.Z., Delgado-Vega A.M., Abelson A.K., Sanchez E., Kelly J.A., Li L., Liu Y., Zhou J., Yan M., Ye Q., Liu S., Xie C., Zhou X.J., Chung S.A., Pons-Estel B., Witte T., de Ramon E., Bae S.C., Barizzone N., Sebastiani G.D., Merrill J.T., Gregersen P.K., Gilkeson G.G., Kimberly R.P., Vyse T.J., Kim I., D'Alfonso S., Martin J., Harley J.B., Criswell L.A., Wakeland E.K., Alarcon-Riquelme M.E., Mohan C. Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J Clin Invest. 2009;119:911–923. doi: 10.1172/JCI36728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tam F.W., Smith J., Morel D., Karkar A.M., Thompson E.M., Cook H.T., Pusey C.D. Development of scarring and renal failure in a rat model of crescentic glomerulonephritis. Nephrol Dial Transplant. 1999;14:1658–1666. doi: 10.1093/ndt/14.7.1658. [DOI] [PubMed] [Google Scholar]
- 14.Smith J., Lai P.C., Behmoaras J., Roufosse C., Bhangal G., McDaid J.P., Aitman T., Tam F.W., Pusey C.D., Cook H.T. Genes expressed by both mesangial cells and bone marrow-derived cells underlie genetic susceptibility to crescentic glomerulonephritis in the rat. J Am Soc Nephrol. 2007;18:1816–1823. doi: 10.1681/ASN.2006070733. [DOI] [PubMed] [Google Scholar]
- 15.Thepot D., Weitzman J.B., Barra J., Segretain D., Stinnakre M.G., Babinet C., Yaniv M. Targeted disruption of the murine junD gene results in multiple defects in male reproductive function. Development. 2000;127:143–153. doi: 10.1242/dev.127.1.143. [DOI] [PubMed] [Google Scholar]
- 16.Tarzi R.M., Cook H.T., Jackson I., Pusey C.D., Lord G.M. Leptin-deficient mice are protected from accelerated nephrotoxic nephritis. Am J Pathol. 2004;164:385–390. doi: 10.1016/S0002-9440(10)63128-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnberg D., Lewis M., Moss J., Xu Y., Botto M., Cook H.T. Complement activation contributes to both glomerular and tubulointerstitial damage in adriamycin nephropathy in mice. J Immunol. 2006;177:4094–4102. doi: 10.4049/jimmunol.177.6.4094. [DOI] [PubMed] [Google Scholar]
- 18.Lai P.C., Smith J., Bhangal G., Chaudhry K.A., Chaudhry A.N., Keith J.C., Jr, Tam F.W., Pusey C.D., Cook H.T. Interleukin-11 reduces renal injury and glomerular NF-kappa B activity in murine experimental glomerulonephritis. Nephron Exp Nephrol. 2005;101:e146–e154. doi: 10.1159/000087938. [DOI] [PubMed] [Google Scholar]
- 19.Saleem M.A., O'Hare M.J., Reiser J., Coward R.J., Inward C.D., Farren T., Xing C.Y., Ni L., Mathieson P.W., Mundel P. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. doi: 10.1681/ASN.V133630. [DOI] [PubMed] [Google Scholar]
- 20.Hernández-Presa M.A., Gómez-Guerrero C., Egido J. In situ non-radioactive detection of nuclear factors in paraffin sections by Southwestern histochemistry. Kidney Int. 1999;55:209–214. doi: 10.1046/j.1523-1755.1999.00226.x. [DOI] [PubMed] [Google Scholar]
- 21.Mezzano S.A., Barria M., Droguett M.A., Burgos M.E., Ardiles L.G., Flores C., Egido J. Tubular NF-kappaB and AP-1 activation in human proteinuric renal disease. Kidney Int. 2001;60:1366–1377. doi: 10.1046/j.1523-1755.2001.00941.x. [DOI] [PubMed] [Google Scholar]
- 22.Sakurai H., Sugita T. c-Jun N-terminal kinase-mediated AP-1 activation in experimental glomerulonephritis in rats. Biochem Mol Biol Int. 1998;45:831–839. doi: 10.1080/15216549800203262. [DOI] [PubMed] [Google Scholar]
- 23.Ma F.Y., Flanc R.S., Tesch G.H., Bennett B.L., Friedman G.C., Nikolic-Paterson D.J. Blockade of the c-Jun amino terminal kinase prevents crescent formation and halts established anti-GBM glomerulonephritis in the rat. Lab Invest. 2009;89:470–484. doi: 10.1038/labinvest.2009.2. [DOI] [PubMed] [Google Scholar]
- 24.Pflegerl P., Vesely P., Hantusch B., Schlederer M., Zenz R., Janig E., Steiner G., Meixner A., Petzelbauer P., Wolf P., Soleiman A., Egger G., Moriggl R., Kishimoto T., Wagner E.F., Kenner L. Epidermal loss of JunB leads to a SLE phenotype due to hyper IL-6 signaling. Proc Natl Acad Sci U S A. 2009;106:20423–20428. doi: 10.1073/pnas.0910371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meixner A., Karreth F., Kenner L., Wagner E.F. JunD regulates lymphocyte proliferation and T helper cell cytokine expression. EMBO J. 2004;23:1325–1335. doi: 10.1038/sj.emboj.7600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eremina V., Baelde H.J., Quaggin S.E. Role of the VEGF-A signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol. 2007;106:32–37. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- 27.Eremina V., Sood M., Haigh J., Nagy A., Lajoie G., Ferrara N., Gerber H.P., Kikkawa Y., Miner J.H., Quaggin S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eremina V., Jefferson J.A., Kowalewska J., Hochster H., Haas M., Weisstuch J., Richardson C., Kopp J.B., Kabir M.G., Backx P.H., Gerber H.P., Ferrara N., Barisoni L., Alpers C.E., Quaggin S.E. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. 2008;358:1129–1136. doi: 10.1056/NEJMoa0707330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Köttgen A., Pattaro C., Boger C.A., Fuchsberger C., Olden M., Glazer N.L. New loci associated with kidney function and chronic kidney disease. Nat Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ding M., Cui S., Li C., Jothy S., Haase V., Steer B.M., Marsden P.A., Pippin J., Shankland S., Rastaldi M.P., Cohen C.D., Kretzler M., Quaggin S.E. Loss of the tumor suppressor Vhlh leads to upregulation of Cxcr4 and rapidly progressive glomerulonephritis in mice. Nat Med. 2006;12:1081–1087. doi: 10.1038/nm1460. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Serum levels of mouse IgG1, IgG2a, IgG2b, and IgG3 and anti-rabbit Ig antibodies were determined in serum samples of WT and Jund-/- mice (n = 8 mice, at least, in each group). There was no significant difference (ns) between the WT and Jund-/- for all of the antibody subclasses tested.
Splenocyte 3H thymidine incorporation 72 hours after exposure to sheep IgG (100 μg/mL) in vitro. Splenocytes were harvested 7 days after preimmunization. n = 6 mice, at least, in each group. ns indicates nonsignificant.
Macrophage activation in WT and Jund-/- macrophages. A: Macrophage activation, assessed by Fc receptor–mediated phagocytosis and oxidization, in WT and Jund-/- BMDMs. BMDMs were stimulated with Fc oxyBURST, and individual data points consisting of 10,000 fluorescence events were collected every 10 seconds in an FACSCalibur. B: Bead phagocytosis measured by the incubation of opsonized beads with WT and Jund-/- BMDMs. C: Relative expression of Nos2, Tnfa, and Ccl2 normalized to Gapdh measured by RT-qPCR in basal and lipopolysaccharide (LPS)–stimulated (100 ng/mL) WT and Jund-/- BMDMs. n = 4 mice, at least, in each group. ns indicates nonsignificant.