

Abstract
This study tests the hypothesis that large porous poly (lactic-co-glycolic acid) (PLGA) microparticles modified with polyethyleneimine (PEI) are viable carriers for pulmonary delivery of prostaglandin E1 (PGE1) used in the treatment of pulmonary arterial hypertension (PAH), a pulmonary vascular disorder. The particles were prepared by a double-emulsion solvent evaporation method with PEI-25 kDa in the internal aqueous phase to produce an osmotic pressure gradient. Polyvinyl alcohol (PVA) was used for external coating of the particles. The particles were examined for morphology, size, aerodynamic diameter, surface area, pore volume and in-vitro release profiles. Particles with optimal properties for inhalation were tested for in-vivo pulmonary absorption, metabolic stability in rat lung homogenates, and acute toxicity in rat bronchoalveolar lavage fluid and respiratory epithelial cells, Calu-3. The micromeritic data indicated that the PEI-modified particles of PGE1 are optimal for inhalation. Incorporation of PEI in the formulations resulted in an increased entrapment efficiency–83.26±3.04% for particles with 1% PVA and 95.48±0.46% for particles with 2% PVA. The amount of cumulative drug released into the simulated interstitial lung fluid was between 50.8±0.76% and 55.36±0.06%. A remarkable extension of the circulation half-life up to 6.0–6.5 hours was observed when the formulations were administered via the lungs. The metabolic stability and toxicity studies showed that the optimized formulations were stable at physiological conditions and relatively safe to the lungs and respiratory epithelium. Overall, this study demonstrates that large porous inhalable polymeric microparticles can be a feasible option for non-invasive and controlled release of PGE1 for treatment of PAH.
Keywords: Prostaglandin E1, Pulmonary Arterial Hypertension, PLGA microparticles, Core modifying polyethyleneimine, pulmonary delivery
1. INTRODUCTION
The lungs are affected by an array of disorders, including infection, inflammation, obstruction, fibrosis, and vascular diseases such as thrombosis and arterial hypertension. Many lung disorders are currently treated by therapeutic agents that are required to be administered by systemic routes such as the parenteral and oral routes (Ewert et al., 2007; Rubin et al., 2002). Because of this systemic administration, the body is exposed to drugs that may harm other vital organs such as the heart and kidney. Such off-target effects in the treatment of lung diseases can be minimized by administering the drugs directly to the lungs. Indeed, the pharmacotherapy of certain pulmonary disorders, including asthma and pulmonary arterial hypertension (PAH), currently involves the use of nebulizers and inhalers for localized delivery of drugs to the lungs (Olschewski et al., 2002; Papi et al., 2007). However, these formulations or delivery systems suffer from a wide range of limitations that include multiple inhalations a day, short duration of action, metabolic instability in the lungs, and drug loss due to premature deposition in the oropharyngeal tract (Lee and Rubin, 2005; Lipworth, 1995). Short duration of action and metabolic instability often stem from the fact that currently marketed inhalable formulations consist of drug dissolved in a mixture of solvents and propellants or plain drug formulated with respirable lactose (Labiris and Dolovich, 2003). These shortcomings can be addressed in two ways: chemical modification of the drug, or reformulation of the drug in controlled-release polymeric carriers. However, the latter approach is preferred because chemical modification often leads to reduction in pharmacological activity. Chemical modification of heparin, for example, has resulted in reduced anti-coagulant activity (Park et al., 2010).
In fact, polymeric particulate carriers have been used for many years to prolong the duration of action and improve the stability of numerous drugs (Lemoine and Preat, 1998; Shive and Anderson, 1997). Of the various polymeric carriers, poly (lactic-co-glycolic acid) (PLGA)-based particles have been extensively investigated for the delivery of drugs via the pulmonary route (Hirota et al., 2010; Ohashi et al., 2009). Moreover, there has been intense interest in the use of large porous PLGA particles for prolongation of the duration of action of inhaled drugs since the publication of Edwards’ seminal paper in 1997. In that paper, Edwards and colleagues proposed that large porous particles with mass densities <0.4 g/cm3 and mean volume diameter >5 μm provide deep lung deposition of the inhaled drug and help bypass clearance mechanisms in the lungs, thereby providing enhanced respirability and prolonged residence time in the lungs (Edwards et al., 1997). In agreement with this study, we and others have shown that porous particles with a density of <1 g/cc release drugs for a longer period of time compared to nonporous and high-density particles (Gupta et al., 2011; Rawat et al., 2008).
Pulmonary arterial hypertension (PAH) is a progressive disease that results from remodeling of the pulmonary vasculature (Gupta and Ahsan, 2010). It is a debilitating disease affecting 50,000–100,000 Americans, with 300–500 new cases diagnosed each year. The current therapeutic strategies for PAH involve the use of short-acting inhalable or injectable formulations of anti-PAH drugs. Unfortunately, the current pharmacotherapeutic approaches for PAH are plagued with many disadvantages, including a requirement of 9 to 12 inhalations a day (Ventavis®, Iloprost inhalation solution), or intravenous or subcutaneous injections (Ricachinevsky and Amantea, 2006). Recently, we showed that PLGA microparticles can be used to overcome the limitations of short duration of action and metabolic instability of an investigational anti-PAH drug, prostaglandin E1 (PGE1) (Gupta et al., 2010). In fact, PGE1 is a potent pulmonary vasodilator with a very short biological half-life, 3–5 minutes; however, it produces systemic side effects when administered intravenously (Meyer et al., 1998). Our published study suggests that it is feasible to develop a long-acting inhalable formulation of PLGA-based microparticles of PGE1 that releases the drug for a period of 8 hours and provides better metabolic stability when compared with plain drug administered via the pulmonary route (Gupta et al., 2010). However, reduced drug loading and poor drug deposition patterns remain problematic for PLGA-based inhalable formulations of this anti-PAH drug. In addition, our lab has very recently demonstrated that PLGA microspheres encapsulating inclusion complex of PGE1 and hydroxypropyl-β-cyclodextrin (PGE1-HPβCD) provide a very significant improvement in the availability of PGE1 in-vivo following pulmonary administration. At the same time, inclusion of HPβCD in the microspheres increases aqueous solubility of PGE1 resulting in better drug release, and also works as a porosigen. However, inclusion of HPβCD could not produce any significant increase in drug entrapment efficiency of the formulations, and also resulted in a significant reduction in biological t1/2 as compared to non-porous PLGA microspheres encapsulating PGE1 (Gupta et al., 2011).
Recently, Rawat et al. showed that incorporation of a porosigen such as polyethyleneimine-25 kDa (PEI-25 kDa) in the aqueous core of PLGA microparticles results in highly porous particles for pulmonary delivery of low molecular weight heparins (Rawat et al., 2008). PEI, a hydrophilic polycation, is known to produce large porous particles with a uniform distribution of pores owing to the osmotic pressure gradient produced between the microparticle core and the external aqueous phase. In addition to facilitating formation of porous particles through the osmotic pressure gradient, PEI is likely to form an electrostatic complex with PGE1 that results in improved solubility and enhanced entrapment of the drug in microparticulate systems. Further, PEI-25 kDa, at therapeutically safe doses, has been reported to work as an absorption enhancer for pulmonary delivery of low molecular weight heparins (Yang et al., 2006). This study therefore tests the hypothesis that PEI-modified large porous microparticles of PGE1 have high drug-loading capacity, an extended drug-release profile, and favorable respirability for deep lung deposition.
2. MATERIALS AND METHODS
2.1. Materials
Poly (lactic-co-glycolic acid) (PLGA) 85:15 with an inherent viscosity of 0.55–0.75 dl/g (average molecular weight = 85.2 kDa) and prostaglandin E1 (PGE1) were obtained from Lactel Absorbable Polymers (Pelham, AL) and Spectrum Chemicals (Gardena, CA), respectively. Polyethyleneimine (PEI) 25 kDa, poly vinyl alcohol (PVA), and dichloromethane (DCM) were purchased from Sigma-Aldrich, Inc. (St Louis, MO). Kits to assay PGE1 and protein were acquired from Assay Designs, Inc. (Ann Arbor, MI) and Pierce Biotechnology (Rockford, IL), respectively. Alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) kits were procured from Pointe Scientific, Inc. (Canton, MI). All other chemicals were of analytical grade and used without any modification.
2.2. Preparation of Core-Modified Large Porous PLGA Microparticles
Core-modified large porous PGE1-loaded PLGA microparticles were prepared by a water- in-oil-in-water (W/O/W) double emulsion/solvent evaporation method after slight modification of a published protocol (Gupta et al., 2010; Rawat et al., 2008). Briefly, 250 mg of PLGA was initially dissolved in 5 mL of DCM (organic phase, OP). The internal aqueous phase (IAP) was prepared by adding 5 mg of PGE1 to 500 μL of deionized water after solubilizing the drug in a minimal quantity of absolute alcohol. PEI 25 kDa, used as a core-modifying agent, was added to IAP at varying concentrations (1.25, 2.5, and 5% w/v; Table 1). The IAP and OP were emulsified (W/O) by probe sonication for 1 minute using a Branson® Sonifier 450 (Branson Ultrasonics Corporation, Danbury, CT). The primary emulsion was then homogenized with 25 mL of PVA solution (external aqueous phase, EAP) at two different concentrations (1% and 2%; Table 1) at 8,000 rpm for 10 minutes with an Ultra-Turrex T-25 Basic (IKA, Wilmington, DE). The resultant W/O/W emulsion was stirred overnight at room temperature to facilitate the removal of the OP. Next day, the formulations were washed by centrifugation with deionized water to remove excess PVA and lyophilized to obtain free flowing powdered microparticles (FreeZone 2.5, Labconco Corporation; Kansas City, MO).
Table 1.
Composition and Polydispersity Indices (PDI) of Different Microspheric Formulations.
| Formulations | IAP:OP:EAP | IAP Composition (w/v) | EAP Composition (w/v) | PDI | Tapped Density (g/cm3) |
|---|---|---|---|---|---|
| Plain-1 | 0.5:5:25 | — | 1% PVA | 5.16±0.021 | 0.36±0.12 |
| PEI-1 | 1.25% PEI | 3.85±0.014 | 0.21±0.03 | ||
| PEI-2 | 2.5% PEI | 1.51±0.006 | 0.15±0.02 | ||
| PEI-3 | 5% PEI | 1.47±0.016 | 0.1±0.003 | ||
|
|
|
||||
| Plain-2 | — | 2% PVA | 3.3560.035 | 0.45±0.11 | |
| PEI-4 | 1.25% PEI | 2.98±0.003 | 0.25±0.02 | ||
| PEI-5 | 2.5% PEI | 1.49±0.003 | 0.2±0.014 | ||
| PEI-6 | 5% PEI | 4.82±0.664 | 0.146±0.012 | ||
IAP, Internal Aqueous Phase; OP, Organic Phase; EAP, External Aqueous Phase; PEI, Polyethyleneimine 25 kDa; PVA, Polyvinyl Alcohol; PDI, Polydispersity Index
2.3. Physical Characterization
2.3.1. Particle Morphology
Particle morphology was examined by using a scanning electron microscope (Hitachi® S-3400N; Hitachi High Technologies America, Inc, Pleasanton, CA). The samples were prepared by sprinkling a small amount of powdered formulation onto a double-sided adhesive tape attached to an aluminum stub. The particles placed on the stub were sputter coated with gold under argon (Emitech K550X; Quorum Technologies Ltd, Kent, UK) and were viewed under the electron microscope. Photomicrographs of the formulations were taken at varying magnifications.
2.3.2. Particle Size
Particle size distribution and mean volume diameters of all the formulations were determined by Tri-laser diffraction technology using a Microtrac® S3500 (Microtrac, Inc; Largo, FL). Samples for wet-fluid sampling were prepared by dispersing 2 mg of microparticulate formulations in 0.2% w/v solution of Tween-80. Polydispersity indices (PDIs) were calculated as a ratio of volume-averaged mean particle size to number-averaged mean diameter. All the formulations were measured in triplicates and the data are presented as mean±SD.
2.3.3. Tapped Density Measurements
The density of microspheric formulations was estimated from tapped density measurements as described previously (Vanbever et al., 1999). An aliquot of microspheres was transferred to a 10 (±0.05) mL graduated cylinder and the initial volume was recorded. The tapped density of the formulations (ρ) was calculated as the ratio between sample weight (g) and the volume (mL) occupied after 100 taps.
2.3.4. Mass Median Aerodynamic Diameter Determination
The mass median aerodynamic diameter (MMAD) was studied in an 8-stage Mark-II non-viable Andersen Cascade Impactor (ACI) (Westech Instruments, Marietta, GA). Briefly, freeze dried microparticulate formulations (10 mg) were filled in size 3, hard gelatin capsules and fired into the cascade impactor via the induction port using a Rotahaler (Spiriva® Handihaler®, Boehringer Ingelheim, Inc., Ridgefield, CT). The impaction was performed at a flow rate of 28.3 L/min for 2 minutes. To prevent bounce or re-entrainment of the particles, glass fiber filter papers were placed in an inverted position on collection plates of all stages 0–7 (Vanbever et al., 1999). The amount of particles deposited at each stage was determined gravimetrically by calculating the differences in the weights of the glass fiber filter papers. Data obtained were plotted on a semilog graph paper and MMAD was determined at 50% of the particle distribution profile. The fine particle fractions (FPF) of the formulations were determined using the following equation:
2.3.5. Entrapment Efficiency
The amount of PGE1 entrapped in the microparticulate formulations was determined by quantifying the amount of PGE1 in the EAP, i.e. unentrapped PGE1 recovered after the first washing of the microparticles. The samples recovered were diluted 104–105 times and were assayed for the unentrapped PGE1 using an ELISA kit (Assay Designs, Ann Arbor, MI) according to the protocol provided by the manufacturer. The kit can reproducibly quantify PGE1 in the range between 4.88 and 5,000 pg/mL. All measurements were performed in triplicates and data are presented as mean±SD.
2.3.6. Surface Area and Pore Size Determination
For determination of surface area and the extent of pore formation, nitrogen physiosorption isotherms were obtained at 77K using a Nova 3200e Surface Area Analyzer (Quantachrome Instrument Corporation, Boynton Beach, FL). For physiosorption isotherm determination, the samples were first degassed for 24 hours at room temperature under vacuum at 75 mm Hg to remove any of the dissolved components from the surface. After completion of degassing, samples were placed on the surface area analyzer in a sample cell with a dewar flask filled with liquid nitrogen underneath the sample cell. After generation of the isotherm, surface areas of all the microparticulate formulations were evaluated by using Brunauer-Emmett-Teller (BET) and Barrett-Joyner-Halenda (BJH) adsorption isotherms. The average pore radius and cumulative pore volumes were calculated from the desorption curve of BJH isotherm from a range of desorption points (Gill et al., 2009). The analysis was performed in triplicates for 24 hours with a 150 s equilibrium interval.
2.4. In-vitro Release Studies in Simulated Interstitial Lung Fluid
In-vitro drug-release studies were performed in a simulated interstitial lung fluid (SILF) prepared based on the “Moss Formula” (Moss, 1979) (Table 2). Briefly, SILF was prepared as per the recipe described in Table 2 (Moss, 1979) and the pH of the fluid was maintained at 7.0–7.2 throughout the experiment. An aliquot of formulations (10 mg) was dispersed in 1 mL of SILF and incubated at 37±1°C under moderate shaking (~200 rpm). To determine the drug release at predetermined time points, samples were centrifuged at 16,000g for 15 minutes at 4°C and the supernatant was collected. To maintain the pH of SILF at 7.0–7.2 and the sink condition, the media was replaced with an equal amount of fresh media during sampling. The drug content in the supernatant was quantified by ELISA. The kit can reproducibly quantify PGE1 in the range between 4.88 and 5,000 pg/mL. All measurements were performed in triplicates and data are presented as mean±SD.
Table 2.
Compositions of Actual Lung Fluids and Simulated Lung Fluids (modified from Moss, 1979).
| Ion | Actual Lung Fluid(mEq) | Simulated Lung Fluid(mEq) |
|---|---|---|
| Calcium, Ca++ | 5.0 | 5.0 |
| Magnesium, Mg++ | 2.0 | 2.0 |
| Potassium, K+ | 4.0 | 4.0 |
| Sodium, Na+ | 145.0 | 145.0 |
|
| ||
| Total Cations | 156.0 | 156.0 |
|
| ||
| Bicarbonate, HCO3− | 31.0 | 31.0 |
| Chloride, Cl− | 114.0 | 114.0 |
| Citrate, H5C6O73− | - | 1.0 |
| Acetate,H3C2O2− | 7.0 | 7.0 |
| Phosphate, HPO42− | 2.0 | 2.0 |
| Sulfate, SO42− | 1.0 | 1.0 |
| Protein | 1.0 | - |
|
| ||
| Total Anions | 156.0 | 156.0 |
|
| ||
| pH | 7.3–7.4 | 7.3–7.4 |
2.5. In-vivo Absorption Studies
Adult male Sprague-Dawley rats (Charles River Laboratories, Charlotte, NC) weighing between 250 g and 300 g were used for studying pulmonary absorption (n = 6–8). Briefly, rats were first anesthetized by an intramuscular (IM) injection of ketamine and xylazine cocktail (90 mg/kg + 10 mg/kg). The optimized formulations (PEI-1, and PEI-4) were administered via the intratracheal route utilizing a microsprayer (Penn-Century, Philadelphia, PA). Microparticulate formulations were administered after dispersion in normal saline at a dose of 120 μg/kg. Three control groups received the following formulations: (i) normal saline via the pulmonary route, (ii) intravenous bolus dose of 80 μg/kg PGE1 via the jugular vein, and (iii) pulmonary dose of 80 μg/kg PGE1. Following administration, blood samples were collected for 24 hours in citrated microcentrifuge tubes containing 10 μg/mL indomethacin as PGE1 synthase inhibitor. The plasma was separated by centrifuging the blood (5,000 rpm for 5 minutes) and stored in separate microcentrifuge tubes at −20°C until further analysis. The plasma levels of PGE1 were determined by using the ELISA kit as described above. The plasma samples from untreated rats were used as negative controls to take into the account the influence of endogenous PGE1 on the analysis.
2.6. Metabolic Degradation Studies in Rat Lung Homogenates
To determine the stability of the optimized formulations (PEI-1 and PEI-4) under physiological conditions and to assess their controlled release properties, metabolic degradation studies were performed in rat lung homogenates according to a protocol established in our laboratory with slight modification (Gupta et al., 2010). Briefly, lungs from healthy adult male Sprague-Dawley rats (300–350 g) were surgically removed and homogenized in 4 volumes of cold homogenization buffer (Bücher medium – 20 mM KH2PO4, 72 mM K2HPO4, 27.6 mM nicotinamide, and 3.6 mM MgCl2, pH 7.4)(Nakano et al., 1973) using a Potter-Elvehjem glass homogenizer. Supernatant collected after centrifugation was used for further studies. For metabolic degradation studies, lung homogenates were incubated with (i) plain PGE1 (10 μg/mL), (ii) PEI-1, or (iii) PEI-4 containing 10 μg PGE1 each for 8 hours in the presence of 2 mM NAD+ at 37°C in an oscillating water bath. The reaction was terminated at predetermined time points by adding 200 μL of 0.1N HCl to the incubation mixture. The amount of drug released in the homogenate samples was determined by PGE1-ELISA. To determine the amount of drug remaining in the formulations, particles in the homogenate were digested and extracted with 5 volumes of DCM. Following the extraction, the organic layer containing DCM with polymer and drug was transferred to a clean test-tube and evaporated in a N2 stream by using N-Evap (Organomation Associates Inc, Berlin, MA). PGE1 was then extracted by vortexing the residue in the test-tube with 1.0 mL of phosphate-buffered saline (PBS, pH 7.2) for at least 30 seconds. The PBS solution thus obtained was assayed for PGE1 content by ELISA. This amount of PGE1 was considered to be the amount of PGE1 not released upon incubation in lung homogenates.
2.7. Bronchoalveolar Lavage Studies
Bronchoalveolar lavage (BAL) fluid studies were performed to establish the safety profile of the formulations (PEI-1 and PEI-4) according to our previously published procedure (Gupta et al., 2010; Hussain and Ahsan, 2005; Thomas et al., 2008). Briefly, four groups of adult male Sprague-Dawley rats (300–400 g) (n = 4) were anesthetized with a cocktail of ketamine and xylazine, and were treated with (i) saline as a negative control, (ii) PEI-1 equivalent to 120 μg/kg PGE1, (iii) PEI-4 equivalent to 120 μg/kg PGE1 and (iv) 0.1% w/v solution of sodium dodecyl sulfate (SDS) as a positive control. The lungs were surgically removed 12 hours post administration from anesthetized rats by exposing the respiratory system by a mid-level incision in the thoracic cavity and severing the abdominal aorta. Excised lungs were carefully cleaned from adjoining tissues/organs and were weighed to investigate the possibility of edema formation. Lungs were lavaged with 5 mL normal saline instilled through the trachea. Saline was left into the lungs for 30 s, withdrawn, re-instilled for 30 s, and finally withdrawn and collected in microcentrifuge tubes. The BAL fluid samples thus collected were centrifuged at 500g for 10 minutes, and the supernatant was collected and stored at −20°C until further analysis (24–48 hours). BAL fluid samples were analyzed for (i) total protein concentration, (ii) ALP and (iii) LDH. All animal studies were performed in accordance with NIH Guidelines for the Care and Use of Laboratory Animals under a protocol approved by Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee (AM-10012).
2.8. Cell Viability Studies
For determining the extent of cytotoxic effects of the optimized formulations on lung epithelial cell lines, cell viability studies were performed by propidium iodide exclusion fluorescence assay using the Calu-3 cell line (Nieminen et al., 1992). Briefly, Calu-3 cells (ATCC, Manassas, VA) were seeded at a density of 20,000 cells/well in a 96-well black microtiter plate (Corning, Inc., Corning, NY) in MEM medium. The cells were allowed to attach overnight in a 5% CO2 incubator at 37°C. All experiments from here onwards were performed in Krebs-Ringer-N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer (KRH) containing 115 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM KH2PO4, 1.2 mM MgSO4, and 25 mM NaHEPES (pH 7.4) (Qian et al., 1997).
Immediately prior to the experiment, media was removed from the wells, cells were washed with KRH buffer twice and 20 μL of test formulations were added at concentrations of 0.5, 1.0, 2.0, and 5.0 mg/mL. Sodium dodecyl sulfate (SDS) at a concentration of 0.1% was used as a positive control. Following a 4-hour incubation at 37°C, test samples were removed, cells were washed with KRH buffer twice and background fluorescence for each well was recorded after adding 100 μL buffer. The cells were then treated with propidium iodide solution (100 μL of 5 μM), incubated at 37°C for 30 minutes and measured for fluorescence (F). Digitonin at a final concentration of 375 μM was added to permeabilize the cells and subsequently label them with propidium iodide. Following addition of digitonin, the fluorescence measurement was repeated every 5 minutes for 15 minutes, which was considered as the final fluorescence (Fmax) and used to calculate cell viability using the following equation (Sarafian et al., 2002; Sarafian et al., 1994).
All fluorescence measurements were performed in a SynergyMX microplate reader (Biotek, Winnoski, VT) using an excitation wavelength (Ex) of 546 nm and an emission wavelength (Em) of 620 nm at a band pass of 20 nm.
2.9. Pharmacokinetic and Data Analysis
Pharmacokinetic parameters for in-vivo absorption studies were determined by using standard non-compartmental extravascular analysis (Kinetica®, version 5.0, Thermo Fisher Scientific, Waltham, MA). Data were analyzed for area under the concentration time curve (AUC0–24h), Cmax, Tmax, and t1/2. All data were analyzed by one-way ANOVA followed by appropriate post hoc analysis (GraphPad Prism version 5.0, GraphPad Software, La Jolla, CA). Values showing p<0.05 were considered significantly different.
3. RESULTS AND DISCUSSION
3.1. Physical Characterization
A series of experiments was performed to study the micromeritics –i.e. morphology, size, density, porosity and pore size–of the particles to be used for depositing PGE1 in the respiratory tract.
3.1.1
First, the morphology of the particles was examined under a scanning electron microscope. All formulations were comprised of spherical particles with a porous surface (Fig. 1). The degree of porosity increased with increasing PEI concentration (5.0%>2.5%>1.25%), which agrees with our previous study that showed that incorporation of PEI in the IAP resulted in very large particles with highly porous surfaces (Rawat et al., 2008). PEI, an osmotically active polycation, increases the porosity by pulling water into the particle core during emulsification. Subsequent removal of this water by lyophilization results in the formation of holes or pores on the particle surface. This assumption agrees with other published studies suggesting that surface porosity results from increased osmotic pressure in the aqueous core (Pistel and Kissel, 2000; Ravivarapu et al., 2000). The concentration of PVA (1% and 2%) used in the EAP also significantly influenced particle morphology. Formulations with 1% PVA (Figs. 1A, B, and C) showed spherical particles with a somewhat dented or bumpy surface, whereas formulations with 2% PVA in the EAP (Fig. 1D, 1E, and 1F) showed uniformly distributed spherical particles with even surfaces. These observations are in agreement with published data that showed that PVA works as an emulsion stabilizer and can affect the hydrophobicity and digestibility of PLGA microparticles by being adsorbed on the particle surface (Jeong et al., 2003).
Figure 1.

SEM of PGE1-loaded large porous PLGA microspheres. (A) PEI-1, (B) PEI-2, (C) PEI-3, (D) PEI-4, (E) PEI-5, and (F) PEI-6. See Table 1 for compositions of different formulations.
3.1.2
The particle size data presented in Fig. 2A suggest that the particle size of various formulations varied depending on PEI concentration in the IAP and PVA concentration in the EAP. The particle size, presented as mean volume diameter–defined as the diameter of a sphere having the same volume as the test particle, increased significantly when the PEI concentration was increased from 10.72±0.04 μM (PEI-1) to 22.53±0.255 μM (PEI-3) and from 7.01±0.009 μM (PEI-4) to 12.26±0.115 μM (PEI-6). The increase in particle size with increasing PEI concentration is because PEI leads to increased porosity (Rawat et al., 2008). However, a reduction in particle size was observed when the PVA concentration was increased from 1% to 2%. PVA stabilizes the emulsion droplets by providing a hydrophilic environment and produces a reduction in particle size (Jeong et al., 2003). The polydispersity index values presented in Table 1 show that the formulations were moderately polydispersed, with polydispersity indices ranging from 0.99 to 3.73.
Figure 2.

Micromeritic profiles of PGE1-loaded large porous PLGA microspheres. (A) Volume- based mean diameter and actual mass median aerodynamic diameter (MMAD); (B) % Fine particle fraction. Data represent mean ± standard deviation (n=3).
3.1.3
The tapped density of particulate formulations can provide important information concerning flowability, particle porosity and particle size distribution. Therefore, tapped density was measured, and a decrease in tapped densities was observed with increasing concentrations of PEI, suggesting that larger but lighter particles were formed. However, no significant differences in tapped density were observed when the PVA concentration was increased from 1% to 2%. The increase in tapped density with the increase in PEI concentration was due to the increased porosity of the particles as observed in SEM photomicrographs.
3.1.4
The mass median aerodynamic diameter (MMAD), the most important particle parameter that determines the flight of the formulation over the route of inhalation, was determined by using an 8-stage non-viable Andersen Cascade Impactor. All formulations showed an aerodynamic diameter between 2.5 μm and 3.5 μm. While no direct correlation between PEI concentration and MMAD was observed, a slight increase in MMAD was observed with increasing PVA concentration despite the smaller particle size (Fig. 2A). Importantly, MMAD was between 1–6 μm, which is considered optimal for efficient deposition in the respiratory tract. In fact, for effective deposition of a particulate drug-delivery carrier, a geometric diameter of 1–5 μm with a particle density of ~1 g/cm3 is desirable (Adjei and Garren, 1990). However, Edwards et al. suggested that particles larger than 5 μm with reduced density (<0.4 g/cm3) can enhance deep lung deposition and provide enhanced bioavailability by prolonging the release period of the drug(Edwards et al., 1997).
3.1.5
The fine particle fraction (FPF) of all the formulations was determined from the fraction of particles that was deposited at stage 3 or lower in the cascade impactor. All formulations showed a FPF in the range of 50.98% to 63.16% (Fig. 2B). Similar to the MMAD data, FPF also showed little or no correlation with PEI concentration, but decreased with the increase in PVA concentration.
3.1.6
Surface area, pore volume, and pore radii were determined using a Nova 3200e surface area analyzer (Gill et al., 2009). The surface area increased as a function of PEI and PVA concentrations. In fact, the surface area data generated from two isotherms, BET and BJH, showed very similar patterns (Fig. 3A). These data agree with the particle size data presented in Fig. 2, which show that the presence of PEI produces a lighter and more porous particle in a concentration-dependent manner. Further, the data are also in consistent with the assumption that increasing the PVA concentration produces less porous, smaller particles. Interestingly, the increase in surface area was directly proportional to the increase in particle size, which contradicts an established fact that surface area is inversely related to particle size. Indeed, mathematically, the external surface area of a non-porous particle decreases with increasing particle size. However, in the case of porous particles, the internal surface area takes into account the porous nature of the particles in addition to the external surface area. For this reason, there was a net increase in the specific surface area with the increase in porosity of the particles, as pointed out by Jeyanthi et al (Jeyanthi et al., 1997). Similar to the surface area data, an increase in the pore volume was observed with the increase in PEI or PVA concentration (Fig. 3B). The pore volume data corroborate the porous character and extent of porosity observed by SEM. The pore radii of the formulations were 27–28 Å, and no significant differences in pore radii were observed among the various formulations (Fig. 3C).
Figure 3.

Physiosorption-based surface characteristics of PGE1-loaded large porous PLGA microspheres. (A) Surface area as determined by BET and BJH adsorption isotherms, (B) Pore volume, and (C) Pore radii. Data represent mean ± standard error of mean (n=3). *Significant differences among the group (1% PVA) (p<0.05); **Significant differences among the group (2% PVA) (p<0.05).
3.2. Entrapment Efficiency
Following the micromeritic analysis, the entrapment efficiencies for all the formulations were evaluated by analyzing drug content in the supernatant recovered after washing the microparticles (Fig. 4A). All PEI-modified formulations showed excellent entrapment efficiency compared to their plain counterparts. In the first set of formulations (1% PVA), the entrapment efficiency was 83.26±3.04% when PEI was used at a concentration of 1.25%. However, the entrapment efficiency was increased to 97–99%, when PEI concentration was increased from 1.25% to 2.5% or 5%. Similar increases in drug loading were observed when the PVA concentration was 2%, although no major differences were observed with the change in PEI concentration. In fact, PEI-modified particles showed a considerable increase in drug loading compared to our previously published study on PLGA particles of PGE1, in which drug entrapment efficiency was between 59.44% and 65.51% (Gupta et al., 2010). Such a dramatic increase in drug loading in PEI-modified particles can be explained on the basis of following two assumptions: First, PEI is a positively charged polymer which forms an electrostatic complex with negatively charged PGE1, resulting in retention of a larger amount of drug in the particle core. Second, both PEI and PVA increase entrapment by enhancing the stability of the primary emulsion (Cirpanli et al., 2005; Lee et al., 1999). Overall, the micromeritic analysis of the particles shows that PEI as a porosigen and PVA as an emulsifier play important roles in controlling the size and porosity of the formulations and that the particles meet the standard for an inhalable formulation. Similar to the influence of PEI and PVA on the physical properties of the particles, these formulation additives play a major role in the loading of drug in PLGA particles.
Figure 4.

(A) Entrapment efficiencies of PGE1-loaded large porous PLGA microspheres, (B) In- vitro release profiles of PGE1 from PLGA microspheres, and (C) In-vitro release patterns showing surface-associated release, burst release, and percent drug released per hour. Data represent mean ± standard deviation (n=3).
3.3. In-vitro Release Studies in Simulated Interstitial Lung Fluid
To access the efficacy of large porous PLGA microspheres in providing prolonged and controlled release of PGE1, the in-vitro release profiles were studied in Simulated Interstitial Lung Fluid (SILF) at 37°C, thus mimicking physiological conditions in the human body. As is evident from Fig. 4B, drug released from all the formulations, except PEI-1, followed a two-step process: an initial burst release phase followed by a nearly zero-order release (Fig. 4B). There was no further release after 10 hours of the study. Formulations containing 1.25% PEI produced a cumulative release of 55.36±0.06% over a 24-hour period.
The amount of drug released from 1.25% PEI-modified particles was 2–5-fold higher than that from plain PLGA particles published in our earlier report on PLGA particles of PGE1 (Gupta et al., 2010). Such a major improvement in drug release can be explained on the basis of the porous nature of the particles, as was observed for porous PLGA particles of low molecular weight heparin (Rawat et al., 2008). In the case of porous particles, a larger amount of release medium comes in contact with the particle surface and facilitates drug release. However, the amount of drug released was dramatically reduced when the PEI concentration was increased. For example, the cumulative drug release for the formulations containing 2.5% and 5% PEI was 35.02±2.31% and 16.59±2.17%, respectively. Similar to the first set of formulations containing 1% PVA, in the second set of formulations containing 2% PVA, a significant increase in drug release was observed for the 1.25% PEI-modified microspheres, whereas the formulations containing 2.5% or 5% PEI showed a reduced drug release. This decrease in cumulative drug release with increasing PEI concentration may stem from the fact that PEI, when used at a higher concentration, forms a strong electrostatic complex with PGE1 that prevents dissociation of the drug from PEI. Further, because of its hydrophobic nature, PVA has been reported to significantly reduce the release of drugs at higher concentrations (Jeong et al., 2003).
The in-vitro release profiles were further analyzed to calculate the following release parameters: (i) surface-associated drug release, i.e., the amount of drug released at zero minutes; (ii) burst release, the amount of drug released in the first 30 minutes; and (iii) the amount of drug released per hour. With increasing PEI concentration, there was a significant increase in the amount of surface-associated drug release (Fig. 4C). The percentages of surface-associated drug released from the first three PEI-modified formulations containing 1% PVA were 1.85±0.73%, 2.86±0.01% and 6.28±0.31%, respectively. Formulations containing 2% PVA showed a similar pattern. Interestingly, PEI-2 (1% PVA, and 2.5% PEI) showed the greatest increase in burst release, followed by PEI-3 and PEI-1. Increase in burst release suggests the presence of greater amounts of drug on the particle surface or in the distal core of the particles. This behavior may have been due to rapid degradation of the particle core during the in-vitro release studies, thereby causing a higher burst release.
The release profiles performed in SILF suggest that PEI-1 and PEI-4 microspheres are likely to produce a prolonged-release effect after inhalation. However, it is possible that some drug molecules diffuse out of the particles and act like a non-particulate formulation. Such diffusion would be minimal since we optimized the porosity of the particles to minimize drug leakage from the particle core or through the porous network of the particle. If the drug had been released all at once, we would not have observed continuous release during in-vitro studies performed in the simulated interstitial lung fluid and/or during metabolic degradation studies described below (Fig. 6). It is also worth noting that SILF is a solution of electrolytes that mimics the electrolytes present in the surfactant-rich fluid released by Type II alveolar cells, which fills the space between alveolar cells and maintains the surface tension of water in the lungs (Daniels and Orgeig, 2003). In terms of electrolyte composition, SILF is virtually identical to actual human lung fluid, although it lacks the proteins present in actual lung fluids. However, in SILF the total ionic contribution from protein is compensated for by citrate ions (Moss, 1979). Because SILF provides an environment that is close to that of the mucosal fluids in the respiratory system, in-vitro release studies in this medium is likely to give a more accurate estimate of the drug that would be released upon pulmonary or intratracheal administration of the formulations. Importantly, there are very few studies that have used SILF for studying the release properties of an inhaled formulation (Cook et al., 2005; Davies and Feddah, 2003). This is the first study to document the release profile of an anti-PAH drug in a simulated lung fluid.
Figure 6.
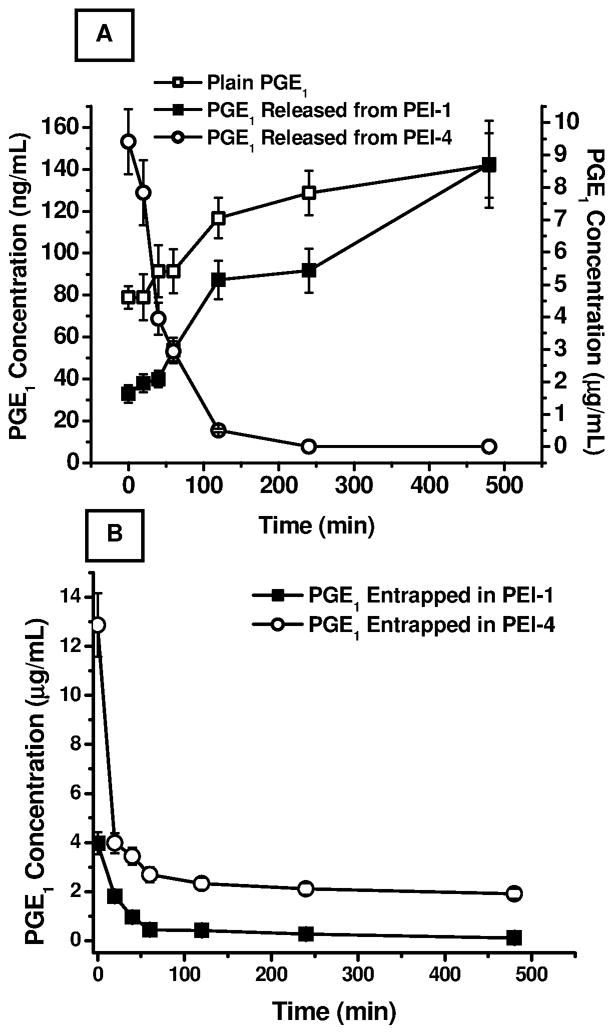
Metabolic degradation of PGE1 in rat lung homogenate. (A) Concentration of PGE1 upon incubation of 10 μg/mL plain PGE1, PEI-1, and PEI-4 containing 10 μg/mL PGE1. (B) The amount of drug extracted from PEI-1 and PEI-4 microspheres incubated in lung homogenates. Data represent mean ± standard deviation (n=3).
3.4. In-vivo Absorption Studies
In-vivo pulmonary drug absorption studies were performed in adult male Sprague-Dawley rats with the formulations that showed optimum micromeritic properties, entrapment and prolonged in-vitro release properties. As we have reported earlier, administration of plain PGE1 (80 μg/kg body weight) via the I.V. route resulted in a rapid rise in PGE1 levels in the plasma, with a Cmax of 80.09±9.37 ng/mL and a half-life (t1/2) of 1.5±0.2 minutes. Pulmonary administration of plain PGE1 showed a Cmax of 56.73±6.2 ng/mL and a half-life of 3.49±0.49 minutes with a relative bioavailability (F) of 0.821±0.15. (Fig. 5A, reproduced from Gupta et al. (Gupta et al., 2010). A rapid rise in plasma drug concentration suggests that intact PGE1 traversed the pulmonary epithelial membrane and reached the systemic circulation in biologically active form (Gupta et al., 2010). Since PGE1 is a small molecule like that of other prostacyclin analogs, inhaled PGE1 is likely to produce its vasodilatory effects directly on the pulmonary arterial wall by diffusing through the outer side (adventitial side) of the pulmonary vascular bed rather than reaching the vasculature upon recirculation from the pulmonary and bronchial artery as discussed previously (Gomberg-Maitland and Olschewski, 2008).
Figure 5.

In-vivo absorption studies. (A) Changes in plasma levels of PGE1 after administration of plain PGE1 (80 μg/kg) via the intravenous and pulmonary routes (n=6, modified from Gupta et al., 2010), and (B) In-vivo performances of the formulations (PEI-1, and PEI-4) after pulmonary administration of PGE1 (120 μg/kg). Data represent the mean ± standard deviation (n=6–8).
Two inhalable microspheric formulations, PEI-1 and PEI-4, were tested in-vivo at a dose of 120 μg/kg body weight. As can be seen in Fig. 5B, pulmonary administration of both PEI-1 and PEI-4 resulted in a significant increase in the half-life of PGE1 compared to the plain drug administered via either the pulmonary or I.V. route. With the increase in half-life of the drug, a concomitant reduction in Cmax was also observed. In fact, compared to plain PGE1 administered via the pulmonary route, a 2.5- to 4-fold reduction in Cmax (PEI-1 – 20.12±3.81 ng/mL, and PEI-4 – 14.51±2.91 ng/mL), and a 100–110 times extension in t1/2 (PEI-1 – 362.3±50.56 min, and PEI-4 – 390.66±55.71 min) were observed with both the microspheric formulations, when administered intratracheally (Table 3). As evident, the Cmax for PEI-1 microspheres was slightly higher than that for PEI-4. This can perhaps be attributed to PEI-1 having more pronounced surface-associated drug release compared to PEI-4, as observed in Figs. 4B and 4C. Importantly, the data from the in-vitro release study provide a good estimate of in-vivo absorption after pulmonary administration.
Table 3.
Pharmacokinetic parameters of PGE1-loaded PLGA microspheric formulations. Data represent mean ± standard deviation (n=6–8).
| Formulation | Cmax (ng/mL) | t1/2 (min) | AUC0–8 (ng/mL* min) | Relative Bioavailability |
|---|---|---|---|---|
| Plain PGE1 I.V.* | 80.09±19.37 | 1.5±0.2 | 723.53±86.4 | -- |
| Plain PGE1 Pulmonary* | 56.73±6.2 | 3.49±0.41 | 594.16±66.1 | 0.821±0.15 |
| PEI-1 | 20.12±3.81 | 362.296±50.56 | 1038.11±180.1 | 0.957±0.13 |
| PEI-4 | 14.51±2.91 | 390.66±55.71 | 973.52±130.2 | 0.897±0.17 |
AUC, Area under the concentration time curve; I.V., intravenous; PEI, Polyethyleneimine 25 kDa
Adapted from Gupta et al., 2010.
The initial rise and decline in the in-vivo absorption profile (Fig. 5B) of microsphere encapsulated PGE1 stems from the fact that a significant amount of drug associated with the particle surface was released soon after administration of the formulation, which is in conjunction with the in-vitro release data (Fig. 4C). In fact, release of surface associated drug results in the availability of more drug early in the absorption phase and such a release pattern is actually desirable for providing an immediate relief from an acute symptom such as hypertension. Nevertheless, the data presented in Fig. 5B reveals that PGE1 was available in the circulation at a concentration of ~7.5 ng/ml at 60 minutes following pulmonary microparticle administration whereas no drug was available in the circulation after 30 minutes of plain PGE1 administration (Fig. 5A). The later time points in Fig. 5B also show that the drug was available in the plasma at a concentration of ~5 ng/ml for about 8 hours, suggesting a continuous release of the drug from the optimized microparticulate formulations. In fact, commercially available PGE1 (Caverject®, currently used in erectile dysfunction) showed a Cmax between 0.089 or 0.102 ng/ml at 30 and 60 minutes following intracavernosal injection of 20 μg drug.
Several factors may have contributed to the reduced Cmax and extended t1/2 of the two formulations. First, because of their porous nature and large geometric diameter, phagocytosis of the particles by alveolar macrophages was delayed or prevented, which allowed for prolonged release of the drug (Edwards et al., 1997). Second, the average biodegradation time for PLGA is about 5–10 weeks, which may also have contributed to the prolonged release of the drug after pulmonary administration. Third, it is also possible that the PVA coating on the microparticle surface prevents polymer degradation by hydrolysis and thus facilitates continuous drug release over a prolonged period (Lee et al., 1999). Fourth, the glass transition temperature of the polymer used also plays an important role in drug release upon polymeric degradation. The glass transition temperature of PLGA 85:15 used in this study is ~45°C, thus making it prone to time-dependant degradation under physiological conditions (Park and Jonnalagadda, 2006). Overall, both large porous formulations showed a significantly extended half-life and excellent bioavailability as compared to our published report with nonporous PLGA microparticles.
3.5. Metabolic Degradation Studies in Rat Lung Homogenates
To determine whether the formulations that showed increased blood levels in the pulmonary absorption studies underwent metabolic degradation in the respiratory epithelium, we investigated the metabolic degradation profiles of PEI-1 and PEI-4 in rat lung homogenates. As discussed elsewhere, lung is a major site of metabolism of PGE1 and 70–80% of the drug is metabolized by oxidation, which is catalyzed by a microsomal enzyme, NAD+-dependent 15- hydroxy prostaglandin dehydrogenase (Nakano et al., 1973). This metabolic pathway is primarily responsible for rapid clearance of PGE1 from the lungs. The metabolic degradation study showed that plain PGE1 (10 μg/mL) underwent rapid degradation in lung homogenate, with little or no drug being detectable after 2 hour’s incubation. The drug concentration decreased from 9.415±1.005 μg/mL at time 0 to 0.505±0.05 μg/mL at 120 minutes. However, when the equivalent amount of PGE1 encapsulated in PLGA microspheres was incubated in lung homogenates, the drug concentration in the homogenate continually increased over the period of the experiment. For PEI-1, the amount of PGE1 released increased from 78.9±5.42 ng/mL at time 0 minutes to 141.9±15.42 ng/mL at 8 hours; for PEI-4, the amount of PGE1 increased from 32.83±4.21 ng/mL at time 0 to 142.42±20.741 ng/mL at 8 hours (Fig. 6A). The presence of PGE1 in the homogenates is an indication of continual release of the drug from the formulations as a result of time-dependent degradation of the polymeric core of the microparticles.
To further confirm the drug degradation in the lung homogenate, we also determined the amount of drug that remained unreleased from the microspheric formulations by extracting PGE1 from the microparticles incubated with the homogenates. As shown in Fig. 6B, with the increase in drug release, a concomitant decrease in the amount of drug in the polymeric microparticles was observed. The amount of drug in PEI-1 microspheres decreased from 3.96±0.45 μg/mL to 0.115±0.0142 μg/mL after 8 hours of incubation. A similar pattern was observed for PEI-4 microspheres: the drug concentration decreased from 12.87±1.3 μg/mL to 1.91±0.21 μg/mL after 8 hours (Fig. 6B). On the whole, in concurrence with the pulmonary absorption profiles, data on metabolic degradation suggest that polymeric microparticles continuously release PGE1 over an extended period and protect the unreleased drug against metabolic degradation.
3.6. Safety Studies
Safety of the formulations was studied in two sets of experiments: analysis of bronchoalveolar lavage and a cell viability study using a propidium iodide assay.
3.6.1
The acute safety of the formulations was studied by analyzing the bronchoalveolar lavage fluid collected from rats treated with two optimized formulations, PEI-1 and PEI-4, and a positive control 0.1% w/v solution of SDS. In addition to determining the levels of injury markers, alkaline phosphatase (ALP) and lactate dehydrogenase (LDH) in BAL fluid, total protein content and wet lung weight were also recorded to investigate accumulation of extracellular fluid in the epithelial cell lining of the respiratory wall upon drug inhalation. Presence of LDH, a cytoplasmic enzyme, in BAL fluid is representative of cell damage and lysis (Henderson et al., 1978). ALP is a membrane-bound lysosomal enzyme, and its presence in the BAL fluid represents alveolar type II cell proliferation in response to type I cell damage (Hussain and Ahsan, 2005). Further, total protein content of the BAL fluid is an important biomarker of permeability of the alveolar capillary barrier and inflammation (Beck et al., 1982). The inflammatory markers, ALP and LDH, are considered as the direct indicators of the inflammation in the respiratory system. In fact, the cough symptoms in humans are initiated by stimulation of two different classes of afferent nerves, myelinated rapidly adapting receptors and nonmyelinated C-fibers with endings in the lungs. Activation of these receptors results in mast cell degranulation that stimulates the release of various inflammatory markers and cause edema (Goldsobel and Chipps, 2010). Based on this fact, it is reasonable to argue that measurement of LDH and ALP levels an acceptable method to evaluate toxicity of inhaled formulations.
Wet lung weights of rats treated with PEI-1 were significantly higher than those of saline-treated animals, but lower than those of 0.1% SDS-treated rats. The increase in wet lung weights caused by PEI-4 was much smaller than that produced by the PEI-1 formulations (Fig. 7A). Further, compared to the saline-treated animals, no formulations produced a remarkable increase in total protein content or elevation in LDH and ALP levels. Both formulations were safe compared to 0.1% SDS. However, these data should be treated with caution because of the acute nature of the study. A long-term study using PEI-1 and PEI-4 would give better insight into the pathological changes that may occur upon repeated administration of the formulations.
Figure 7.
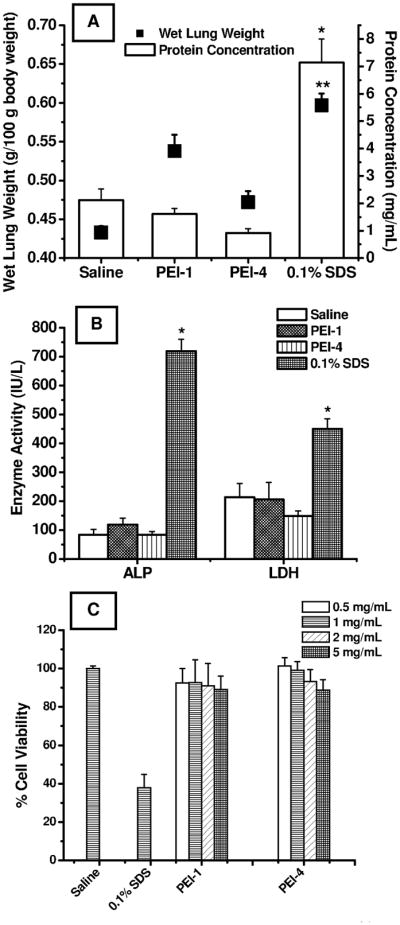
Acute in-vivo toxicity studies of optimized formulations PEI-1 and PEI-4 in bronchoalveolar lavage (BAL) fluid after 12 hours of administration. (A) Corrected wet lung weights (g/100 g body weight) and total protein concentration (mg/mL). *Means are significantly different from (p<0.05) (protein concentration), and **Means are significantly different (p<0.05) (wet lung weight). (B) Levels of lactate dehydrogenase (LDH) (IU/L), and alkaline phosphatase (ALP) (IU/L). *Means are significantly different (p<0.05). Data represent mean ± standard deviation (n=4). (C) Acute cytotoxicity studies to determine in-vitro safety profiles of the optimized formulations, PEI-1, and PEI-4, by propidium iodide fluorescence exclusion assay in the Calu-3 respiratory epithelial cell line at 0.5, 1.0, 2.0, and 5.0 mg/mL of microspheric formulations. Data represent mean ± standard deviation (n=16).
3.6.2
The cell viability study was performed using a propidium iodide exclusion fluorescence assay in Calu-3 cells, a lung epithelial cell line, a widely used model for studying transport and safety of inhaled drug or formulation adjuvants (Foster et al., 2000). Propidium iodide is a fluorescent dye and an intercalating agent that binds to double-stranded nucleic acids of non-viable cells by intercalating between bases. It is a membrane-impermeable molecule and excluded from viable cells (Nieminen et al., 1992; Pavlik et al., 1985). When bound to nucleic acids, propidium iodide causes a red shift and about a 20-to 30-fold increase in fluorescence compared to background. The cell viability data presented in Fig. 7C show high levels of cell viability when incubated with saline. In fact, saline-treated cells were 100% viable, whereas SDS-treated cells showed a viability of 37.79±7.03%. However, neither formulation produced a major reduction in cell viability. In the case of PEI-1, cell viability was reduced from 92.49±7.61% to 89.13±6.85% when the concentration of the formulation was increased from 0.5 mg/mL to 5 mg/mL. A similar reduction in cell viability was observed with PEI-4-treated cells at concentrations between 0.5 and 5 mg/mL. As is evident from the data, PEI, a polycationic absorption enhancer, when encapsulated in the polymeric particles, exhibited moderate cytotoxicity toward Calu-3 cells compared to SDS treated cells. This can be attributed to its reduced charge density upon complexation with PGE1. Although the cell viability studies suggest that both formulations are safe when treated with a bronchial epithelial cell line, caution should be exercised in translating the data to the overall safety of the formulation.
4. CONCLUSIONS
In summary, this study investigates in-vitro release profiles of polymeric PLGA microspheres of PGE1 in a simulated lung fluid. PEI-modified microspheres can enhance the drug payload and produce large porous particles. The resulting microspheres meet micromeritical standards of inhaled particles. The microspheres provided an extended biological half-life in addition to offering protection against metabolic degradation, suggesting that these formulations can overcome important limitations of existing therapies for pulmonary hypertension, a pulmonary vascular disorder. Currently, studies are underway in our laboratory to investigate the efficacy of these formulations in producing sustained vasodilation of pulmonary arteries in a rodent animal model of pulmonary hypertension.
Acknowledgments
The authors sincerely thank Dr. Louisa Hope-Weeks, Department of Chemistry, Texas Tech University, Lubbock, TX, for her help with particle analysis using the Microtrac S3500 and Nova 3200e Surface Area Analyzer; and Mr. Charles Linch at the Department of Medical Photography and Electron Microscopy, Texas Tech University Health Sciences Center, Lubbock, TX, for his help with the scanning electron microscopy experiments. This work was supported by an American Recovery and Reinvestment Act Fund, NIH 1R15HL103431 (FA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adjei A, Garren J. Pulmonary delivery of peptide drugs: effect of particle size on bioavailability of leuprolide acetate in healthy male volunteers. Pharm Res. 1990;7:565–569. doi: 10.1023/a:1015853824722. [DOI] [PubMed] [Google Scholar]
- Beck BD, Brain JD, Bohannon DE. An in vivo hamster bioassay to assess the toxicity of particulates for the lungs. Toxicol Appl Pharmacol. 1982;66:9–29. doi: 10.1016/0041-008x(82)90057-6. [DOI] [PubMed] [Google Scholar]
- Cirpanli Y, Unlu N, Calis S, Hincal AA. Formulation and in-vitro characterization of retinoic acid loaded poly (lactic-co-glycolic acid) microspheres. J Microencapsul. 2005;22:877–889. doi: 10.1080/02652040500273878. [DOI] [PubMed] [Google Scholar]
- Cook RO, Pannu RK, Kellaway IW. Novel sustained release microspheres for pulmonary drug delivery. J Control Release. 2005;104:79–90. doi: 10.1016/j.jconrel.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Daniels CB, Orgeig S. Pulmonary surfactant: the key to the evolution of air breathing. News Physiol Sci. 2003;18:151–157. doi: 10.1152/nips.01438.2003. [DOI] [PubMed] [Google Scholar]
- Davies NM, Feddah MR. A novel method for assessing dissolution of aerosol inhaler products. Int J Pharm. 2003;255:175–187. doi: 10.1016/s0378-5173(03)00091-7. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R. Large porous particles for pulmonary drug delivery. Science. 1997;276:1868–1871. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- Ewert R, Opitz CF, Wensel R, Winkler J, Halank M, Felix SB. Continuous intravenous iloprost to revert treatment failure of first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Clin Res Cardiol. 2007;96:211–217. doi: 10.1007/s00392-007-0490-3. [DOI] [PubMed] [Google Scholar]
- Foster KA, Avery ML, Yazdanian M, Audus KL. Characterization of the Calu-3 cell line as a tool to screen pulmonary drug delivery. Int J Pharm. 2000;208:1–11. doi: 10.1016/s0378-5173(00)00452-x. [DOI] [PubMed] [Google Scholar]
- Gill SK, Shobe AM, Hope-Weeks LJ. Synthesis of cobalt oxide aerogels and nanocomposite systems containing single-walled carbon nanotubes. Scanning. 2009;31:132–138. doi: 10.1002/sca.20147. [DOI] [PubMed] [Google Scholar]
- Goldsobel AB, Chipps BE. Cough in the pediatric population. J Pediatr. 2010;156:352–358. doi: 10.1016/j.jpeds.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. European Respiratory Journal. 2008;31:891–901. doi: 10.1183/09031936.00097107. [DOI] [PubMed] [Google Scholar]
- Gupta V, Ahsan F. Inhalational therapy for pulmonary arterial hypertension: current status and future prospects. Crit Rev Ther Drug Carrier Syst. 2010;27:313–370. doi: 10.1615/critrevtherdrugcarriersyst.v27.i4.20. [DOI] [PubMed] [Google Scholar]
- Gupta V, Davis M, Hope-Weeks LJ, Ahsan F. PLGA Microparticles Encapsulating Prostaglandin E1-Hydroxypropyl-β-cyclodextrin (PGE1-HPβCD) Complex for the Treatment of Pulmonary Arterial Hypertension (PAH) Pharm Res. 2011 doi: 10.1007/s11095-011-0409-6. In Press. [DOI] [PubMed] [Google Scholar]
- Gupta V, Rawat A, Ahsan F. Feasibility study of aerosolized prostaglandin E1 microspheres as a noninvasive therapy for pulmonary arterial hypertension. J Pharm Sci. 2010;99:1774–1789. doi: 10.1002/jps.21946. [DOI] [PubMed] [Google Scholar]
- Henderson RF, Damon EG, Henderson TR. Early damage indicators in the the lung I. Lactate dehydrogenase activity in the airways. Toxicol Appl Pharmacol. 1978;44:291–297. doi: 10.1016/0041-008x(78)90191-6. [DOI] [PubMed] [Google Scholar]
- Hirota K, Hasegawa T, Nakajima T, Inagawa H, Kohchi C, Soma G, Makino K, Terada H. Delivery of rifampicin-PLGA microspheres into alveolar macrophages is promising for treatment of tuberculosis. J Control Release. 2010;142:339–346. doi: 10.1016/j.jconrel.2009.11.020. [DOI] [PubMed] [Google Scholar]
- Hussain A, Ahsan F. State of insulin self-association does not affect its absorption from the pulmonary route. Eur J Pharm Sci. 2005;25:289–298. doi: 10.1016/j.ejps.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Jeong YI, Song JG, Kang SS, Ryu HH, Lee YH, Choi C, Shin BA, Kim KK, Ahn KY, Jung S. Preparation of poly(DL-lactide-co-glycolide) microspheres encapsulating all-trans retinoic acid. Int J Pharm. 2003;259:79–91. doi: 10.1016/s0378-5173(03)00207-2. [DOI] [PubMed] [Google Scholar]
- Jeyanthi R, Mehta RC, Thanoo BC, DeLuca PP. Effect of processing parameters on the properties of peptide-containing PLGA microspheres. J Microencapsul. 1997;14:163–174. doi: 10.3109/02652049709015330. [DOI] [PubMed] [Google Scholar]
- Labiris NR, Dolovich MB. Pulmonary drug delivery. Part I: physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol. 2003;56:588–599. doi: 10.1046/j.1365-2125.2003.01892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Oh JT, Jang MH, Chung SI. Quantitative analysis of polyvinyl alcohol on the surface of poly(D, L-lactide-co-glycolide) microparticles prepared by solvent evaporation method: effect of particle size and PVA concentration. J Control Release. 1999;59:123–132. doi: 10.1016/s0168-3659(98)00185-0. [DOI] [PubMed] [Google Scholar]
- Lee SH, Rubin LJ. Current treatment strategies for pulmonary arterial hypertension. J Intern Med. 2005;258:199–215. doi: 10.1111/j.1365-2796.2005.01542.x. [DOI] [PubMed] [Google Scholar]
- Lemoine D, Preat V. Polymeric nanoparticles as delivery system for influenza virus glycoproteins. J Control Release. 1998;54:15–27. doi: 10.1016/s0168-3659(97)00241-1. [DOI] [PubMed] [Google Scholar]
- Lipworth BJ. New perspectives on inhaled drug delivery and systemic bioactivity. Thorax. 1995;50:105–110. doi: 10.1136/thx.50.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J, Theilmeier G, Van Aken H, Bone HG, Busse H, Waurick R, Hinder F, Booke M. Inhaled prostaglandin E1 for treatment of acute lung injury in severe multiple organ failure. Anesth Analg. 1998;86:753–758. doi: 10.1097/00000539-199804000-00015. [DOI] [PubMed] [Google Scholar]
- Moss OR. Simulants of lung interstitial fluid. Health Phys. 1979;36:447–448. [PubMed] [Google Scholar]
- Nakano J, Prancan AV, Morsy NH. Metabolism of prostaglandin E1 in stomach, jejunum chyle and plasma of the dog and the rat. Jpn J Pharmacol. 1973;23:355–361. doi: 10.1254/jjp.23.355. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Gores GJ, Bond JM, Imberti R, Herman B, Lemasters JJ. A novel cytotoxicity screening assay using a multiwell fluorescence scanner. Toxicol Appl Pharmacol. 1992;115:147–155. doi: 10.1016/0041-008x(92)90317-l. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Kabasawa T, Ozeki T, Okada H. One-step preparation of rifampicin/poly(lactic-co-glycolic acid) nanoparticle-containing mannitol microspheres using a four-fluid nozzle spray drier for inhalation therapy of tuberculosis. J Control Release. 2009;135:19–24. doi: 10.1016/j.jconrel.2008.11.027. [DOI] [PubMed] [Google Scholar]
- Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, Nikkho S, Speich R, Hoeper MM, Behr J, Winkler J, Sitbon O, Popov W, Ghofrani HA, Manes A, Kiely DG, Ewert R, Meyer A, Corris PA, Delcroix M, Gomez-Sanchez M, Siedentop H, Seeger W. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med. 2002;347:322–329. doi: 10.1056/NEJMoa020204. [DOI] [PubMed] [Google Scholar]
- Papi A, Canonica GW, Maestrelli P, Paggiaro P, Olivieri D, Pozzi E, Crimi N, Vignola AM, Morelli P, Nicolini G, Fabbri LM. Rescue use of beclomethasone and albuterol in a single inhaler for mild asthma. N Engl J Med. 2007;356:2040–2052. doi: 10.1056/NEJMoa063861. [DOI] [PubMed] [Google Scholar]
- Park JW, Jeon OC, Kim SK, Al-Hilal TA, Moon HT, Kim CY, Byun Y. Anticoagulant efficacy of solid oral formulations containing a new heparin derivative. Mol Pharm. 2010;7:836–843. doi: 10.1021/mp900319k. [DOI] [PubMed] [Google Scholar]
- Park PIP, Jonnalagadda S. Predictors of glass transition in the biodegradable polylactide and poly-lactide-co-glycolide polymers. J Appl Polym Sci. 2006;100:1983–1987. [Google Scholar]
- Pavlik EJ, Flanigan RC, van Nagell JR, Jr, Hanson MB, Donaldson ES, Keaton K, Doss B, Bartmas J, Kenady DE. Esterase activity, exclusion of propidium iodide, and proliferation in tumor cells exposed to anticancer agents: phenomena relevant to chemosensitivity determinations. Cancer Invest. 1985;3:413–426. doi: 10.3109/07357908509039802. [DOI] [PubMed] [Google Scholar]
- Pistel KF, Kissel T. Effects of salt addition on the microencapsulation of proteins using W/O/W double emulsion technique. J Microencapsul. 2000;17:467–483. doi: 10.1080/026520400405723. [DOI] [PubMed] [Google Scholar]
- Qian T, Nieminen AL, Herman B, Lemasters JJ. Mitochondrial permeability transition in pH-dependent reperfusion injury to rat hepatocytes. Am J Physiol. 1997;273:C1783–1792. doi: 10.1152/ajpcell.1997.273.6.C1783. [DOI] [PubMed] [Google Scholar]
- Ravivarapu HB, Lee H, DeLuca PP. Enhancing initial release of peptide from poly(d,l-lactide-co-glycolide) (PLGA) microspheres by addition of a porosigen and increasing drug load. Pharm Dev Technol. 2000;5:287–296. doi: 10.1081/pdt-100100543. [DOI] [PubMed] [Google Scholar]
- Rawat A, Majumder QH, Ahsan F. Inhalable large porous microspheres of low molecular weight heparin: in vitro and in vivo evaluation. J Control Release. 2008;128:224–232. doi: 10.1016/j.jconrel.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricachinevsky CP, Amantea SL. Treatment of pulmonary arterial hypertension. J Pediatr (Rio J) 2006;82:S153–165. doi: 10.2223/JPED.1556. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, Pulido T, Frost A, Roux S, Leconte I, Landzberg M, Simonneau G. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Kouyoumjian S, Tashkin D, Roth MD. Synergistic cytotoxicity of Delta(9)-tetrahydrocannabinol and butylated hydroxyanisole. Toxicol Lett. 2002;133:171–179. doi: 10.1016/s0378-4274(02)00134-0. [DOI] [PubMed] [Google Scholar]
- Sarafian TA, Vartavarian L, Kane DJ, Bredesen DE, Verity MA. bcl-2 expression decreases methyl mercury-induced free-radical generation and cell killing in a neural cell line. Toxicol Lett. 1994;74:149–155. doi: 10.1016/0378-4274(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Shive MS, Anderson JM. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- Thomas C, Rawat A, Bai S, Ahsan F. Feasibility study of inhaled hepatitis B vaccine formulated with tetradecylmaltoside. J Pharm Sci. 2008;97:1213–1223. doi: 10.1002/jps.21069. [DOI] [PubMed] [Google Scholar]
- Vanbever R, Mintzes JD, Wang J, Nice J, Chen D, Batycky R, Langer R, Edwards DA. Formulation and physical characterization of large porous particles for inhalation. Pharm Res. 1999;16:1735–1742. doi: 10.1023/a:1018910200420. [DOI] [PubMed] [Google Scholar]
- Yang T, Hussain A, Bai S, Khalil IA, Harashima H, Ahsan F. Positively charged polyethylenimines enhance nasal absorption of the negatively charged drug, low molecular weight heparin. J Control Release. 2006;115:289–297. doi: 10.1016/j.jconrel.2006.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]