

Abstract
Comorbidities that promote the progression of Alzheimer's disease (AD) remain to be uncovered and evaluated in animal models. Because elderly individuals are vulnerable to viral and bacterial infections, these microbial agents may be considered important comorbidities that could potentiate an already existing and tenuous inflammatory condition in the brain, accelerating cognitive decline, particularly if the cellular and molecular mechanisms can be defined. Researchers have recently demonstrated that triggering inflammation in the brain exacerbates tau pathological characteristics in animal models. Herein, we explore whether inflammation induced via viral infection, compared with inflammation induced via bacterial lipopolysaccharide, modulates AD-like pathological features in the 3xTg-AD mouse model and provide evidence to support the hypothesis that infectious agents may act as a comorbidity for AD. Our study shows that infection-induced acute or chronic inflammation significantly exacerbates tau pathological characteristics, with chronic inflammation leading to impairments in spatial memory. Tau phosphorylation was increased via a glycogen synthase kinase-3β–dependent mechanism, and there was a prominent shift of tau from the detergent-soluble to the detergent-insoluble fraction. During chronic inflammation, we found that inhibiting glycogen synthase kinase-3β activity with lithium reduced tau phosphorylation and the accumulation of insoluble tau and reversed memory impairments. Taken together, infectious agents that trigger central nervous system inflammation may serve as a comorbidity for AD, leading to cognitive impairments by a mechanism that involves exacerbation of tau pathological characteristics.
Alzheimer's disease (AD) is a progressive neurodegenerative disorder and the leading cause of dementia, afflicting >35 million individuals worldwide. The AD brain displays several characteristic pathological features, including the buildup of amyloid plaques composed of amyloid-β (Aβ), which can also accumulate intracellularly, and neurofibrillary tangles composed of hyperphosphorylated tau protein.1 Neuronal loss, dystrophic neurites, and dendritic spine loss are other critical changes that are well documented in AD. In addition, inflammation, as evidenced by reactive glial cells surrounding amyloid plaques, is consistently observed in the AD brain.2–4
The factors and molecular mechanisms that affect the pathogenesis of AD still remain largely unknown, although it is widely accepted that this disorder is multifactorial. Certain factors and insults, such as hypoxia, brain ischemia, and stress, that dysregulate brain homeostasis and physiological functions may increase the susceptibility of developing AD (as comorbid factors).5,6 Thus, decades of research in epidemiology and postmortem AD brains has suggested that viral or bacterial infections may contribute to the onset of AD.7,8 With improved quantitative and analytical methods, several viral and bacterial genes, including herpes simplex virus, Chlamydia pneumoniae, and Helicobacter pylori, have been isolated from AD brains; in some reports,9–11 the presence of these genes in AD brains is statistically significantly higher than in non-AD brains. Evidence from both in vivo and in vitro studies indicates that infections significantly exacerbate AD-like pathological changes, suggesting that infection-mediated alterations (ie, altered immune response) in the brain may increase the susceptibility of developing AD later in life.12,13 Brain inflammatory responses may contribute to this pathogenic process.14–16
Neuroinflammation in the AD brain likely plays both beneficial and harmful roles.17 For example, chronic inflammation and cytokine up-regulation induce tau hyperphosphorylation in prepathological 3xTg-AD mice.15 In addition, studies18–21 indicate that inflammatory processes are involved in clearing or degrading Aβ depositions. The deficiency of CCR2, a chemokine receptor, impairs microglia accumulation and increases Aβ deposition in amyloid precursor protein (APP)-transgenic mice, indicating a role for microglia in regulating Aβ accumulation.22,23 On the other hand, chronic lipopolysaccharide (LPS)–induced neuroinflammation increases intraneuronal Aβ load in transgenic mice,16,24 possibly through the release of proinflammatory cytokines and other toxic species25,26 and the subsequent exacerbation of AD-related pathological features.27,28 Collectively, infection and neuroinflammation may well be linked to AD and may play key roles in the accelerated onset and development of the disease.
In this study, we investigated the role that viral and bacterial infections have on the development of the AD phenotype in the 3xTg-AD mouse model. Viral infection by mouse hepatitis virus (MHV) or LPS to mimic a bacterial infection induced robust, but transient, neuroinflammation; exacerbated tau pathological characteristics; and compromised cognitive function in aged 3xTg-AD mice. LPS injection caused an increase in tau phosphorylation and its partition to the detergent-insoluble fraction, indicating a buildup of aggregated tau in neurons; the aberrant activation of glycogen synthase kinase (GSK)-3β was concomitantly detected in these mice. GSK-3β appears to be one of the main cellular mediators that is activated by infection-induced inflammation, underlying the increased tau pathological characteristics. To determine whether GSK-3β was a necessary mediator of the inflammation-induced changes in tau, we treated mice with lithium, a potent GSK-3β inhibitor, and found that its inhibition reversed both the tau hyperphosphorylation and its shift into the insoluble fraction. Significantly, treatment with lithium also led to an improvement in the cognitive phenotype. Together, our data strongly suggest that viral- or bacterial-mediated infections may act as critical comorbid factors and that tau pathological features are accelerated.
Materials and Methods
Animals
3xTg-AD and nontransgenic (NonTg) mice were maintained on a 12-hour light-dark cycle and had free access to food and water. In this study, 11- to 13-month-old 3xTg-AD or age- and strain-matched NonTg mice were used.
LPS Injections of Aged Mice
LPS (from Escherichia coli 055:B5; Sigma, St Louis, MO) was dissolved in 0.9% NaCl at a concentration of 0.1 mg/mL. LPS was administered i.p. to 12-month-old 3xTg-AD or NonTg mice at a dose of 0.5 mg/kg body weight twice per week for 6 weeks [n = 9 (four females and five males) for 3xtg-AD mice and n = 12 (six females and six males) for NonTg mice)]. A control group of mice received injections in the same manner with 0.9% saline only [n = 10 (six females and four males) for 3xTg-AD mice and n = 9 (four females and five males) for NonTg mice)]. The amount of LPS injected was adjusted according to weight weekly. Mice were euthanized 48 hours after the last injection and perfused with ice-cold PBS, and their brains were isolated. Half of the brain was fixed in 4% paraformaldehyde, and the other half was snap frozen in dry ice and stored at −80°C.
LPS and Lithium Treatment of Aged 3xTg-AD Mice
Twelve-month-old 3xTg-AD mice were divided into four groups: group 1, received standard rodent chow and saline injections [n = 6 (four females and two males)]; group 2, received standard rodent chow and LPS injections [n = 6 (four females and two males)]; group 3, received lithium chloride (2 g/kg) containing rodent chow (AIN-76A; Research Diets, New Brunswick, NJ) and saline injections [n = 6 (four females and two males)]; and group 4, received lithium chloride containing rodent chow (AIN-76A) and LPS injections [n = 6 (four females and two males)]. LPS (0.5 mg/kg) or saline injections were given twice a week for 6 weeks, as previously described. No obvious weight loss was observed during the 6-week period, and lithium intake was estimated to be 6 to 10 mg/d per mouse based on the assumption that a mouse consumes 3 to 5 g/d of chow. During the last week of injections, mice underwent behavioral testing, as described later. Mice were euthanized 4 days after the last injection and perfused with ice-cold PBS, and their brains were isolated. Half of the brain was fixed in 4% paraformaldehyde, and the other half was snap frozen in dry ice and stored at −80°C.
MHV Infection
To evaluate the impact of MHV infection on AD pathological characteristics, 11- to 13-month-old 3xTg-AD or NonTg mice were infected with the neuroadapted JHM strain of MHV suspended in HBSS or received an injection of HBSS alone as sham controls.29,30 Mice were anesthetized by i.p. injection of ketamine (80 to 100 mg/kg; Phoenix, St. Joseph, MO) and xylazine (5 to 10 mg/kg; MP Biomedicals, LLC, Aurora, OH), diluted in sterile HBSS. Anesthetized mice were injected intracranially with 500 plaque-forming units of the neurotrophic MHV strain J2.2-V.1 (provided by John Fleming, M.D., University of Wisconsin, Madison, WI), diluted in 30 μL of sterile HBSS (n = 34 to 37).31–33 Sham control animals were injected with 30 μL HBSS alone and did not develop any behavioral deficits.
Flow Cytometric Analysis of the Monocyte Population
Mice were euthanized 3, 7, and 10 days after MHV infection (n = 6 to 9); and infiltrating leukocytes were immunophenotyped by fluorescence-activated cell sorted staining to define the surface antigens using established methods.29 Brains were removed and stored on ice in 5 mL Dulbecco's modified Eagle's media until processing. The tissue was transferred to a sterile Petri dish and mashed into a single-cell suspension. The cell suspension was transferred to a 15-mL conical tube, and a medium (Percoll; GE Healthcare, Uppsala, Sweden) was added for a final concentration of 30%. The medium (70% Percoll), 1 mL, was underlain; and tubes were centrifuged at 1100 × g for 30 minutes at 4°C. Live cells were collected from the interface, washed twice, and stained for flow cytometric analysis. Isolated cells were Fc blocked with anti-CD16/32, 1:200 (BD Biosciences, San Jose, CA), and immunophenotyed with fluorescent antibodies (BD Biosciences) specific for the following cell surface markers: CD4 (L3T4), CD8a (53-6.7), CD45 (30-F11; eBiosciences, San Diego, CA), I-A/I-E (M5/114.15.2), and F4/80 (CI:A3-1; AbD Serotec, Raleigh, NC). Appropriate isotype antibodies were used for each antibody. Cells were run on a flow cytometer (FACSCalibur; BD Biosciences) and analyzed with software (FlowJo; TreeStar, OR). Frequency data are presented as the percentage of positive cells within the gated population. Total cells were calculated by multiplying these values by the total number of live cells isolated.
Intracellular Cytokine Staining
Intracellular staining for interferon-γ was performed in cells isolated from the brains of MHV-infected control mice and transgenic mice. Cells were stimulated with 5 μmol/L peptide from a control antigen or from the MHV spike glycoprotein (S510, residues 510 to 518). Stimulated cells were incubated for 6 hours at 37°C in media containing Golgi/Stop (Cytofix/Cytoperm kit; BD Biosciences), at which point cells were Fc blocked with anti-CD16/32, 1:200 (BD Biosciences). Cells were then stained with fluorescent antibodies (BD Biosciences) for the following cell surface markers: CD4 (L3T4), CD8a (53-6.7), and CD45 (30-F11; eBiosciences).
Measurement of Spinal Cord Pathological Characteristics
Spinal cords were obtained from experimental groups at day 14 post infection (p.i.) and fixed by immersion overnight in 10% normal buffered formalin, after which portions of tissue were embedded in paraffin. Spinal cords (7-μm sections) were stained with Luxol fast blue and analyzed by light microscopy. Demyelination was scored as follows: 0, no demyelination; 1, mild inflammation accompanied by loss of myelin integrity; 2, moderate inflammation with increasing myelin damage; 3, numerous inflammatory lesions accompanied by a significant increase in myelin stripping; and 4, intense areas of inflammation accompanied by numerous phagocytic cells engulfing myelin debris. Slides containing stained spinal cord sections were blinded and scored.
Immunoblotting
Frozen brain halves were homogenized in tissue protein extraction reagent (Pierce, Rockford, IL), protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN), and phosphatase inhibitors (5 mmol/L sodium fluoride and 50 μmol/L sodium orthovanadate). Homogenates were centrifuged at 100,000 x g for 1 hour at 4°C. Supernatants were collected as the detergent-soluble fraction. Pellets were resuspended in 70% formic acid and homogenized. After centrifugation at 100,000 x g for 1 hour at 4°C, the resulting supernatants were saved as the formic acid fraction. Protein concentrations were determined by the Bradford method. Equal amounts of protein (20 to 50 μg, depending on the protein of interest) were separated by SDS-PAGE on a 10% Bis-Tris gel (Invitrogen, Carlsbad, CA), transferred to 0.45-μm polyvinylidene difluoride membranes, and blocked for 1 hour in 5% (v/v) nonfat milk in Tris-buffered saline (pH 7.5) supplemented with 0.2% Tween 20. Fractions were immunoblotted with antibodies that recognize APP, total tau (HT7), total endogenous tau (Dako, Carpinteria, CA), phosphorylated tau [AT8, Ser202/Thr205; AT180, Thr231/Ser235; AT100, Thr212/Ser214; and paired helical filament (PHF-1), Ser396/Ser404 (Pierce)], p35/p25 (Santa Cruz Biotechnologies, Santa Cruz, CA), cdk5 (Calbiochem, La Jolla, CA), total GSK-3αβ or phospho-GSK-3β (Ser9) (both from Cell Signaling, Beverly, MA), or total GSK-3β (BD Biosciences). Antibody against β-actin was used as a loading control. Formic acid fractions were neutralized by mixing equal amounts of sample, 10N NaOH, and neutralizing buffer (1 mol/L Tris base and 0.5 mol/L Na2HPO). Equal amounts of protein in the neutralized sample were separated on SDS-PAGE gels in the same manner as the soluble fractions. Quantification of band intensity was measured using software (Scion Image) and was normalized with glyceraldehyde-3-phosphate dehydrogenase, β-actin, or total tau (HT7) levels (for phosphorylated tau analyses).
Immunohistochemistry
Fixed brain halves were sliced on a vibratome at 50 μmol/L thickness. Before overnight incubation with primary antibody, sections were quenched with 3% hydrogen peroxide plus 10% methanol, permeabilized with 0.1% Triton X-100 Tris-buffered saline, and blocked in solution containing 3% bovine serum albumin. After incubation with primary antibody in Tris-buffered saline containing 3% serum overnight, slices were washed with 0.1% Triton X-100 Tris-buffered saline and incubated with the appropriate secondary antibody. The presence of secondary antibody in tissue was revealed by reaction with diaminobenzidine. Certain antigens required special conditions. Aβ staining required pretreatment with 90% formic acid. CD45 antigen required removal of detergent during antibody incubations.
Images of stained hippocampus, entorhinal cortex, and amygdala were acquired by a digital camera (Axiocam) connected to a microscope (Axioskop 50) (Carl Zeiss MicroImaging, Thornwood, NY) and software (AxioVision 4.6). Aβ plaques with a diameter >10 μm were counted in three to four random fields of the CA1 hippocampus in each animal to quantitatively analyze the plaque number. For the analysis of phosphorylated tau-bearing neurons, AT8- or PHF-1–positive neurons in the subiculum and CA1 hippocampus were counted in each animal.
Aβ Enzyme-Linked Immunosorbent Assay
To measure Aβ levels, equal amounts of protein (200 μg) from soluble fractions were loaded directly onto enzyme-linked immunosorbent assay (ELISA) plates. Equal amounts of protein (200 μg) from formic acid fractions were diluted 1:20 in neutralization buffer (1 mol/L Tris base and 0.5 mol/L Na2HPO) before loading. Before loading, antibody mAβ20.1 at a concentration of 25 μg/mL in coating buffer (0.1 mol/L NaCO3 buffer, pH 9.6) was coated onto immunoplates (Nunc, Naperville, IL); and plates were blocked with 3% bovine serum albumin. Synthetic Aβ standards of both Aβ40 and Aβ42 were made in antigen capture buffer [20 mmol/L NaH2PO4, 2 mmol/L EDTA, 0.4 mol/L NaCl, 0.5 g (in 1 L total volume) of 3-([3-cholamidopropyl]dimethylammonio)-1-propanesulfonate, and 1% bovine serum albumin (pH 7.0)] and loaded onto ELISA plates, along with soluble and formic acid fractions. Samples and standards were loaded in duplicate, and plates were incubated overnight at 4°C. Plates were washed and probed with either horseradish peroxidase–conjugated anti–Aβ35-40 (MM32-13.1.1 for Aβ1-40) or anti–Aβ35-42 (MM40-21.3.4 for Aβ1-42) overnight at 4°C. The chromogen used was tetramethylbenzidine, and 30% O-phosphoric acid was used to stop the reaction. The concentration of samples was determined from readings at 450 nm.
Cytokine ELISA
To detect levels of IL-1β and IL-6 in homogenized brains, ELISA kits were purchased (Pierce) and the protocol provided by the manufacturer was followed. Briefly, samples were incubated with biotinylated antibodies against IL-1β and IL-6 on precoated plates. After incubation with a streptavidin–horseradish peroxidase solution, tetramethylbenzidine chromogen was applied and the manufacturer-supplied stop solution was used. The concentration of samples was determined by reading at 450 nm.
Kinase Assay
Brain samples were immunoprecipitated with protein G–agarose and GSK-3β antibody or protein A–agarose and cdk5 antibody. A reaction mixture containing 20 mmol/L 4-morpholinepropanesulfonic acid (pH 7.2), 5 mmol/L MgCl2, 1 mmol/L sodium orthovanadate, 5 mmol/L NaF, 100 μmol/L ATP, 2.5 μCi (γ-32P)ATP, and 0.2 mmol/L cdk5 substrate (Calbiochem) for the cdk5 kinase assay or 0.2 mmol/L GSK-3β substrate (Calbiochem) for the GSK-3β assay. After allowing the reaction to proceed for 1 hour at 30°C, the supernatant was placed on P81 phosphocellulose squares (Upstate, Waltham, MA). After washing in 0.3% phosphoric acid, squares were counted in a scintillation counter to determine the kinase activity.
Behavior Testing (Morris Water Maze)
The apparatus used for the water maze task was a circular aluminum tank (1.2-m diameter), painted white and filled with water maintained at 22°C to 24°C. The maze was located in a room containing several simple visual extramaze cues. To reduce stress, mice were placed on the platform for 10 seconds before the first training trial. Mice were trained to swim to a 14-cm diameter circular clear Plexiglas platform submerged 1.5 cm beneath the surface of the water and invisible to the mice while swimming. The platform location was selected randomly for each mouse but was kept constant for each individual mouse throughout training. For each trial, the mouse was placed into the tank at one of four designated starting points in a pseudorandom order. Mice were allowed to find and escape onto the submerged platform. If a mouse failed to find the platform within 60 seconds, the mouse was manually guided to the platform and allowed to remain there for 10 seconds. Afterward, each mouse was placed into a holding cage under a warming lamp for 25 seconds until the start of the next trial. To ensure that memory differences were not because of lack of task learning, mice were given four trials a day for as many days as were required to train the lithium-untreated and lithium-treated 3xTg-AD mice to criterion (<20 seconds mean escape latency before the first probe trial was run). To control for overtraining, probe trials were run for each group, both as soon as they reached group criterion and after all groups had reached criterion. Retention of the spatial training was assessed 1.5 hours and again 24 hours after the last training trial. In the probe trials, the platform was removed from the pool and mice were monitored by a ceiling-mounted camera directly above the pool during the 60-second period. All trials were recorded for subsequent analysis. The parameters measured during the probe trial included the following: i) time spent in the target quadrant (the quadrant where the platform was located), ii) latency to cross the platform location, and iii) number of platform location crosses. Target quadrants varied between mice within a group to control for potential differences in the salience of extramaze cues.
Statistical Analysis
All data were analyzed using one-way analysis of variance, with a posttest including a Dunnett, Bonferroni, or Newman-Keuls post hoc test or an unpaired t-test. P ≤ 0.05 was considered statistically significant.
Results
Acute Neuroinflammation Induced by MHV Infection Exacerbates AD-Like Pathological Characteristics in Aged 3xTg-AD Mice
We first examined a temporal profile of central nervous system (CNS) immune responses after an acute viral infection in the CNS of mice. Both NonTg and 3xTg-AD mice were infected with MHV via a single unilateral injection into the hippocampus, and this induced a robust neuroinflammatory response characterized by increased infiltration of macrophages (CD45High or F4/80+), activation of microglia (CD45Low, F4/80+, or major histocompatibility complex class II positive), and infiltration of T cells (CD4+ or CD8+) (see Supplemental Figure S1, A–C, at http://ajp.amjpathol.org). There was no significant difference in mortality between infected 3xTg-AD (25.4%) and NonTg (20.6%) mice. These infected mice successfully controlled viral replication, and viral titers within the brains were reduced to lower than detectable levels (approximately 100 plaque-forming units per gram of tissue) by day 12 p.i. (see Supplemental Figure S1D at http://ajp.amjpathol.org). These findings indicate that no differences exist between aged 3xTg-AD and age-matched NonTg mice in limiting viral replication within the CNS. In support of this observation, no differences were detected in interferon-γ secretion by resident cells within the CNS, as measured by major histocompatibility complex class II staining on resident microglia (see Supplemental Figure S1B at http://ajp.amjpathol.org), and in virus-specific T cell infiltration in the CNS, as measured by interferon-γ secretion by peptide-stimulated T cells (see Supplemental Figure S1E at http://ajp.amjpathol.org).
Given the evidence that both NonTg and 3xTg-AD mice exhibited the same immune responses against MHV infection, we investigated how the acute CNS insult and subsequent immune responses affected the long-lasting neuropathological changes in the brain and spinal cord. Thus, we examined neuropathological changes at 4 weeks after MHV injection, when no residual short-term immune responses were detected in the brain. We found detectable demyelination in the spinal cord of both NonTg and 3xTg-AD mice that received MHV injection, but not in sham-injected mice (see Supplemental Figure S2 at http://ajp.amjpathol.org). Some mice also showed phenotypic signs of motor impairments; therefore, no cognitive tests were conducted in MHV-injected mice. Based on these results, we further examined detailed neuropathological changes and underlying mechanisms in 3xTg-AD mice at earlier points.
At 2 and 4 weeks after the MHV injection in aged 3xTg-AD mice, we analyzed the effect on Aβ and tau pathological characteristics. Although we detected a significant increase in β-C-terminal fragment (CTF) (C99 fragment), but not in α-CTF (C83 fragment), 2 weeks after infection (Figure 1A; see also Supplemental Figure S3 at http://ajp.amjpathol.org), the overall Aβ pathological features, as measured by quantitative ELISA and immunostaining, were not altered at both points (Figure 1, B and C). However, marked exacerbation of the tau pathological characteristics in the brain was observed in MHV-infected 3xTg-AD mice, but not in sham-injected 3xTg-AD mice (Figure 1, A and D; see also Supplemental Figure S3 at http://ajp.amjpathol.org). MHV-injected NonTg (control) mice did not develop any obvious accumulation of Aβ or tau in the hippocampus (Figure 1E). The exacerbation of tau pathological features after MHV infection in 3xTg-AD mice was associated with the aberrant activation of GSK-3β, as measured by the steady-state levels of phospho–GSK-3β at Ser9 and increased cdk5/p25 in the brain at both points (Figure 1A; see also Supplemental Figure S3 at http://ajp.amjpathol.org).
Figure 1.
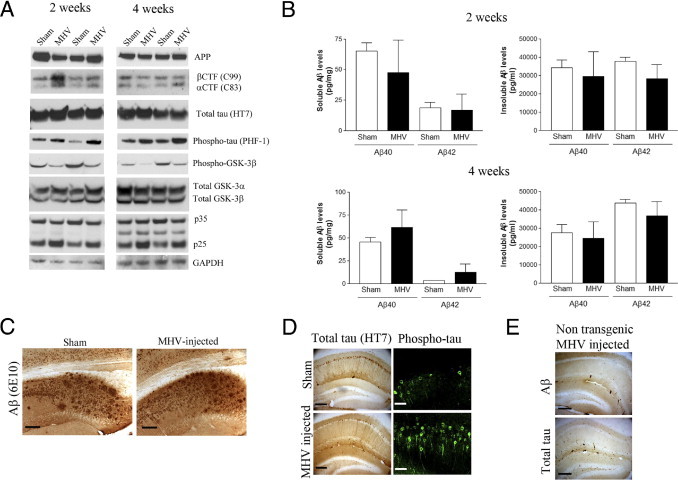
A single MHV injection selectively exacerbates tau pathological characteristics in an aged 3xTg-AD mouse. A: Immunoblot analyses of APP processing and tau pathological features 2 or 4 weeks after MHV infection. Densitometric analyses of these bands are shown in Supplemental Figure S3 (available at http://ajp.amjpathol.org). B: Quantitative analysis of soluble and insoluble Aβ in the brain of sham- or MHV-treated 3xTg-AD mice. No significant difference was observed in Aβ levels (n = 5). C: Representative Aβ plaque burden in the hippocampal region of sham- or MHV-treated 3xTg-AD mice (4 weeks after injection). D: Representative immunostaining of tau in the hippocampus. Total tau (HT7) and phospho-tau (phosphorylated at Ser202/Thr205). E: Representative Aβ and tau immunohistological staining on NonTg mice that received a single MHV injection. No obvious Aβ or tau accumulation is detected (n = 5). Scale bars: 100 μm (C); 200 μm (D, left); 30 μm (D, right); 200 μm (E). CTF indicates; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Long-Term LPS Injection Exacerbates Tau Pathological Features in Aged 3xTg-AD Mice
Next, we sought to examine how sustained brain inflammation affected AD-like pathological characteristics. Because viral infection into the brain was efficiently cleared within 10 to 14 days, as previously described, we used an alternative approach, systemically injecting LPS repeatedly as a mimic of bacterial infection. This treatment paradigm has induced robust inflammation in the brain of several mouse models, including prepathological 3xTg-AD mice, but not aged mice.15,16,34 Aged 3xTg-AD mice were injected with LPS for 6 weeks, which led to a marked increase in brain inflammation, as indicated by more CD45+ infiltrated macrophages and microglia and by elevated levels of proinflammatory cytokines (see Supplemental Figure S4, A–C, at http://ajp.amjpathol.org).
Despite evidence of a chronic inflammatory response in the brain, the plaque load appeared similar between the LPS-injected and saline-treated 3xTg-AD mice because we did not detect any difference in the staining pattern for Aβ plaque pathological features using antibody 6E10 (Figure 2, C and D). APP steady-state levels were unchanged in the LPS-treated mice, indicating that APP processing was likely unchanged (Figure 2A). Furthermore, we quantitatively analyzed Aβ1-40 and Aβ1-42 levels in both the soluble and insoluble (ie, formic acid) fractions by ELISA and found that Aβ levels were not significantly different between LPS- and saline-treated groups (Figure 2B). Based on a previous study15 in which LPS was injected into prepathological 3xTg-AD mice, and the results previously described, including use of a single MHV infection or long-term LPS injections into aged 3xTg-AD mice, we found that chronic inflammation does not appear to significantly affect the onset or progression of Aβ pathological characteristics.
Figure 2.
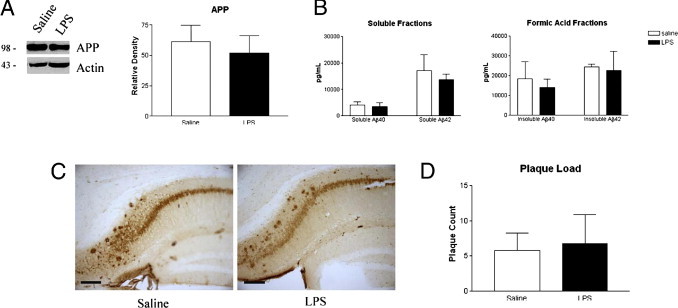
Aβ load is not altered in LPS-treated 3xTg-AD mice. A: APP levels by Western blot analysis using 6E10 antibody appear unchanged between LPS- and saline-treated mice. B: Aβ load, as determined by ELISA, is not significantly different between LPS- and saline-treated mice. C: Representative 6E10 immunoreactivity in saline- and LPS-treated mice reveals similar levels of both intraneuronal and extracellular Aβ. Scale bar = 100 μm. D: Plaque counts between saline- and LPS-treated mice are not significantly different.
Unlike the effect on Aβ, chronic inflammation significantly decreased tau steady-state levels in the detergent-soluble fraction by approximately 50% (Figure 3, A and B). Despite decreases in soluble tau levels, higher levels of phospho-tau were observed in the soluble fraction of the LPS-treated mice (Figure 3, A and B). Specifically, phosphorylation at the AT100 (Thr212/Ser214) site was significantly increased twofold compared with saline controls. In the LPS-treated mice, phosphorylation at Ser214 was significantly increased by 30%, whereas phosphorylation at Thr212 was decreased. Studies35 have indicated that phosphorylation at Ser214 may inhibit further phosphorylation at Thr212, producing the pattern of phosphorylation observed herein. Favoring phosphorylation at Ser214 is also consistent with an increase in reactivity with the AT100 epitope because phosphorylation at Ser214 is required for formation of the AT100 epitope.35 In addition, AT8+ neurons were markedly increased (>10 times) in mice exposed to chronic inflammation (Figure 3, C and D).
Figure 3.
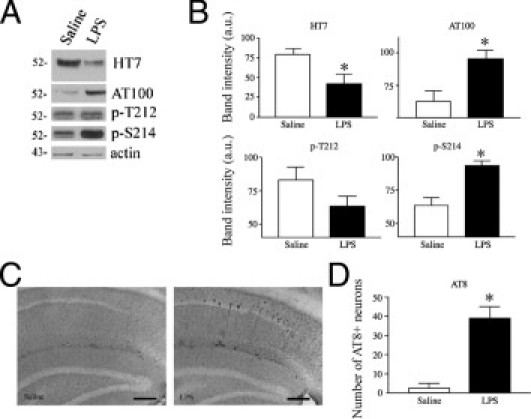
Phospho-tau is increased in LPS-treated 3xTg-AD mice. A: Mice with chronic inflammation have reduced levels of tau by immunoblot, as evident with antibody HT7. Phospho-tau, as evident with antibody AT100 [Thr212 (T212) or Ser214 (S214)], is increased in LPS-treated mice. B: Quantification of the immunoblots shows that total tau levels, as measured by HT7, are significantly decreased in the soluble fraction, whereas phospho-tau is significantly increased (*P < 0.05, n = 5). C: Representative AT8 immunoreactivity showing increased levels of AT8 reactivity in LPS-treated mice. Scale bar = 100 μm. D: The number of AT8 immunoreactive cells in the subiculum and CA1 hippocampus is significantly increased in LPS-treated mice (*P < 0.05, n = 5).
Because chronic inflammation appears to be exacerbating tau pathological features, and tau may be shifting to the insoluble fraction, we examined tau levels in the insoluble formic acid fraction. In LPS-treated mice, we observed a significant 50% increase in detergent-insoluble tau (Figure 4, A and B). To determine whether this shift in tau to the insoluble fraction was associated with the development of more advanced tau pathological features, we examined levels of PHF-1–immunoreactive tau, which were significantly increased by 100% in the formic acid fractions of LPS-treated mice (Figure 4, A and B). In addition, the number of PHF-1–immunoreactive neurons was significantly increased by 2.5 times in LPS-treated mice (Figure 4C). However, we were not able to detect any AD-like neurofibrillary tangles in these mice, suggesting that increased accumulation of insoluble tau in LPS-treated mice could be pretangle tau, such as protofibrillar or oligomeric intermediates of tau, which may eventually turn into neurofibrillary tangle–like structures.
Figure 4.
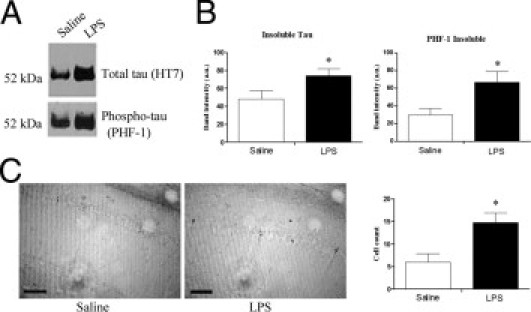
Insoluble tau and phospho-tau is increased in LPS-treated 3xTg-AD mice. A: Representative HT7 and PHF-1 immunoblot of formic acid fractions shows increased levels of tau and phosphorylated tau in LPS-treated mice. B: Quantification of HT7 and PHF-1 immunoblot shows significantly increased levels of tau and PHF-1–positive tau in the formic acid fraction of LPS-treated mice (*P < 0.05, n = 5). C: Representative PHF-1 reactivity shows increased levels of PHF-1 in LPS-treated mice. PHF-1–immunoreactive neurons in the subiculum and CA1 hippocampus are significantly increased in LPS-treated mice (*P < 0.05, n = 5). Scale bar = 100 μm.
Next, we investigated whether the exacerbation of tau pathological characteristics was mediated by aberrant activation of GSK-3β and cdk5. Previously, aberrant cdk5/p25 activation exacerbated tau phosphorylation in prepathological young 3xTg-AD mice.15 Interestingly, in this study, we observed a significant 50% decrease in levels of p35 and p25 in the LPS-injected aged 3xTg-AD mice (Figure 5, A and B). On the other hand, we detected a significant decrease in levels of GSK-3β phosphorylated at Ser9 in LPS-treated mice (Figure 5, A and B). Radioactive kinase assays further confirmed that GSK-3β activity was significantly increased by 25.2% in mice exposed to chronic inflammation, whereas there was no significant difference in cdk5 activity between saline- and LPS-treated mice (Figure 5C). This discrepancy between young and aged 3xTg-AD mice may be the result of a different temporal activation pattern of cdk5 and GSK-3β in the brain. At least in the 3xTg-AD mouse model, cdk5/p25 activation was detected at a younger age, peaked at approximately 12 months, and then decreased during aging, whereas GSK-3β activity increased in an age-dependent manner up to the age of 15 months (see Supplemental Figure S5, A and B, at http://ajp.amjpathol.org). Our findings collectively show that infection-induced inflammatory responses in the brain play a key role in exacerbating tau pathological features through dysregulated kinase activity, particularly mediated by GSK-3β.
Figure 5.
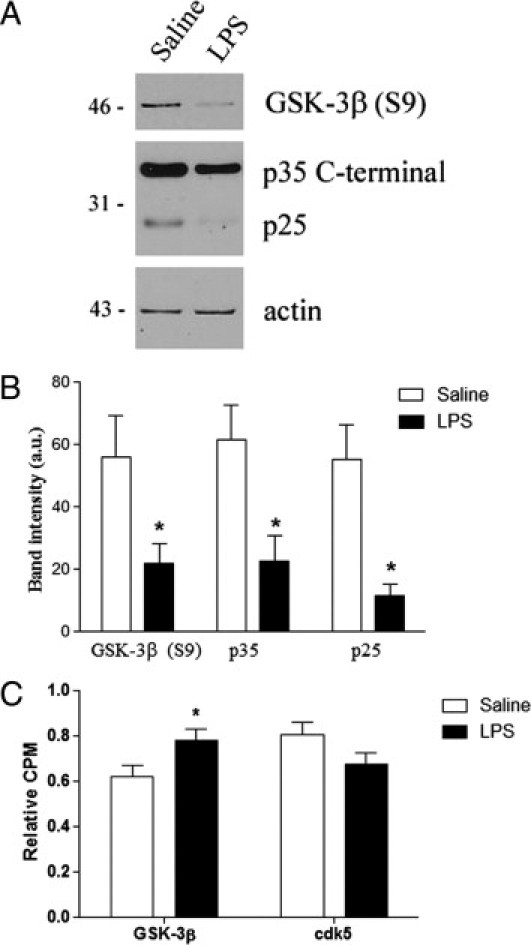
GSK-3β–dependent mechanism underlies the increase in phospho-tau in 3xTg-AD mice with advanced pathological features. A: Representative immunoblots showing decreased levels of GSK-3β phosphorylated at Ser9, p35, and p25 in LPS-treated mice. B: Quantification of immunoblots reveals significantly decreased levels of GSK-3β (S9), p35, and p25 in LPS-treated mice (*P < 0.05, n = 5). a.u. indicates arbitrary unit. C: Kinase assay for GSK-3β activity shows significant increased levels of GSK-3β activity in LPS-treated mice (*P < 0.05, n = 10).
Inhibiting GSK-3β Attenuates Tau Pathological Features in an Aged 3xTg-AD Mouse
To confirm that increased GSK-3β activity underlies the enhanced phosphorylation of tau induced by LPS in older mice, we treated LPS-injected mice with lithium, a well-established potent inhibitor of GSK-3β. Similar to the previous experiments, LPS (0.5 mg/kg) was injected twice a week for 6 weeks into 12-month-old 3xTg-AD mice. One group of mice was maintained on normal feed, whereas another group was given lithium-enriched feed. Previously, this amount of lithium in the feed has inhibited GSK-3β activity.36 A cohort of mice was subjected to behavioral tasks, mice were sacrificed after the 6-week period, and their brains were harvested.
Treating mice with lithium reversed several pathological effects of chronic inflammation. In particular, GSK-3β, phosphorylated at Ser9, was significantly increased in lithium-treated mice, indicating successful inhibition of GSK-3β activity (Figure 6, A and B). We observed a significant threefold increase in detergent-soluble tau levels in lithium-treated mice, along with a significant threefold decrease in tau phosphorylated at AT8 in the soluble fraction (Figure 6, A and B). In addition, in the insoluble fraction, PHF-1–positive tau was significantly decreased threefold compared with mice treated with LPS only (Figure 6, C and D). Lithium treatment appeared to reverse the LPS-induced tau pathological characteristics without altering the Aβ load or the inflammatory responses, as measured by IL-1β and IL-6 (Figure 7, A–C).
Figure 6.
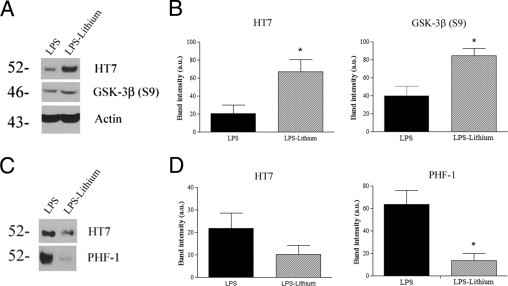
Lithium cotreatment rescues LPS-induced tau pathological characteristics. A: Representative HT7 immunoblots showing increased tau levels in the detergent-soluble fractions of mice treated with both LPS and lithium versus mice treated with only LPS. GSK-3β (S9) is also increased in mice treated with LPS and lithium. B: Quantification of immunoblots shows significantly increased levels of soluble HT7 and GSK-3β (S9) in mice treated with both LPS and lithium. *P < 0.05, n = 6. C: Representative HT7 and PHF-1 immunoblots showing decreased tau levels in the formic acid fractions of mice treated with both LPS and lithium. D: Quantification of immunoblots shows significantly decreased levels of PHF-1–positive tau (*P < 0.05, n = 6).
Figure 7.
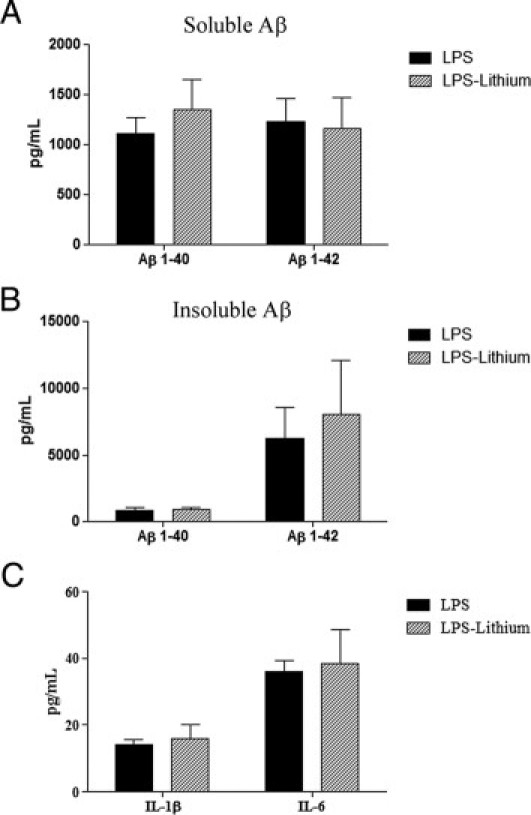
Aβ and cytokine load is unchanged between LPS-treated mice and mice treated with both LPS and lithium. Detergent-soluble (A) and formic acid–soluble (B) Aβ levels by ELISA are unchanged. C: Levels of IL-1β and IL-6, as measured by ELISA, are unchanged.
Chronic Inflammation Induces Learning and Memory Impairments
Chronic inflammation in the brains of LPS-treated mice induced learning and memory impairments. In the Morris Water Maze (MWM) task, the LPS-treated mice took longer to reach criterion, indicating impairments in learning (Figure 8A). Mice with elevated inflammation also exhibited a significant increase in escape latency to cross the platform location and decrease the number of crosses of the platform location at both 1.5- and 24-hour probe trials (Figure 8, B and C). In comparison, age-matched NonTg mice that received LPS in the same manner also exhibited a delayed latency to cross the platform location at the 24-hour probe trial, yet to a much lower degree than that in LPS-treated 3xTg-AD mice (see Supplemental Figure S6A at http://ajp.amjpathol.org). The number of platform crosses was, unlike 3xTg-AD mice, not different between sham and LPS treatments in NonTg mice (see Supplemental Figure S6B at http://ajp.amjpathol.org). Endogenous tau phosphorylation and insoluble tau levels were concomitantly increased in LPS-treated NonTg mice (see Supplemental Figure S6C at http://ajp.amjpathol.org). Our data suggested that cognition was much more affected in 3xTg-AD mice, possibly because of the presence of pathological mutant tau and the exacerbation of tau pathological characteristics. These results were, in part, confirmed by the fact that lithium treatment reversed the behavioral impairments induced by chronic inflammation. Mice treated with both lithium and LPS performed comparably to saline-treated animals in the MWM. In contrast, LPS-treated mice perform significantly worse than saline controls. In addition, mice treated with both lithium and LPS have significantly less latency to cross the platform compared with LPS-treated mice in the probe trials (Figure 8, B and C).
Figure 8.
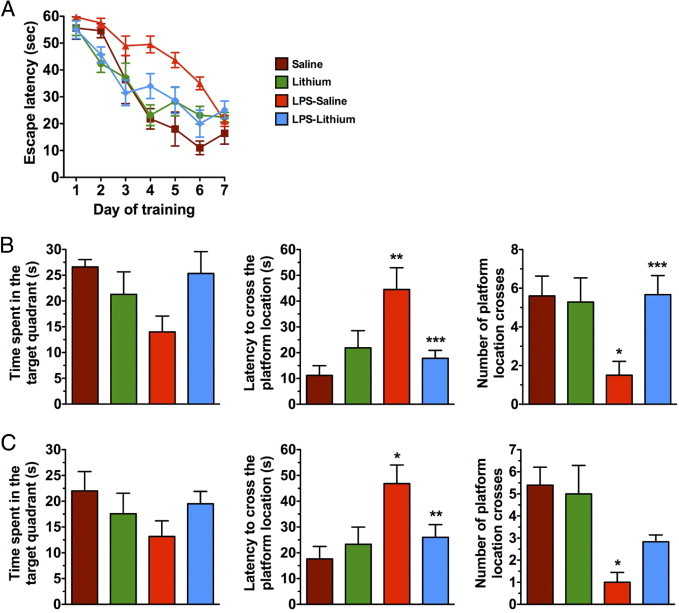
Cognitive impairments induced by LPS treatment are prevented by lithium cotreatment in aged 3xTg-AD mice. A: Acquisition curve during the training of MWM. LPS-treated mice require more training to reach criterion. B: LPS-treated mice have a significantly increased latency to cross the platform location in the 1.5-hour probe trial. LPS-treated mice cotreated with lithium perform significantly better than mice treated with LPS only. *P < 0.05 or **P < 0.01 versus the saline-treated group, and ***P < 0.05 versus the LPS-treated group. C: LPS-treated mice are also impaired in the 24-hour probe trial with significantly increased latency to cross the platform and significantly fewer platform crosses versus saline-treated controls. *P < 0.05 versus the saline-treated group, and **P < 0.05 versus the LPS-treated group.
Discussion
The present study demonstrates that acute or chronic viral or bacterial infections that modulate brain inflammatory responses markedly affect the development of tau pathological features in a mouse model of AD. The inflammation-mediated exacerbation of tau pathological features leads to impairments in cognition that are effectively blocked by inhibiting GSK-3β activity. Previously, young prepathological 3xTg-AD mice exhibited increased tau phosphorylation in CA1 neurons after LPS-induced inflammation,15 suggesting that sustained brain inflammation triggers tau pathological features. This pathological role of inflammation is further supported by another study37 showing that inflammation precedes tau tangle formation in a mouse model of tauopathy. The present study provides novel evidence that chronic inflammation, as triggered by viral infection or LPS injection, initiates and contributes to the progression of tau pathological features and cognitive impairment in aged 3xTg-AD mice. We observed increased tau phosphorylation at several epitopes, including Ser202/Thr205 (AT8) and Ser396/Ser404 (PHF-1), and increased detergent-insoluble tau accumulation in LPS-injected mice. Interestingly, however, we were not able to detect tanglelike pathological features of tau in these mice at this age. The increase in detergent-insoluble tau or somatodendritic tau accumulation after the administration of infectious agents may be mediated by increased formation of pretangle tau, such as protofibrillar or oligomeric tau intermediates, that may play an important role in neuronal loss, synaptic dysfunction, and cognitive impairments.38 These changes in tau pathological characteristics were associated with dysregulation of GSK-3β kinase activity, consistent with numerous mouse models that show that GSK-3β becomes the primary modulator of tau phosphorylation as mice age.39–41 Interestingly, in contrast to previous observations in young prepathological 3xTg-AD mice, we find a significant decrease in p35, a coregulator of cdk5, without an increase in the generation of p25; cdk5 activity was decreased in older mice with chronic inflammation. As mice age, cdk5 becomes a regulator of GSK-3β activity, with studies39,41 showing that increased cdk5 activity can induce phosphorylation of GSK-3β at the Ser9 site, leading to inhibition of GSK-3β activity. GSK-3β is not considered a direct substrate for cdk5, but cdk5 may mediate its actions through either up-regulation of the phosphoinositide 3-kinase/Akt pathway or inhibition of protein phosphatases.41,42 Thus, the decreased cdk5 activity observed in older mice with chronic inflammation may be partly responsible for the significant increase in GSK-3β activity observed.
Dysregulation of kinase activity and subsequent tau hyperphosphorylation have been implicated to be important in the development of tau aggregates and ultimately the classic tangles observed in the AD brain.43 We demonstrate that inflammation is, in part, responsible for altered kinase activity in the brain, further exacerbating tau pathological features in neurons. The inhibition of GSK-3β by lithium and subsequent blockade of tau phosphorylation attenuated detergent-insoluble tau and improved cognitive function in these mice. Previously, no significant rescue effect of lithium was found on cognition of aged (16 months) 3xTg-AD mice.44 The discrepancy with our present study may be, in part, because of the ages of mice, which significantly affect both AD-like neuropathological and cognitive impairments. The age we used in this study (11 to 13 months) may still be young enough so that cognitive impairments can be reversed by modulating pathological changes. Thus, our data indicate that tau phosphorylation is an important process for the formation of pretangle tau intermediates and tau aggregation or neurofibrillary tangles. In addition, our data suggest that GSK-3β hyperactivity and the subsequent increase in phospho-tau accumulation may partly contribute to the early cognitive impairment in 3xTg-AD mice. Hyperphosphorylation of tau is known to reduce its stability, which can, in turn, cause axonal transport deficits.45,46 The integrity of the axonal transport system is crucial for maintaining proper synapse function.47 Deficits in synapse function secondary to the hyperphosphorylation of tau may underlie the cognitive dysfunction observed in mice with chronic inflammation. A recent study48 showed that inhibiting GSK-3β activity or genetically reducing GSK-3β levels prevents memory reconsolidation. In contrast, our data show that long-lasting elevated GSK-3β activity impairs learning and memory, consistent with a study49 showing memory impairments on short-term hyperactivation of GSK-3β. Thus, although normal activity of GSK-3β may be necessary for memory consolidation, hyperactive or long-lasting active GSK-3β and consequent hyperphosphorylation of tau is likely detrimental to learning and memory.
Our study particularly focuses on the pathological involvement of GSK-3β on tau pathological characteristics after chronic inflammation, and lithium was used as an inhibitor for GSK-3β. Lithium may activate other beneficial pathways to facilitate tau degradation. Lithium is known to inhibit protein phosphatase 2A to maintain the phosphorylation of Akt and GSK-3β.50,51 A recent study52 demonstrates that lithium also activates autophagy by inhibiting inositol monophosphatase. Autophagy has degraded aberrant protein aggregates in cytosol and attenuated disease conditions.53,54 How phosphorylated tau is effectively degraded after lithium treatment is still an open question.
In conclusion, we find that viral infection or LPS-mediated changes in brain inflammation can exacerbate both tau pathological characteristics and cognitive decline in the aged 3xTg-AD mice. The immune responses are altered and become dysfunctional during aging.55 Therefore, infectious agents may not be as detrimental in young individuals when the immune responses can be properly regulated, but this becomes more deleterious in older individuals. However, the LPS injection used in our study does not completely mimic bacterial infections, and some of its course of action may be different. For example, the course of duration is much shorter compared with typical bacterial infections, and the lack of bacterial foci may influence the host immune responses. Despite these differences, LPS is still widely used as a surrogate for bacterial infection, and its actions on inflammatory responses are robust and comparable to bacterial infections. Overall, we believe this work is significant because it shows that certain microbial infections may act as comorbid factors for AD, which, based on the evidence we observed herein, appear to act mainly by affecting tau pathological features and exacerbating cognitive decline.
Footnotes
Supported by grants from the National Institutes of Health: NIH/National Institute of Arthritis and Musculoskeletal and Skin DiseasesK99AR054695 (M.K.), NIH/National Institute on Aging (NIA) R01AG20335 (F.M.L.), and Program Project grant AG00538 (F.M.L.). Aβ antibodies were provided by the University of California, Irvine, Alzheimer's Disease Research Center, funded by NIH/NIA grant P50AG16573.
M.S. and M.K. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.02.012.
Supplementary data
Infiltration of inflammatory monocytes in the brain after MHV infection in NonTg and aged 3xTg-AD mice. NonTg and 3xTg-AD mice, aged 11 to 13 months, received a single intrahippocampal injection of MHV. After infection, NonTg and 3xTg-AD mice had increased infiltrating macrophages (CD45HighF4/80+) (A), activation of resident microglia (frequency and geomean of I-A/I-E+, gated on CD45LowF4/80+ cells) (B), and increased CD4- and CD8+ T cells (C). Sham infection of NonTg and 3xTg-AD mice showed that the baseline level of neuroinflammation in both groups was low before infection (solid and dashed lines, respectively; A–C). NonTg and 3xTg-AD mice quickly controlled viral replication lower than detectable levels (D) and developed robust virus-specific immune responses (E). A red bar indicates the mean. A broken line shows the assay limit (150 plaque-forming units per gram) (D). In these experiments, n = 2 mice for sham treatment (baseline measurement, A–C) and n = 6 to 9 mice for MHV injection.
A single MHV injection causes demyelination in the spinal cord. Neuropathological changes in the spinal cord were examined 4 weeks after the MHV injection. Clear demyelinations and cell infiltrations were observed in MHV-injected mice (dashed circles), whereas sham-injected mice showed a normal and intact structure.
Quantitative analyses of APP processing and tau-related proteins in the brains of MHV-injected mice. Densitometric analyses were performed in the immunoblots shown in Figure 5A. *P < 0.05 or **P < 0.01 versus sham-treated mice (n = 5 per group).
Neuroinflammation is increased in aged LPS-treated 3xTg-AD mice. A: Representative CD45 immunoreactivity in saline-injected 3xTg-AD mice. B: Representative CD45 immunoreactivity in LPS-treated mice. C: Quantification of the pixel density of CD45 immunoreactivity shows mice with chronic inflammation have increased activated microglia (P < 0.05, n = 5). D: IL-1β and IL-6 levels, as determined by ELISA, in LPS-treated mice and saline-injected controls. Levels of IL-6 are significantly higher in LPS-treated mice (P < 0.05, n = 5).
Temporal activation pattern of cdk5 and GSK-3β in the brain of 3xTg-AD mice. A: Representative immunoblot analysis of temporal pattern of phospho–GSK-3β and p35/p25 levels in the brain of 3xTg-AD mice (n = 4). B: Quantitative analysis of p25/p35 ratio and phospho–GSK-3β levels from the immunoblot data. *P < 0.05 or **P < 0.01 versus data from 3-month-old 3xTg-AD mice.
Effect of long-term LPS treatment on NonTg mice. NonTg mice, aged 11 to 13 months, received 0.5 mg/kg LPS or saline twice a week for 6 weeks (n = 9 to 12). At the end of the treatment, all mice were tested by MWM. Latency to cross-platform location (A) and number of platform crosses (B) at the 24-hour probe trial are shown. For comparison, data from 3xTg-AD mice are also included. *P < 0.05 versus the saline-treated NonTg group, and #P < 0.05 versus the saline-treated 3xTg-AD group. C: Immunoblot analysis of tau in detergent-soluble and detergent-insoluble fractions. Graphs represent densitometric analysis of phospho-tau (AT8) in the soluble fraction and total tau in the insoluble fraction (n = 9 to 12). *P < 0.05 versus the saline group.
References
- 1.Selkoe D.J. Toward a remembrance of things past: deciphering Alzheimer disease. Harvey Lect. 2003;99:23–45. [PubMed] [Google Scholar]
- 2.Itagaki S., McGeer P.L., Akiyama H., Zhu S., Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989;24:173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 3.McGeer P.L., Rogers J., McGeer E.G. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- 4.Rogers J., Lue L.F. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer's disease. Neurochem Int. 2001;39:333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- 5.Rothman S.M., Mattson M.P. Adverse stress, hippocampal networks, and Alzheimer's disease. Neuromolecular Med. 2010;12:56–70. doi: 10.1007/s12017-009-8107-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viswanathan A., Rocca W.A., Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. 2009;72:368–374. doi: 10.1212/01.wnl.0000341271.90478.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holmes C., Cotterell D. Role of infection in the pathogenesis of Alzheimer's disease: implications for treatment. CNS Drugs. 2009;23:993–1002. doi: 10.2165/11310910-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Honjo K., van Reekum R., Verhoeff N.P. Alzheimer's disease and infection: do infectious agents contribute to progression of Alzheimer's disease? Alzheimers Dement. 2009;5:348–360. doi: 10.1016/j.jalz.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 9.Balin B.J., Gerard H.C., Arking E.J., Appelt D.M., Branigan P.J., Abrams J.T., Whittum-Hudson J.A., Hudson A.P. Identification and localization of Chlamydia pneumoniae in the Alzheimer's brain. Med Microbiol Immunol. 1998;187:23–42. doi: 10.1007/s004300050071. [DOI] [PubMed] [Google Scholar]
- 10.Kountouras J., Tsolaki M., Gavalas E., Boziki M., Zavos C., Karatzoglou P., Chatzopoulos D., Venizelos I. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;66:938–940. doi: 10.1212/01.wnl.0000203644.68059.5f. [DOI] [PubMed] [Google Scholar]
- 11.Itzhaki R.F., Wozniak M.A. Herpes simplex virus type 1 in Alzheimer's disease: the enemy within. J Alzheimers Dis. 2008;13:393–405. doi: 10.3233/jad-2008-13405. [DOI] [PubMed] [Google Scholar]
- 12.Little C.S., Hammond C.J., MacIntyre A., Balin B.J., Appelt D.M. Chlamydia pneumoniae induces Alzheimer-like amyloid plaques in brains of BALB/c mice. Neurobiol Aging. 2004;25:419–429. doi: 10.1016/S0197-4580(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 13.Wozniak M.A., Itzhaki R.F., Shipley S.J., Dobson C.B. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429:95–100. doi: 10.1016/j.neulet.2007.09.077. [DOI] [PubMed] [Google Scholar]
- 14.Brugg B., Dubreuil Y.L., Huber G., Wollman E.E., Delhaye-Bouchaud N., Mariani J. Inflammatory processes induce beta-amyloid precursor protein changes in mouse brain. Proc Natl Acad Sci U S A. 1995;92:3032–3035. doi: 10.1073/pnas.92.7.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kitazawa M., Oddo S., Yamasaki T.R., Green K.N., LaFerla F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer's disease. J Neurosci. 2005;25:8843–8853. doi: 10.1523/JNEUROSCI.2868-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheng J.G., Bora S.H., Xu G., Borchelt D.R., Price D.L., Koliatsos V.E. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–145. doi: 10.1016/s0969-9961(03)00069-x. [DOI] [PubMed] [Google Scholar]
- 17.Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- 18.Ard M.D., Cole G.M., Wei J., Mehrle A.P., Fratkin J.D. Scavenging of Alzheimer's amyloid beta-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 19.Akiyama H., Schwab C., Kondo H., Mori H., Kametani F., Ikeda K., McGeer P.L. Granules in glial cells of patients with Alzheimer's disease are immunopositive for C-terminal sequences of beta-amyloid protein. Neurosci Lett. 1996;206:169–172. doi: 10.1016/s0304-3940(96)12474-5. [DOI] [PubMed] [Google Scholar]
- 20.Herber D.L., Roth L.M., Wilson D., Wilson N., Mason J.E., Morgan D., Gordon M.N. Time-dependent reduction in Abeta levels after intracranial LPS administration in APP transgenic mice. Exp Neurol. 2004;190:245–253. doi: 10.1016/j.expneurol.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Herber D.L., Mercer M., Roth L.M., Symmonds K., Maloney J., Wilson N., Freeman M.J., Morgan D., Gordon M.N. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2:222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- 22.El Khoury J., Toft M., Hickman S.E., Means T.K., Terada K., Geula C., Luster A.D. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 23.Wyss-Coray T., Yan F., Lin A.H., Lambris J.D., Alexander J.J., Quigg R.J., Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J.W., Lee Y.K., Yuk D.Y., Choi D.Y., Ban S.B., Oh K.W., Hong J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Griffin W.S., Sheng J.G., Royston M.C., Gentleman S.M., McKenzie J.E., Graham D.I., Roberts G.W., Mrak R.E. Glial-neuronal interactions in Alzheimer's disease: the potential role of a “cytokine cycle” in disease progression. Brain Pathol. 1998;8:65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akiyama H., Barger S., Barnum S., Bradt B., Bauer J., Cole G.M., Cooper N.R., Eikelenboom P., Emmerling M., Fiebich B.L., Finch C.E., Frautschy S., Griffin W.S., Hampel H., Hull M., Landreth G., Lue L., Mrak R., Mackenzie I.R., McGeer P.L., O'Banion M.K., Pachter J., Pasinetti G., Plata-Salaman C., Rogers J., Rydel R., Shen Y., Streit W., Strohmeyer R., Tooyoma I., Van Muiswinkel F.L., Veerhuis R., Walker D., Webster S., Wegrzyniak B., Wenk G., Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y., Liu L., Barger S.W., Griffin W.S. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–1611. doi: 10.1523/JNEUROSCI.23-05-01605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintanilla R.A., Orellana D.I., Gonzalez-Billault C., Maccioni R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Stiles L.N., Hardison J.L., Schaumburg C.S., Whitman L.M., Lane T.E. T cell antiviral effector function is not dependent on CXCL10 following murine coronavirus infection. J Immunol. 2006;177:8372–8380. doi: 10.4049/jimmunol.177.12.8372. [DOI] [PubMed] [Google Scholar]
- 30.Walsh K.B., Lodoen M.B., Edwards R.A., Lanier L.L., Lane T.E. Evidence for differential roles for NKG2D receptor signaling in innate host defense against coronavirus-induced neurological and liver disease. J Virol. 2008;82:3021–3030. doi: 10.1128/JVI.02032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lane T.E., Asensio V.C., Yu N., Paoletti A.D., Campbell I.L., Buchmeier M.J. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol. 1998;160:970–978. [PubMed] [Google Scholar]
- 32.Totoiu M.O., Nistor G.I., Lane T.E., Keirstead H.S. Remyelination, axonal sparing, and locomotor recovery following transplantation of glial-committed progenitor cells into the MHV model of multiple sclerosis. Exp Neurol. 2004;187:254–265. doi: 10.1016/j.expneurol.2004.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang F.I., Stohlman S.A., Fleming J.O. Demyelination induced by murine hepatitis virus JHM strain (MHV-4) is immunologically mediated. J Neuroimmunol. 1990;30:31–41. doi: 10.1016/0165-5728(90)90050-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McAlpine F.E., Lee J.K., Harms A.S., Ruhn K.A., Blurton-Jones M., Hong J., Das P., Golde T.E., LaFerla F.M., Oddo S., Blesch A., Tansey M.G. Inhibition of soluble TNF signaling in a mouse model of Alzheimer's disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34:163–177. doi: 10.1016/j.nbd.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng-Fischhofer Q., Biernat J., Mandelkow E.M., Illenberger S., Godemann R., Mandelkow E. Sequential phosphorylation of tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- 36.Kitazawa M., Trinh D.N., LaFerla F.M. Inflammation induces tau pathology in inclusion body myositis model via glycogen synthase kinase-3beta. Ann Neurol. 2008;64:15–24. doi: 10.1002/ana.21325. [DOI] [PubMed] [Google Scholar]
- 37.Yoshiyama Y., Higuchi M., Zhang B., Huang S.M., Iwata N., Saido T.C., Maeda J., Suhara T., Trojanowski J.Q., Lee V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Sahara N., Maeda S., Takashima A. Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res. 2008;5:591–598. doi: 10.2174/156720508786898442. [DOI] [PubMed] [Google Scholar]
- 39.Wen Y., Planel E., Herman M., Figueroa H.Y., Wang L., Liu L., Lau L.F., Yu W.H., Duff K.E. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28:2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oddo S., Caccamo A., Cheng D., Jouleh B., Torp R., LaFerla F.M. Genetically augmenting tau levels does not modulate the onset or progression of Abeta pathology in transgenic mice. J Neurochem. 2007;102:1053–1063. doi: 10.1111/j.1471-4159.2007.04607.x. [DOI] [PubMed] [Google Scholar]
- 41.Plattner F., Angelo M., Giese K.P. The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem. 2006;281:25457–25465. doi: 10.1074/jbc.M603469200. [DOI] [PubMed] [Google Scholar]
- 42.Fu A.K., Fu W.Y., Cheung J., Tsim K.W., Ip F.C., Wang J.H., Ip N.Y. Cdk5 is involved in neuregulin-induced AChR expression at the neuromuscular junction. Nat Neurosci. 2001;4:374–381. doi: 10.1038/86019. [DOI] [PubMed] [Google Scholar]
- 43.Arnaud L., Robakis N.K., Figueiredo-Pereira M.E. It may take inflammation, phosphorylation and ubiquitination to “tangle” in Alzheimer's disease. Neurodegener Dis. 2006;3:313–319. doi: 10.1159/000095638. [DOI] [PubMed] [Google Scholar]
- 44.Caccamo A., Oddo S., Tran L.X., LaFerla F.M. Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol. 2007;170:1669–1675. doi: 10.2353/ajpath.2007.061178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alonso A.D., Grundke-Iqbal I., Barra H.S., Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Evans D.B., Rank K.B., Bhattacharya K., Thomsen D.R., Gurney M.E., Sharma S.K. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau's ability to promote microtubule assembly. J Biol Chem. 2000;275:24977–24983. doi: 10.1074/jbc.M000808200. [DOI] [PubMed] [Google Scholar]
- 47.Mandelkow E.M., Stamer K., Vogel R., Thies E., Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 48.Kimura T., Yamashita S., Nakao S., Park J.M., Murayama M., Mizoroki T., Yoshiike Y., Sahara N., Takashima A. GSK-3beta is required for memory reconsolidation in adult brain. PLoS One. 2008;3:e3540. doi: 10.1371/journal.pone.0003540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Zhang J.X., Du X.X., Zhao L., Tian Q., Zhu L.Q., Wang S.H., Wang J.Z. Temporal correlation of the memory deficit with Alzheimer-like lesions induced by activation of glycogen synthase kinase-3. J Neurochem. 2008;106:2364–2374. doi: 10.1111/j.1471-4159.2008.05578.x. [DOI] [PubMed] [Google Scholar]
- 50.Cross D.A., Alessi D.R., Cohen P., Andjelkovich M., Hemmings B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 51.Mora A., Sabio G., Risco A.M., Cuenda A., Alonso J.C., Soler G., Centeno F. Lithium blocks the PKB and GSK3 dephosphorylation induced by ceramide through protein phosphatase-2A. Cell Signal. 2002;14:557–562. doi: 10.1016/s0898-6568(01)00282-0. [DOI] [PubMed] [Google Scholar]
- 52.Sarkar S., Floto R.A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L.J., Rubinsztein D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005;170:1101–1111. doi: 10.1083/jcb.200504035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fornai F., Longone P., Cafaro L., Kastsiuchenka O., Ferrucci M., Manca M.L., Lazzeri G., Spalloni A., Bellio N., Lenzi P., Modugno N., Siciliano G., Isidoro C., Murri L., Ruggieri S., Paparelli A. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2008;105:2052–2057. doi: 10.1073/pnas.0708022105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heiseke A., Aguib Y., Riemer C., Baier M., Schatzl H.M. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109:25–34. doi: 10.1111/j.1471-4159.2009.05906.x. [DOI] [PubMed] [Google Scholar]
- 55.Jimenez S., Baglietto-Vargas D., Caballero C., Moreno-Gonzalez I., Torres M., Sanchez-Varo R., Ruano D., Vizuete M., Gutierrez A., Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Infiltration of inflammatory monocytes in the brain after MHV infection in NonTg and aged 3xTg-AD mice. NonTg and 3xTg-AD mice, aged 11 to 13 months, received a single intrahippocampal injection of MHV. After infection, NonTg and 3xTg-AD mice had increased infiltrating macrophages (CD45HighF4/80+) (A), activation of resident microglia (frequency and geomean of I-A/I-E+, gated on CD45LowF4/80+ cells) (B), and increased CD4- and CD8+ T cells (C). Sham infection of NonTg and 3xTg-AD mice showed that the baseline level of neuroinflammation in both groups was low before infection (solid and dashed lines, respectively; A–C). NonTg and 3xTg-AD mice quickly controlled viral replication lower than detectable levels (D) and developed robust virus-specific immune responses (E). A red bar indicates the mean. A broken line shows the assay limit (150 plaque-forming units per gram) (D). In these experiments, n = 2 mice for sham treatment (baseline measurement, A–C) and n = 6 to 9 mice for MHV injection.
A single MHV injection causes demyelination in the spinal cord. Neuropathological changes in the spinal cord were examined 4 weeks after the MHV injection. Clear demyelinations and cell infiltrations were observed in MHV-injected mice (dashed circles), whereas sham-injected mice showed a normal and intact structure.
Quantitative analyses of APP processing and tau-related proteins in the brains of MHV-injected mice. Densitometric analyses were performed in the immunoblots shown in Figure 5A. *P < 0.05 or **P < 0.01 versus sham-treated mice (n = 5 per group).
Neuroinflammation is increased in aged LPS-treated 3xTg-AD mice. A: Representative CD45 immunoreactivity in saline-injected 3xTg-AD mice. B: Representative CD45 immunoreactivity in LPS-treated mice. C: Quantification of the pixel density of CD45 immunoreactivity shows mice with chronic inflammation have increased activated microglia (P < 0.05, n = 5). D: IL-1β and IL-6 levels, as determined by ELISA, in LPS-treated mice and saline-injected controls. Levels of IL-6 are significantly higher in LPS-treated mice (P < 0.05, n = 5).
Temporal activation pattern of cdk5 and GSK-3β in the brain of 3xTg-AD mice. A: Representative immunoblot analysis of temporal pattern of phospho–GSK-3β and p35/p25 levels in the brain of 3xTg-AD mice (n = 4). B: Quantitative analysis of p25/p35 ratio and phospho–GSK-3β levels from the immunoblot data. *P < 0.05 or **P < 0.01 versus data from 3-month-old 3xTg-AD mice.
Effect of long-term LPS treatment on NonTg mice. NonTg mice, aged 11 to 13 months, received 0.5 mg/kg LPS or saline twice a week for 6 weeks (n = 9 to 12). At the end of the treatment, all mice were tested by MWM. Latency to cross-platform location (A) and number of platform crosses (B) at the 24-hour probe trial are shown. For comparison, data from 3xTg-AD mice are also included. *P < 0.05 versus the saline-treated NonTg group, and #P < 0.05 versus the saline-treated 3xTg-AD group. C: Immunoblot analysis of tau in detergent-soluble and detergent-insoluble fractions. Graphs represent densitometric analysis of phospho-tau (AT8) in the soluble fraction and total tau in the insoluble fraction (n = 9 to 12). *P < 0.05 versus the saline group.