

Abstract
Many individuals infected with hepatitis C virus (HCV) develop a chronic infection, and of those who are treated with pegylated interferon and ribavirin (RBV), many do not respond. While the nucleoside analog RBV improves treatment outcome, and will likely be an important component of therapy with next-generation viral inhibitors, RBV's mechanism is controversial. Most of RBV's proposed mechanisms require RBV import into cells. Therefore, we explored whether host-based RBV resistance develops through reduced cellular uptake, akin to chemotherapy resistance in some cancers. We examined the effect of host-based RBV resistance on HCV replication in cultured hepatoma Huh7.5 liver cells and whether RBV resistance develops in HCV patients. When Huh7.5 cells were exposed to RBV, resistance developed through reduced RBV uptake via the ENT1 nucleoside transporter and antiviral efficacy was reduced. The uptake defect in RBV-resistant cells was specific to RBV, since transport of another ENT1 substrate, cytidine, was unaffected. Importantly, RBV uptake significantly declined in HCV patient peripheral blood mononuclear cells (PBMCs) following 4 weeks of therapy. Furthermore, maintenance of RBV uptake correlated with rapid treatment response. Our results uncovered a novel form of antiviral drug resistance and suggest that host-based RBV resistance develops in HCV patients undergoing therapy and that maintenance of RBV uptake may contribute to rapid viral clearance.
INTRODUCTION
Current treatment for hepatitis C virus (HCV) infection is a combination of pegylated alpha interferon (IFN) and ribavirin (RBV), a guanosine nucleoside analog. Despite a significant research effort, only half of patients infected with HCV genotype 1 respond to therapy (50). IFN clearly plays an important role in HCV treatment response. The addition of RBV to IFN therapy doubled response rates, illustrating the importance of RBV in HCV treatment (37). Efforts to replace RBV with taribavirin, a RBV prodrug formerly known as viramidine, have been unsuccessful (1). Although HCV protease and polymerase inhibitors are promising new antivirals, RBV will continue to be a critical part of combination therapy because eliminating RBV from telaprevir-IFN therapy significantly reduced response rates (26, 43).
Patient treatment response is based on viral RNA levels, and the ultimate goal is to achieve a sustained virological response (SVR; defined as undetectable HCV RNA for at least 24 weeks posttherapy) (22). Patients with a rapid virological response (RVR; undetectable HCV RNA by week 4) are more likely to achieve SVR (17, 42). Lack of an early virological response (EVR; ≥2-log decline in HCV RNA by week 12) is generally an indicator of those who will not respond to therapy (22). Additionally, several viral and host factors are associated with a positive treatment response, such as low baseline viral loads, female gender, lack of comorbidities such as HIV coinfection, low initial IFN-stimulated-gene (ISG) expression, and specific alleles of the IL-28B gene (15, 21, 22).
While RBV plays a crucial role in treatment, its antiviral mechanism against HCV is unclear, with five proposed mechanisms. First, RBV is phosphorylated by host kinases, and RBV monophosphate can reduce host GTP pools through inhibition of IMP dehydrogenase (IMPDH), which is responsible for de novo GTP synthesis (44). Therefore, viral replication may be inhibited due to a lack of available nucleotides. Second, RBV triphosphate (RTP) may directly inhibit the viral RNA-dependent RNA polymerase (8, 35). Third, RTP can be incorporated into the viral genome, which can increase mutation rates (3, 5, 6, 48). Since RNA viruses have high error frequencies, increased mutation rates can induce viral error catastrophe (9). Fourth, it has been suggested that RBV acts indirectly against HCV by shifting the immune response from a humoral TH2 response to a cell-mediated TH1 response (40, 46). Fifth, RBV enhances ISG expression (16). Nearly all proposed mechanisms require import of RBV into the cell for antiviral activity. Therefore, we have focused on understanding host-based mechanisms of RBV uptake and resistance.
Nucleoside transporters are involved in the cellular import of natural and synthetic nucleosides and mediate RBV uptake (8). They consist of two subtypes, the equilibrative nucleoside transporters ENT1 to -4, which mediate facilitated bidirectional diffusion of nucleosides, and the concentrative nucleoside transporters CNT1 to -3, which can transport nucleosides against a concentration gradient (30). Numerous studies have linked nucleoside analog chemotherapy drug resistance to decreased nucleoside transporter expression (18, 20, 45). ENT1 is generally accepted as the primary RBV transporter; however, CNT2, CNT3, and ENT2 may also transport RBV in various cell types (13, 28, 39, 51). Our previous work determined that ENT1 was the primary RBV transporter in Huh7 liver cells. RBV-resistant (RBVR) Huh7 cells were generated by passage in increasing concentrations of RBV, and resistance was mediated by reduced RBV transport through ENT1 (27). As a result of decreased RBV import, RBVR Huh7 cells supported robust poliovirus replication in the presence of RBV compared to RBV-sensitive (RBVS) Huh7 cells, suggesting that the level of RBV import impacts antiviral efficacy.
In this study, we examined whether reduced cellular import of RBV influences HCV replication in vitro and treatment failure in HCV patients. Our results suggest that RBV resistance promotes robust HCV replication in the presence of RBV. Moreover, peripheral blood mononuclear cells (PBMCs) obtained from patients exhibited declines in RBV uptake as therapy progressed, suggesting that patients undergoing combination IFN-RBV therapy may develop resistance to RBV. Importantly, rapid treatment response correlated with maintenance of RBV uptake, suggesting that maintaining basal RBV uptake levels may improve treatment efficacy.
MATERIALS AND METHODS
Cells and viruses.
Cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (Huh7.5 human hepatoma cells kindly provided by C. Rice [2]) or 10% calf serum (HeLa human cervical carcinoma cells) and 1% penicillin-streptomycin. HCV stocks were generated in Huh7.5 cells using infectious JFH-1 (genotype 2a) from a synthetic clone kindly provided by M. Gale (to be described elsewhere) (49). Supernatants from infected cells were concentrated using Centricon Plus-70 filters (Milipore) according to the manufacturer's recommendation. Viral titers were determined using a focus-forming assay (10). Mahoney serotype 1 poliovirus was generated in HeLa cells as previously described (27).
Generation of RBVR Huh7.5 cells and viability assay.
RBVR Huh7.5 cells were generated as previously described (41). Briefly, cells were initially passaged in 100 μM RBV for approximately 4 weeks, and the RBV concentration was subsequently increased in 100 μM increments every 2 to 4 weeks until a concentration of 400 μM RBV was achieved. Viability and resistance were measured using a crystal violet viability assay previously described in conjunction with viable-cell counts following a 3-day exposure to increasing concentrations of RBV (41). For the crystal violet viability assay, cells were passaged at the indicated drug concentrations, split every 1 to 3 days for 7 to 10 days, and then visualized via crystal violet staining. Similar methods were employed for crystal violet viability assays using RBV and guanosine (Guo) and included equimolar concentrations (0, 100, or 400 μM) of RBV and Guo or Guo alone. For 3-day viability assays, subconfluent cells were treated in duplicate with increasing concentrations of RBV or RBV and Guo. Viable-cell numbers were determined on day 3 using trypan blue exclusion and normalized as a percentage of the level for the no-drug control. The RBV-resistant phenotype was stable, with cells retaining resistance despite 8 weeks of culture in the absence of RBV.
Nucleoside uptake assays and transfection.
The [3H]RBV uptake assay was conducted as described previously (27). Cells were treated with medium containing 5 μM RBV (containing approximately 1% [3H]RBV as a tracer; Moraveck, Brea, CA) for 30 min. [3H]cytidine uptake assays were performed in a similar manner (5 μM cytidine with less than 1% [3H]cytidine as a tracer; Moraveck), with uptake measured after 5, 15, and 30 min. For assays with the inhibitor nitrobenzylmercaptopurine riboside (NBMPR; Sigma), cells were pretreated with NBMPR for 15 min and incubated with RBV for 5 min or cytidine for 15 min. In phloridzin (Sigma) experiments, the pretreatment period was reduced to 5 min. The [3H]RBV uptake assay was modified for use in PBMCs. PBMCs from HCV patients were quickly thawed, resuspended in RPMI medium, and incubated for a 6-hour recovery period at 37°C prior to the assay. Viable-cell numbers were determined postrecovery using trypan blue exclusion. Cells were resuspended in DMEM at a concentration determined to be within the linear range of the assay (described below) and incubated with 5 μM RBV (containing 1% [3H]RBV) for 15 min. Uptake values (cpm/cell) were normalized to the level for a control sample assayed in parallel. The control sample consisted of healthy donor PBMCs frozen into aliquots to allow normalization for experiments performed on different days. Normalized values were obtained by calculating the average number of cpm/cell for each sample to a percentage of the average number of cpm/cell for the control sample (average number of cpm/cell for HCV PBMCs divided by average number of cpm/cell for healthy donor control PBMCs). The control number of cpm/cell was set to 100%. The absolute values for the control sample ranged from 2.50 × 10−4 to 9.97 × 10−4 cpm/cell (average, 5.34 × 10−4; standard deviation [SD], 1.67 × 10−4). For assays with the ENT inhibitor NBMPR, PBMCs were pretreated for 15 min at 37°C in the presence of 0, 15, or 100 μM NBMPR, followed by a 15-min RBV uptake assay as described above. For assays using healthy donor PBMCs, following a 3-hour recovery period, cells were cultured in RPMI medium containing 0 μM or 10 μM RBV for 7 days, and RBV uptake was quantified on day 7. To determine the linear range of the assay, PBMCs were resuspended in DMEM, in duplicate, at various concentrations (total number of PBMCs/ml medium). Each was then treated with 5 μM RBV (1% of which was 3H labeled) for 15 min, and numbers of cpm in lysates were quantified with a scintillation counter. The amount of RBV uptake linearly correlated with the number of cells used in the assay in the range of 1.0 × 105 to 1.2 × 106 cells (GraphPad Prism 5.03; linear regression analysis).
Nucleoside transporter transfections were conducted as previously described using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendation (27). Plasmid DNA (pcDNA 3.1, pENT1, and pCNT3) was provided by K. Giacomini (UCSF). Transfection efficiency was monitored using pLacZ-transfected cells and ranged from 20 to 40%.
RBV export assays.
The [3H]RBV uptake assay was modified to examine RBV export. Cells were incubated at 37°C with medium containing 5 μM RBV (with 1% [3H]RBV as a tracer) for 1 h. The cells were then rinsed twice with ice-cold DMEM to remove all noninternalized RBV, and the medium was replaced with either cold DMEM (time zero sample) or warm DMEM (samples for all other time points). The cells were then placed back at 37°C for various time intervals except for time zero plates, which were immediately processed. The remainder of the assay was performed similarly to standard RBV uptake assays except that the cells were harvested at 0, 0.5, 1, 2, and 4 h after removal of [3H]RBV-containing medium.
Real-time RT-PCR and immunoblot analysis.
Total RNA was isolated from RBVR and RBVS Huh7.5 cells as previously described (27). The relative levels of ENT1 and glyceraldehyde-3-phosphate dehyrdrogenase (GAPDH) RNA were quantified using one-step real-time reverse transcriptase PCR (RT-PCR) and the comparative method as described previously (27). Reactions were conducted using an ABI 7500 sequence detection system (Applied Biosystems) and analyzed using ABI Sequence Detection 1.3 detection software.
Immunoblot analysis was conducted using prior methods (27), using ENT1 Rabbit polyclonal immunoglobulin (Ig) (Abgent) and goat anti-rabbit IgG horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz). For analysis of GAPDH, blots were probed with goat anti-human GAPDH (V-18, Santa Cruz) diluted in 5% bovine serum albumin (BSA; Fisher) in phosphate-buffered saline–Tween (PBS-T)–0.15 M NaCl and donkey anti-goat IgG-HRP (Santa Cruz).
Viral infections.
In the experiments represented in Fig. 4A and B, cells were pretreated for 2 h with 0 μM RBV, 15 μM RBV, 50 μM RBV, 100 μM RBV, 100 μM Guo, or 100 μM RBV and 100 μM Guo, infected at a multiplicity of infection (MOI) of 0.05, and given fresh drug-containing medium after infection. After 72 h, viral titers from cell supernatants were determined using a focus-forming assay (10). For the serial passage infections represented in Fig. 5A and B, cells were infected at an MOI of 0.1 for 3 days and expanded, and on day 6, the supernatant was harvested for determination of titers and to begin a new infection in freshly plated cells (passage 2) and so forth. Infections continued through completion of six passages, representing 36 days of viral culture.
Fig. 4.

Impact of RBV resistance on HCV replication in vitro. (A) Three-day HCV infections in the absence or presence of high concentrations of RBV and guanosine. Cells were treated with 0 or 100 μM RBV and/or guanosine (Guo) and infected with HCV for 3 days. Titers were determined in triplicate at the end of each infection, and results are shown in focus forming units per ml (FFU/ml) with SD. Results for one of three representative experiments are shown (P = 0.002; unpaired, two-tailed, Student's t test). (B) Three-day HCV infections in the absence or presence of low RBV concentrations. Infections were performed as described for panel A, except that 0, 15, or 50 μM RBV was used.
Fig. 5.

HCV and poliovirus yields during serial passage in the presence of RBV. Serial HCV infections performed in the presence of 0, 15, or 50 μM RBV in Huh7.5 cells (A) or RBVR Huh7.5 cells (B). To begin passage 1, cells were infected at a MOI of 0.1 and expanded at day 3. On day 6, the supernatant from each plate was used for viral titer analysis and for initiation of a new infection (passage 2). Serial infections continued for six passages, and the experiment was performed twice. The titer results from one representative experiment, performed in triplicate, are shown with SD. Serial poliovirus infections were performed in the presence of 0, 50, 100, or 800 μM RBV in HeLa cells (C), Huh7.5 cells (D), or RBVR Huh7.5 cells (E). To begin passage 1, cells were infected at an MOI of 1, and cell-associated virus was harvested at 5 h postinfection. This virus was quantified by a plaque assay using HeLa cells and used as an inoculum for passage 2, and so on. Passage 6 virus stocks were serially passaged for approximately 6 more cycles of replication, yielding passage 12 virus stocks.
For the poliovirus experiments represented in Fig. 5, 1 × 106 cells were pretreated with medium containing 0 μM, 50 μM, 100 μM, or 800 μM RBV for 30 min, followed by infection with Mahoney poliovirus at an MOI of 1, in the presence of drug-containing medium. After 5 h, cells were harvested and resuspended in PBS, and cell-associated virus was released by freeze-thawing. Viral yield was quantified by a plaque assay using HeLa cells. This cycle was repeated five times to generate single-cycle passages 1 through 6. To determine whether the extended multicycle passage of these viruses would culminate in viral titer reduction, passage 6 viruses were used to inoculate fresh cells for multicycle passages. A total of 1 × 105 PFU of each passage 6 virus was used to infect 1 × 106 fresh cells in the presence or absence of RBV for 24 h, and virus-containing supernatants were harvested upon observation of cytopathic effects. These supernatants were then used to infect fresh cells in the presence or absence of RBV for 12 h, followed by an additional 12-hour infection cycle, ending with the harvest of cell-associated virus. The viruses from the final passage represent ∼36 h of viral infection cycles, representing approximately 6 cycles of viral replication. These final viruses were quantified by a plaque assay with HeLa cells, and the data are displayed as passage “12” viruses in Fig. 5C, D, and E.
Fig. 1.

Characterization of RBVR Huh7.5 cells. (A) Crystal violet viability assay. RBVR and RBVS Huh7.5 cells were passaged for 10 days in medium containing 0 μM or 400 μM RBV and stained with crystal violet for visualization. (B) RBV uptake assay. Cells were incubated with 3H-labeled RBV, and uptake was quantified by scintillation counting. The average from two experiments is shown as the number of counts per minute (cpm) per cell (P = 0.001). (C) RBV uptake following nucleoside transporter transfection. Cells were transfected with plasmids expressing ENT1, CNT3, or vector. The RBV uptake assay was performed at 24 h posttransfection (hpt). Results for one representative experiment, out of three, are shown. Asterisks denote P values of ≤0.01. (D and E) Inhibition of endogenous RBV uptake. (D) Cells were pretreated with the ENT inhibitor NBMPR, followed by the RBV uptake assay (P = 0.04). (E) Cells were pretreated with phloridzin, followed the RBV uptake assay. (F) Phloridzin control. Huh7.5 cells were transfected with the CNT3 expression plasmid or vector, and at 24 hpt, a RBV uptake assay was performed in the presence of phloridzin (P = 0.01). (G) Real-time RT-PCR analysis of ENT1 RNA levels. The average relative fold expression from three experiments is shown. (H) ENT1 immunoblot analysis. Results are shown for one of two representative experiments, with GAPDH as a loading control. S, RBVS Huh7.5 cells; R, RBVR Huh7.5 cells. Error bars for all panels (B to G) represent SD, with significance determined using unpaired, two-tailed Student t tests.
Subjects and PBMC processing.
Blood samples from eight healthy donors and 16 genotype 1 HCV-infected patients were collected, and PBMCs were isolated by Ficoll centrifugation. Isolated PBMCs were stored in a standard freezing mixture (15% fetal bovine serum, 4.3 μM HEPES, 1.4 mM l-glutamine, and 10% dimethyl sulfoxide [DMSO] in RPMI medium) and stored in liquid nitrogen following preliminary freezing at −80°C in an isopropanol bath. All patients received 800 to 1,200 mg/day RBV (per body weight) in combination with pegylated-IFN-α2a (Pegasys; F. Hoffman-La Roche, Nutley, NJ) at a dose of 180 μg subcutaneously for 48 weeks. Blood was collected prior to the onset of treatment and at 14 and 28 days posttreatment. Serum HCV RNA levels on day 28 were used to determine rapid treatment response. Rapid response was defined as undetectable HCV-RNA in branch DNA (bDNA) analysis (<615 RNA units [IU]/ml) or PCR analysis (<50 IU/ml). Signed consent was obtained from all HCV-infected patients and healthy donors, and all research was preapproved by the UT Southwestern institutional review board (IRB). HCV patients were included if they were receiving HCV treatment and were willing to provide consent (UT Southwestern IRB protocols 102005-009 [to M. Jain] and 082007-080 [to J. Pfeiffer]). For healthy donors, the only exclusion criteria were based on general health and willingness to participate and provide consent (UT Southwestern IRB protocol 082006-081 [to J. Pfeiffer]).
Statistical analysis.
Statistical significance for RBVR and RBVS Huh7.5 experiments, excluding real-time RT-PCR analysis, were determined using an independent two-sample Student t test. In assays using PBMCs, the Student t test was two tailed and either paired (for comparison within a donor) or unpaired (for comparison between donors).
RESULTS
RBVR Huh7.5 cells with reduced RBV uptake.
We previously demonstrated that host-based RBV resistance can develop and reduce RBV antiviral efficacy in a poliovirus model; however, the effect on HCV was unknown (27). Therefore, we generated RBVR human hepatoma cells (Huh7.5), permissive for HCV replication (2), by passage in increasing concentrations of RBV. Viability assays confirmed resistance in the RBVR cells when exposed to concentrations of RBV that were toxic to the RBVS parental cells (Fig. 1A). The RBVR cells also exhibited reduced uptake of radiolabeled RBV ([3H]RBV), suggesting that reduced RBV uptake enhances cellular survival in the presence of RBV (Fig. 1B).
To dissect the mechanism of RBV resistance, nucleoside transporter overexpression and inhibition studies were conducted. We previously demonstrated that the nucleoside transporter ENT1 mediated RBV transport and resistance in Huh7 liver cells (27). Although the Huh7.5 cells used in this study were derived from Huh7 cells, resistance to nucleoside analogs can develop through various pathways (53); therefore, the mechanism of resistance was analyzed further. Overexpression of ENT1, or the concentrative nucleoside transporter CNT3, restored RBV uptake in RBVR cells (Fig. 1C). We previously demonstrated that overexpression of CNT1 or CNT2 was unable to enhance RBV uptake in Huh7 cells (27). Additionally, although CNT3 overexpression increased RBV uptake, CNT3 was not expressed in Huh7 cells (27). To examine the activity of endogenous transporters, inhibition assays were per- formed. The addition of nitrobenzylmercaptopurine riboside (NBMPR), an inosine analog known to specifically inhibit ENT1 at low concentrations (30), decreased RBV uptake in RBVS cells to levels observed in RBVR cells (Fig. 1D). No significant reduction in RBV uptake was observed with a higher concentration of NBMPR (100 μM), which inhibits both ENT1 and ENT2 (34, 52). While the literature indicates that NBMPR treatment specifically inhibits ENT-mediated transport, we cannot exclude the remote possibility that another NBMPR-sensitive RBV transporter exists (34, 52). Inhibition of concentrative transport through phloridzin treatment, an inhibitor of sodium-dependent nucleoside transport (25, 47), did not reduce RBV uptake in cells, unless they expressed CNT3 via transfection (Fig. 1E and F). This was expected, because our prior work determined that endogenous CNT3 RNA was undetectable via real-time RT-PCR analysis, but CNT3 was capable of transport if exogenously expressed through transfection (27). Analysis of ENT1 protein and RNA levels revealed no significant difference between resistant and sensitive cells (Fig. 1G and H), suggesting that reduced ENT1 activity and/or transporter mislocalization may contribute to RBV resistance rather than reduced ENT1 levels. Alternatively, alterations in RBV phosphorylation could contribute to RBV resistance through apparent reduction in RBV uptake since ENT1 transport is equilibrative and nonphosphorylated nucleosides are exported.
To determine whether reduced RBV phosphorylation and subsequent RBV export contribute to the development of resistance, we measured RBV export in RBVS and RBVR Huh7.5 cells. RBVR and RBVS cells were exposed to RBV-containing medium for 1 h to allow accumulation of RBV phosphate derivatives, and the maintenance of cellular RBV was quantified. As expected, RBVS cells initially accumulated more intracellular RBV than RBVR cells during constant RBV exposure (Fig. 2, time zero). When both cell lines were placed in RBV-free medium, RBV export was greater in RBVS cells. At 4 h after RBV removal, RBVR cells retained 44% of baseline RBV, while RBVS cells retained only 29% of baseline RBV, demonstrating that RBVR cells have reduced RBV export (Fig. 2). Therefore, the reduced RBV uptake in RBVR cells is not due to increased export. Moreover, these results suggest that RBV catabolism via dephosphorylation is not increased in RBVR cells. Taken together, these results indicate that reduced RBV uptake in RBVR cells was due to reduced ENT1 activity rather than altered RBV metabolism within the cells.
Fig. 2.
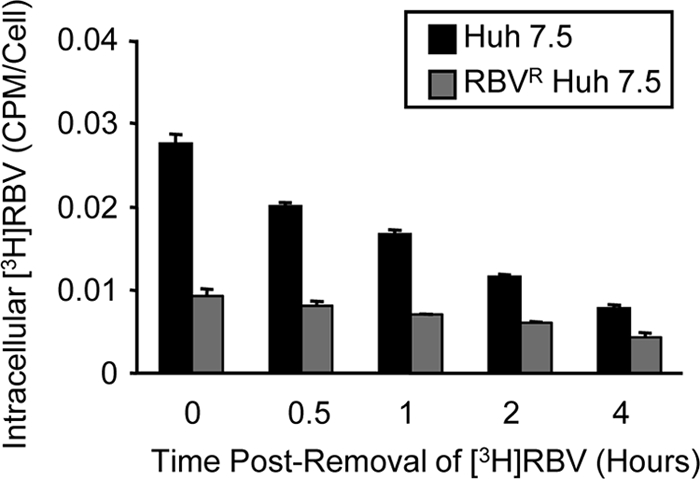
RBV retention in RBVR and RBVS Huh7.5 cell lines. RBVR and RBVS Huh7.5 cells were incubated for 1 h in medium containing 3H-labeled RBV. The amount of nonexported, intracellular RBV was determined at 0, 0.5, 1, 2, and 4 h after removal of RBV-containing medium. Results are shown for one of two representative experiments performed in duplicate, with SD. The difference between the 0- and 4-hour data sets was statistically significant for each cell line (P < 0.02; Student's t test).
Because ENT1-mediated RBV uptake and export were reduced in RBVR Huh7.5 cells, we sought to determine whether transport of other ENT1 nucleoside substrates was reduced. To determine whether the alteration was RBV specific, cytidine uptake was examined. Cytidine is a natural nucleoside transported by ENT1. The transport of [3H]cytidine in RBVS and RBVR Huh7.5 cells was examined. No significant difference in cytidine import was observed after 15 or 30 min (Fig. 3A). Since the affinity of ENT1 for cytidine is much higher than for RBV, it was possible that differences in cytidine import were masked by extended incubation periods (36). When cytidine import was examined after 5 min, both RBVR and RBVS Huh7.5 cells again displayed similar levels of cytidine uptake. Conversely, there was a significant difference in the level of RBV import over the same period (Fig. 3B). Therefore, RBVR cells maintain cytidine uptake despite reduced RBV uptake.
Fig. 3.
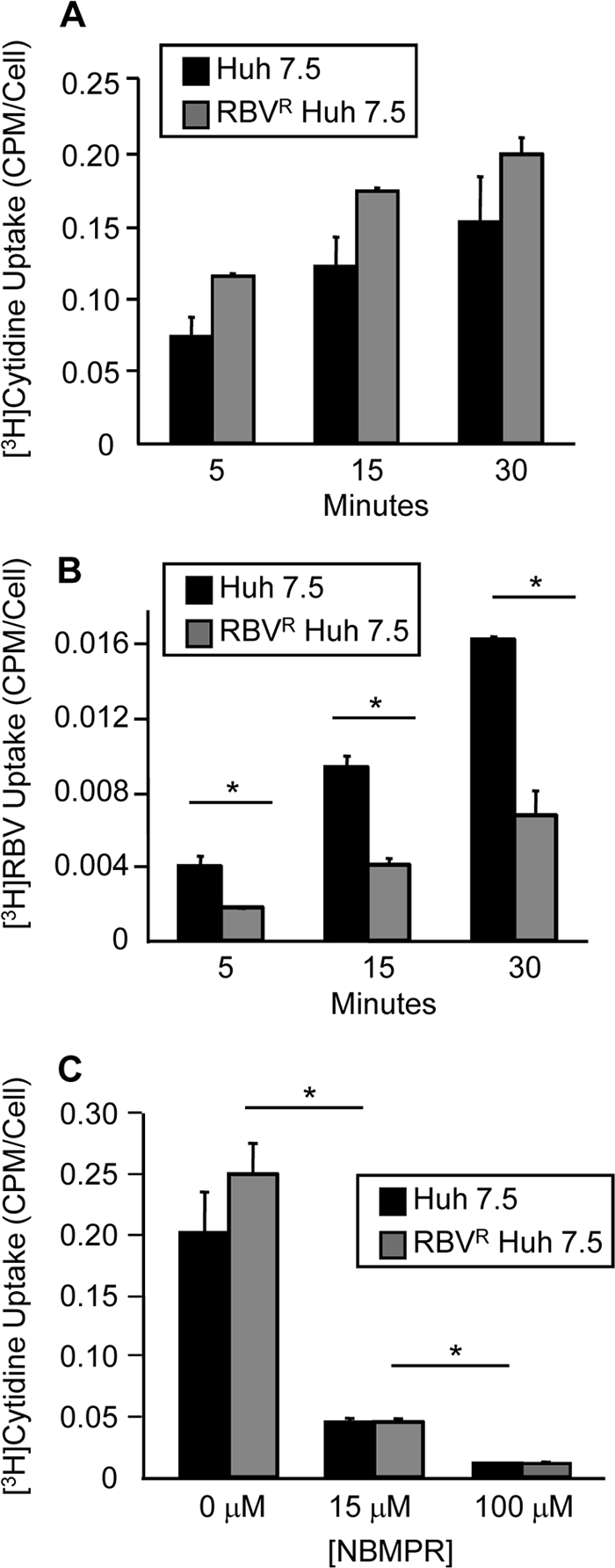
Cytidine transport in RBVR and RBVS Huh7.5 cells. (A) Cytidine uptake assay. Cells were incubated with 3H-labeled cytidine, and uptake was quantified at 5, 15, and 30 min postincubation by scintillation counting. The average from two experiments is shown as the number of cpm/cell. (B) RBV uptake was determined as previously described at 5, 15, and 30 min. The average from two experiments is shown. (C) Inhibition of endogenous cytidine uptake. Cells were pretreated with NBMPR, followed by the cytidine uptake assay. Error bars for all panels represent SD, with significance determined using unpaired, two-tailed Student t tests (P < 0.05; Student's t test).
Despite the fact that cytidine affinity for ENT1 is 10-fold higher than for ENT2, it was possible that ENT2 or alternate transporters could contribute to cytidine uptake, masking ENT1 transport (36, 53). Therefore, cytidine uptake was examined in RBVR and RBVS cells during endogenous equilibrative transport inhibition through NBMPR treatment (Fig. 3C). A low concentration of NBMPR, 15 μM, which inhibits ENT1-mediated transport, significantly reduced cytidine uptake, suggesting that the majority of cytidine uptake occurred through ENT1. Taken together, these results suggest that RBV resistance is mediated through altered RBV-specific ENT1 transport mechanisms and indirectly provide evidence against ENT1 mislocalization.
HCV replication in RBVS and RBVR Huh7.5 cells.
With the development of RBVR Huh7.5 cells, we were in a unique position to examine the effects of RBV on HCV replication, potential mechanisms of RBV action, and the consequences of RBV resistance. HCV infections were performed in RBVR and RBVS Huh7.5 cells in the presence or absence of 100 μM RBV. RBV treatment induced a significant viral titer reduction in RBVS cells (Fig. 4A). RBVR cells displayed no titer reduction in the presence of RBV, despite having only a 2-fold RBV uptake defect, suggesting that a modest reduction in RBV uptake can have a major impact on viral replication. Reduced cell health from RBV exposure may have reduced viral replication in RBVS cells. Studies have shown that guanosine supplementation can counter RBV-induced toxicity by restoring host GTP pools (33). Although IMPDH inhibition cannot completely account for RBV's antiviral activity, the addition of guanosine can increase cell health through restoration of nucleotide pool balance (32, 33). Therefore, infections were performed in the RBVS cells in the presence of equimolar concentrations of RBV and guanosine. Supplementation with guanosine restored viral titers to levels observed in the absence of RBV (Fig. 4A). Since 100 μM RBV likely exceeds most physiological concentrations, we wondered whether viral titers would be reduced at lower RBV concentrations. Infections were conducted using 15 μM RBV, to mimic an achievable plasma concentration in patients, as well as 50 μM (Fig. 4B) (14, 31). No titer reductions were observed at either concentration, indicating that reduced cell health from RBV exposure may have inhibited HCV replication at high RBV concentrations.
We considered the possibility that a 72-hour infection period was insufficient to observe RBV antiviral effects, especially if RBV-induced mutagenesis plays a role. RBV-induced mutagenesis requires multiple cycles of viral replication for mutations to accumulate and lead to observable antiviral effects (5–7). To examine RBV's effect on HCV following extended exposure, HCV was serially passaged in RBVS and RBVR cells in the absence or presence of 15 or 50 μM RBV. Each 6-day passage was followed by supernatant harvest and infection of naïve cells, with titer analysis after each passage. No significant RBV-mediated titer reduction was observed in either RBVS or RBVR cells (Fig. 5A and B).
To ensure that low-level mutation accumulation and error catastrophe were not occurring during the viral RBV passages, we examined whether error catastrophe occurs for poliovirus under similar conditions. Poliovirus is known to undergo mutation accumulation and error catastrophe during RBV passage (5, 6). We reasoned that if poliovirus does not undergo error catastrophe in RBVS and RBVR Huh7.5 cells, then HCV is extremely unlikely to undergo error catastrophe under similar conditions. We performed 6 serial single cycle passages and 6 additional pooled viral passages of poliovirus in RBVS Huh7.5, RBVR Huh7.5, or HeLa cells in the presence or absence of RBV. HeLa cells were previously shown to support RBV-mediated poliovirus error catastrophe (5, 6). In accordance with published data, 800 μM RBV induced a dramatic decline in poliovirus titers in HeLa cells and viral extinction occurred by passage three (Fig. 5C) (5, 6, 41). When poliovirus infections were carried out in RBVR and RBVS Huh7.5 cells, error catastrophe did not occur, despite continued RBV exposure over 12 passages (Fig. 5C and D). Since passage in 800 μM RBV failed to induce poliovirus error catastrophe in RBVR and RBVS Huh7.5 cells, it is unlikely that passage in 50 μM RBV induced HCV error catastrophe in the same cell lines. These results suggest that, at high concentrations, RBV may act indirectly against HCV, potentially through toxicity-mediated mechanisms.
RBVS and RBVR Huh7.5 viability during RBV exposure.
To further investigate the effects of RBV on cell survival and toxicity, cellular viability assays were performed in the presence of RBV and guanosine. Cells were exposed to RBV for 7 days, or for 3 days to mimic the infection conditions used in the experiments represented in Fig. 4, and cellular viability was determined by crystal violet staining and trypan blue exclusion assays. Guanosine enhanced cellular viability in RBVS cells at high concentrations of RBV (400 μM) (Fig. 6A). Additionally, RBV induced a 50% decline in RBVS cell numbers at an estimated concentration of 70 to 80 μM in the presence of guanosine and 40 to 50 μM in the absence of guanosine (Fig. 6B). RBVR cells never reached 50% population loss, despite treatment with 800 μM RBV (Fig. 6B and data not shown). The combination of RBV's known cytostatic effect in conjunction with toxicity likely contributed to decreased cell survival in RBVS cells (24). Nevertheless, cell health could not be completely restored with guanosine treatment, suggesting that mechanisms other than IMPDH-induced alterations in nucleotide pools contribute to the adverse effect of RBV on cellular health.
Fig. 6.
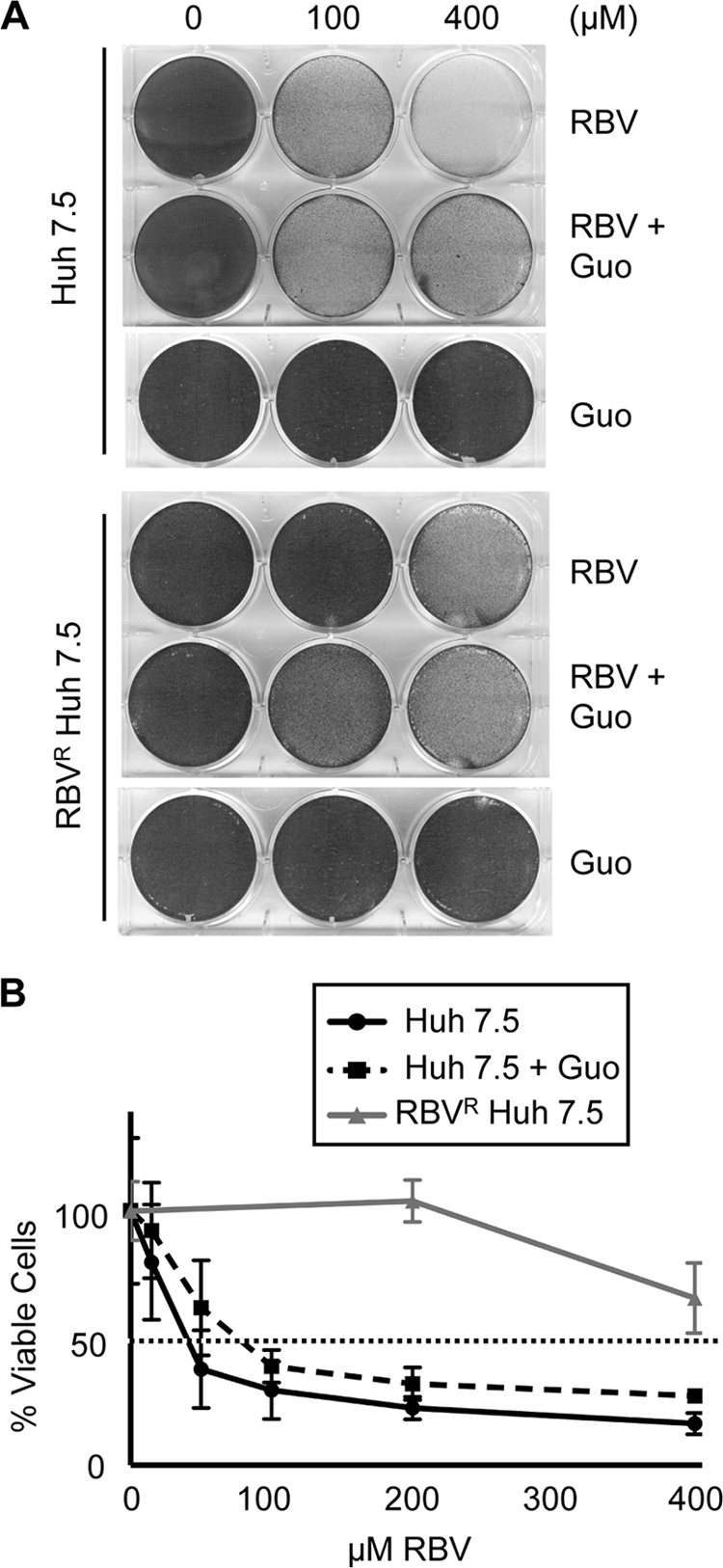
Cellular viability studies. (A) Cells were passaged with the indicated concentrations of RBV and Guo for 7 days. On day 7, viable cells were visualized using crystal violet staining. Results are shown for one of two representative experiments. (B) Huh7.5 cells were passaged at various concentrations of RBV, or RBV and Guo at equimolar concentrations, for 3 days. On day 3, viable-cell counts were obtained using trypan blue exclusion and normalized to a percentage of the no-drug control (0 μM). Shown is the combined average from two experiments (for RBVS cells) or one of three representative experiments (for RBVR cells). Error bars indicate SD.
RBV uptake in PBMCs from healthy donors and HCV patients.
Our cell culture results suggest that RBV resistance develops in many cultured cell lines and impacts antiviral efficacy (27); however, the development of host-based RBV resistance has yet to be described in vivo. Therefore, we determined whether host-based resistance to RBV develops using PBMCs from healthy donors and HCV-infected patients undergoing combination IFN-RBV therapy (see Table 1 for patient data). We began by examining whether ex vivo exposure to a physiologically relevant concentration of RBV can reduce RBV uptake in PBMCs from healthy donors. PBMCs were cultured in the absence or presence of 10 μM RBV for 7 days, and RBV uptake was quantified immediately after RBV exposure. Short-term, low-concentration RBV exposure significantly reduced RBV uptake in PBMCs from healthy donors (Fig. 7A to C). RBV uptake declined in PBMCs from every donor examined (Fig. 7C). Given that HCV patients receive IFN-RBV treatment for 24 to 48 weeks, we hypothesized that RBV uptake declines over time. We quantified RBV uptake in PBMCs isolated from HCV genotype 1-infected patients before therapy (day 0) and after 14 and 28 days of pegylated IFN-RBV therapy. PBMC RBV uptake was significantly reduced at days 14 and 28, compared with the level for day 0 (Fig. 8A). To ascertain whether host-based resistance could play a role in treatment response, we compared RBV uptake in patients with rapid response (rapid responders [RRs]; viral clearance by day 28) to RBV uptake in patients with slow response or no response (nonrapid responders [non-RRs]). We chose to examine rapid treatment response since it is highly predictive of SVR (17, 42). Interestingly, RRs maintained significantly higher levels of RBV uptake than non-RRs (Fig. 8B and C). These results suggest that RBV resistance develops in vivo and that maintenance of basal uptake levels correlates with rapid clearance of HCV.
Table 1.
Patient data
| Patient | HCV genotype | Responsea | Gender | Race | Age (yr) | HIV status | Wt (kg) | RBV dose (mg) | RBV dose (mg/kg) | % RBV uptake (day 28 level/day 0 level)c |
|---|---|---|---|---|---|---|---|---|---|---|
| 70 | 1a | RR | Male | White | 35 | + | 81.8 | 1,200 | 14.7 | 122.0 |
| 65 | 1a | RR | Male | White | 49 | + | 76.1 | 1,200 | 15.8 | 116.7 |
| 41 | 1a | RR | Male | White | 38 | + | 70.1 | 800b | 11.4 | 102.9 |
| 64 | 1a | RR | Male | White | 39 | + | 80.8 | 1,200 | 14.9 | 97.9 |
| 69 | 1a | Non-RR | Male | White | 38 | + | 93.9 | 1,200 | 12.8 | 95.5 |
| 12 | 1a | RR | Male | White | 49 | + | 79.7 | 1,000 | 12.6 | 77.7 |
| 63 | 1a | RR | Male | Black | 47 | + | 67.2 | 1,000 | 14.9 | 74.5 |
| 68 | 1b | Non-RR | Male | White | 32 | + | 69.0 | 1,000 | 14.5 | 71.1 |
| 51 | 1b | Non-RR | Male | Black | 43 | + | 118.7 | 1,200 | 10.1 | 66.4 |
| 7 | 1a | Non-RR | Female | White | 36 | + | 85.1 | 1,200 | 14.1 | 66.1 |
| 66 | 1b | Non-RR | Female | Black | 56 | + | 94.0 | 1,200 | 12.8 | 65.0 |
| 45 | 1a | Non-RR | Female | Black | 59 | + | 74.6 | 1,000 | 13.4 | 63.7 |
| 53 | 1a | Non-RR | Male | White | 37 | − | 70.5 | 1,000 | 14.2 | 63.7 |
| 61 | 1a | RR | Male | White | 44 | + | 69.2 | 1,000 | 14.5 | 54.8 |
| 58 | 1a | Non-RR | Female | Black | 45 | + | 87.6 | 1,200 | 13.7 | 50.6 |
| 49 | 1b | Non-RR | Female | Black | 54 | + | 79.2 | 1,200 | 15.2 | 37.2 |
RR, rapid response; non-RR, nonrapid response.
Received a lower RBV dose due to inclusion in a separate study comparing the efficacies of RBV.
The value shown is the amount of RBV uptake in PBMCs isolated at day 28 posttherapy relative to the amount of RBV uptake prior to therapy on day 0 (see Fig. 8).
Fig. 7.

RBV uptake in PBMCs from healthy donors. (A) PBMCs from healthy donors were cultured in 0 μM or 10 μM RBV for 7 days, followed by the RBV uptake assay. Shown is the average number of cpm/cell (n = 8; P = 0.01; paired, two-tailed Student t test). Horizontal lines within each group represent the mean. (B) Values from panel A graphed to visualize pre- and posttreatment values. (C) Data from panel A, normalized to 0 μM RBV values, highlighting trends following ex vivo treatment with 10 μM RBV, with 0 μM values set to 100%.
Fig. 8.

RBV uptake in PBMCs from HCV patients undergoing IFN/RBV therapy. (A) RBV uptake in HCV patient PBMCs isolated prior to the onset of treatment (day 0) and at 14 or 28 days posttreatment was quantified. To account for assay variability on different days, the average number of cpm/cell was normalized to RBV uptake values from an aliquoted healthy donor PBMC control sample assayed in parallel (number of cpm/cell for the patient sample divided by number of cpm/cell for the control) (n = 16 for day 0 versus day 28 [P = 0.001]; n = 10 for day 0 versus day 14 [P = 0.009] [paired, two-tailed Student t tests]). (B) RBV uptake before and after 28 days of treatment for each HCV patient, with treatment response indicated (RR, rapid responder; non-RR, nonrapid responder; n = 16). (C) Day 28 RBV uptake values from panel B categorized by treatment response (n = 7 and 9 for RRs and non-RRs, respectively; P = 0.01 in unpaired, two-tailed Student t test). Horizontal lines within each group represent the mean.
While our results suggest that cultured Huh7.5 cells develop RBV resistance through reduced activity of the ENT1 nucleoside transporter, it was unknown whether ENT1 plays a role in RBV transport and resistance in primary human cells. Therefore, we examined ENT1 activity in PBMCs from healthy donors and HCV patients using the inhibitor NBMPR. Treatment with 15 μM NBMPR, which specifically inhibits ENT1 activity (30), reduced RBV uptake in PBMCs by 56 to 70% (Fig. 9). Treatment with 100 μM NBMPR, which inhibits ENT1 and ENT2 (34, 52), yielded similar results. Therefore, ENT1 is a major RBV transporter in primary human PBMCs. Taken together, these results suggest that ENT1-mediated RBV uptake plays an important role in HCV therapy and treatment response.
Fig. 9.

RBV uptake in PBMCs in the presence of the ENT inhibitor NBMPR. PBMCs from a healthy donor or HCV patients were pretreated with the ENT inhibitor NBMPR, followed by the RBV uptake assay. Results for one representative assay performed in duplicate are shown; error bars indicate SD. All 15 and 100 μM NBMPR values are significantly lower than the matched 0 μM NBMPR control value (P < 0.03; unpaired, two-tailed Student t test).
DISCUSSION
Although the concept of host-based resistance to nucleoside analog therapy is well documented in cancer studies, it is generally not considered in antiviral therapy resistance. Nucleoside and deoxynucleoside analogs are routinely used in chemotherapy, and their cytotoxicity can cause severe side effects and the development of resistance (29, 53). Chemotherapy nucleoside analog resistance can occur through increased export, altered phosphorylation, and/or reduced import (53). Nearly all proposed mechanisms of action for RBV require RBV import. Therefore, there is a need to understand factors that impact cellular RBV import and the development of host-based RBV resistance.
In an effort to determine whether reduced cellular import of RBV can contribute to HCV treatment failure, we began by modeling the development of RBV resistance using a cell culture system. In accordance with our previous results (27), resistance developed in Huh7.5 cells through reduced ENT1-mediated RBV uptake. The specific mechanism by which ENT1 is altered remains to be completely elucidated but appears to be specific to RBV import. RBVS and RBVR cells exhibit similar ENT1 RNA and protein levels (Fig. 1). Given the increased level of RBV export within sensitive cells, it is unlikely that RBVR cells acquired resistance through increased RBV catabolism (Fig. 2). Additionally, RBVR cells demonstrate no defect in transport of the naturally occurring substrate cytidine (Fig. 3), suggesting that RBVR cells can selectively reduce import of the guanosine analog RBV while maintaining transport of naturally occurring nucleosides. Furthermore, these results suggest that ENT1-mediated RBV resistance is not the result of ENT1 mislocalization from the plasma membrane, which would reduce both RBV and cytidine import. Posttranslational modifications of ENT1 protein or mutations in the ENT1 sequence could account for altered ENT1 transporter activity, while maintaining similar protein and RNA levels. A polymorphism within the transmembrane domain of human CNT3 has been shown to alter transport specificity for several nucleoside-derived drugs (11, 12). Several single-nucleotide polymorphisms (SNPs) within the ENT1 gene (SLC29A1) have been reported (38). One of the SNPs (rs760370A → G) correlated with rapid treatment response in HCV patients, further supporting the role of ENT1-mediated RBV uptake in HCV treatment response. Mutations in SLC29A1 could alter splicing, which could affect ENT1 protein stability, localization, and activity. Splice variants that inactivate ENT1 or alter RBV uptake have been identified (4, 19). Although no differences were found in ENT1 RNA or protein levels in the RBVS and RBVR cells examined here, subtle changes such as mutations or altered splicing could have occurred in RBVR cells which may have been masked in our ENT1 protein and RNA analysis. Despite this, the restoration of RBV uptake in RBVR cells following ENT1 overexpression and the recapitulation of RBVR uptake levels in RBVS cells during inhibition of endogenous ENT1 transport indicates that RBV uptake is primarily ENT1-mediated in Huh7.5 cells. Alterations in RBV-specific ENT1 binding and/or transport may contribute to the development of resistance in RBVR Huh7.5 cells.
The effect of RBV import on HCV replication was also investigated. HCV titers were significantly reduced in RBVS cells in the presence of 100 μM RBV, in contrast to the level for RBVR cells which supported HCV replication (Fig. 4A). RBV may exert its antiviral effects indirectly as a result of reduced cell health since viral replication was restored in the presence of guanosine and HCV replication was not inhibited at low concentrations of RBV (15 and 50 μM) (Fig. 4). If RBV were to primarily act through mutagenesis, viral titers would decline in serial passage infections, as has been observed with poliovirus (5, 6). HCV titers were not reduced in RBVS or RBVR cells following a total of 36 days of viral culture in the presence of 15 or 50 μM RBV. Moreover, RBV failed to induce characteristic viral titer reductions in Huh7.5 cells during poliovirus infections, a virus known to undergo RBV-induced error catastrophe (5, 6). Overall, our results indicate that RBV may inhibit HCV replication indirectly, potentially through adverse effects on cell health.
RBV-induced toxicity may arise through a variety of mechanisms, including IMPDH inhibition and other mechanisms that have yet to be elucidated. Clinically, RBV can induce hemolytic anemia from the accumulation of RTP within erythrocytes (23). However, the ability of RBV to interfere with cellular processes is not limited to erythrocytes. RBV is a known cytostatic agent, at least partially through IMPDH inhibition and reduced GTP levels (33, 44). Indeed, when we examined cellular viability after RBV exposure, the addition of guanosine partially restored viability in RBVS cells (Fig. 4). The fact that viability was not completely restored suggests RBV can negatively impact cell health through additional mechanisms and is not solely dependent on IMPDH inhibition. RBV may induce low-level toxicity or stress responses which can then indirectly limit viral replication.
Finally, our results indicate that host-based resistance to RBV may occur in HCV patients undergoing combination IFN-RBV therapy. In all cases examined, RBV exposure reduced RBV uptake in PBMCs from healthy donors (Fig. 7). Declines in RBV uptake occurred after only 7 days of culture with a low, clinically relevant plasma concentration of RBV (10 μM). RBV uptake was also reduced in PBMCs from patients undergoing combination IFN-RBV therapy (Fig. 8), suggesting that the development of resistance is a general phenomenon and not limited to cell culture. Perhaps most interesting, we found that PBMCs from patients who responded rapidly to treatment (RRs) had significantly higher RBV uptake at day 28 than PBMCs from patients who were slow responders or nonresponders. These results suggest that maintenance of basal RBV uptake levels may improve treatment response. While HCV replicates in hepatocytes, we used PBMCs for this study due to their high viability and ease of sample acquisition at multiple time points. In PBMCs, RBV uptake was reduced following ex vivo and in vivo exposure to RBV. Inhibition studies suggested that ENT1 is a major RBV transporter in PBMCs (Fig. 9). Because treatment response correlated with RBV uptake in PBMCs, either RBV uptake is similarly affected in liver cells or PBMC function is altered by RBV treatment, impacting innate or adaptive antiviral responses in the liver. Further research aimed at understanding the precise mechanisms by which RBV can affect cellular processes is warranted and may improve our understanding of RBV's antiviral activity against HCV.
In conclusion, our results uncovered a novel form of host-based antiviral drug resistance that correlates with HCV treatment response. The data suggest that RBV toxicity drives the development of host-based resistance through reduced cellular uptake. Notably, our results also suggest that patients undergoing combination IFN-RBV therapy develop resistance in vivo, with significantly reduced cellular RBV uptake over time. RBV uptake in PBMCs could be used as a diagnostic tool to predict treatment response. The knowledge that host-based RBV resistance may impact treatment outcome suggests that the development of drugs specifically targeted to counter the development of resistance may improve HCV treatment response.
ACKNOWLEDGMENTS
We thank Michael Gale for the JFH-1 HCV infectious clone, Charlie Rice for Huh7.5 cells, Kathleen Giacomini for the nucleoside transporter expression plasmids, and Andrea Erickson and Bruce Doxey for assistance with experiments. We thank Lara Alder, Nahid Attar, Tianna Petersen, Robert Kreymer, and Saharnaz Salari for assistance with patient samples and data and William M. Lee, Pinghui Feng, and Tawanda Gumbo for helpful advice and comments on the manuscript.
This work was supported by the Pew Scholars Program, the Lizanell and Colbert Coldwell Foundation, pilot grants from the American Cancer Society and the North and Central Texas Clinical and Translational Science Initiative through the NIH, and a Research Scholar Grant from the American Cancer Society (RSG-11-059-01-MPC).
Footnotes
Published ahead of print on 4 May 2011.
REFERENCES
- 1. Benhamou Y., et al. 2009. A phase III study of the safety and efficacy of viramidine versus ribavirin in treatment-naive patients with chronic hepatitis C: ViSER1 results. Hepatology 50:717–726 [DOI] [PubMed] [Google Scholar]
- 2. Blight K. J., McKeating J. A., Rice C. M. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bougie I., Bisaillon M. 2003. Initial binding of the broad spectrum antiviral nucleoside ribavirin to the hepatitis C virus RNA polymerase. J. Biol. Chem. 278:52471–52478 [DOI] [PubMed] [Google Scholar]
- 4. Cai J., et al. 2008. Two distinct molecular mechanisms underlying cytarabine resistance in human leukemic cells. Cancer Res. 68:2349–2357 [DOI] [PubMed] [Google Scholar]
- 5. Crotty S., Cameron C. E., Andino R. 2001. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. U. S. A. 98:6895–6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crotty S., et al. 2000. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6:1375–1379 [DOI] [PubMed] [Google Scholar]
- 7. Dixit N. M., Layden-Almer J. E., Layden T. J., Perelson A. S. 2004. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 432:922–924 [DOI] [PubMed] [Google Scholar]
- 8. Dixit N. M., Perelson A. S. 2006. The metabolism, pharmacokinetics and mechanisms of antiviral activity of ribavirin against hepatitis C virus. Cell Mol. Life Sci. 63:832–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Domingo E., Holland J. J. 1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol. 51:151–178 [DOI] [PubMed] [Google Scholar]
- 10. Erickson A. K., Seiwert S., Gale M., Jr 2008. Antiviral potency analysis and functional comparison of consensus interferon, interferon-alpha2a and pegylated interferon-alpha2b against hepatitis C virus infection. Antivir. Ther. 13:851–862 [PMC free article] [PubMed] [Google Scholar]
- 11. Errasti-Murugarren E., Cano-Soldado P., Pastor-Anglada M., Casado F. J. 2008. Functional characterization of a nucleoside-derived drug transporter variant (hCNT3C602R) showing altered sodium-binding capacity. Mol. Pharmacol. 73:379–386 [DOI] [PubMed] [Google Scholar]
- 12. Errasti-Murugarren E., Molina-Arcas M., Casado F. J., Pastor-Anglada M. 2010. The human concentrative nucleoside transporter-3 C602R variant shows impaired sorting to lipid rafts and altered specificity for nucleoside-derived drugs. Mol. Pharmacol. 78:157–165 [DOI] [PubMed] [Google Scholar]
- 13. Errasti-Murugarren E., Pastor-Anglada M., Casado F. J. 2007. Role of CNT3 in the transepithelial flux of nucleosides and nucleoside-derived drugs. J. Physiol. 582:1249–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feld J. J., Hoofnagle J. H. 2005. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature 436:967–972 [DOI] [PubMed] [Google Scholar]
- 15. Feld J. J., et al. 2010. Ribavirin improves early responses to peginterferon through improved interferon signaling. Gastroenterology 139:154–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feld J. J., et al. 2007. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology 46:1548–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferenci P., et al. 2005. Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40 KD)/ribavirin. J. Hepatol. 43:425–433 [DOI] [PubMed] [Google Scholar]
- 18. Fotoohi A. K., Lindqvist M., Peterson C., Albertioni F. 2006. Involvement of the concentrative nucleoside transporter 3 and equilibrative nucleoside transporter 2 in the resistance of T-lymphoblastic cell lines to thiopurines. Biochem. Biophys. Res. Commun. 343:208–215 [DOI] [PubMed] [Google Scholar]
- 19. Fukuchi Y., Furihata T., Hashizume M., Iikura M., Chiba K. 2010. Characterization of ribavirin uptake systems in human hepatocytes. J. Hepatol. 52:486–492 [DOI] [PubMed] [Google Scholar]
- 20. Galmarini C. M., et al. 2002. Potential mechanisms of resistance to cytarabine in AML patients. Leuk. Res. 26:621–629 [DOI] [PubMed] [Google Scholar]
- 21. Ge D., et al. 2009. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 461:399–401 [DOI] [PubMed] [Google Scholar]
- 22. Ghany M. G., Strader D. B., Thomas D. L., Seeff L. B. 2009. Diagnosis, management, and treatment of hepatitis C: an update. Hepatology 49:1335–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gish R. G. 2006. Treating HCV with ribavirin analogues and ribavirin-like molecules. J. Antimicrob. Chemother. 57:8–13 [DOI] [PubMed] [Google Scholar]
- 24. Graci J. D., Cameron C. E. 2006. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 16:37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Griffith D. A., Jarvis S. M. 1996. Nucleoside and nucleobase transport systems of mammalian cells. Biochim. Biophys. Acta 1286:153–181 [DOI] [PubMed] [Google Scholar]
- 26. Hezode C., et al. 2009. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N. Engl. J. Med. 360:1839–1850 [DOI] [PubMed] [Google Scholar]
- 27. Ibarra K. D., Pfeiffer J. K. 2009. Reduced ribavirin antiviral efficacy via nucleoside transporter-mediated drug resistance. J. Virol. 83:4538–4547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jarvis S. M., Thorn J. A., Glue P. 1998. Ribavirin uptake by human erythrocytes and the involvement of nitrobenzylthioinosine-sensitive (es)-nucleoside transporters. Br. J. Pharmacol. 123:1587–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jordheim L. P., Dumontet C. 2007. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim. Biophys. Acta 1776:138–159 [DOI] [PubMed] [Google Scholar]
- 30. Kong W., Engel K., Wang J. 2004. Mammalian nucleoside transporters. Curr. Drug Metab. 5:63–84 [DOI] [PubMed] [Google Scholar]
- 31. Lindahl K., Stahle L., Bruchfeld A., Schvarcz R. 2005. High-dose ribavirin in combination with standard dose peginterferon for treatment of patients with chronic hepatitis C. Hepatology 41:275–279 [DOI] [PubMed] [Google Scholar]
- 32. Loustaud-Ratti V., Rousseau A., Marquet P., Denis F., Alain S. 2009. Ribavirin in chronic hepatitis C: past and future. Expert Rev. Anti Infect. Ther. 7:249–253 [DOI] [PubMed] [Google Scholar]
- 33. Lowe J. K., Brox L., Henderson J. F. 1977. Consequences of inhibition of guanine nucleotide synthesis by mycophenolic acid and virazole. Cancer Res. 37:736–743 [PubMed] [Google Scholar]
- 34. Lum P. Y., Ngo L. Y., Bakken A. H., Unadkat J. D. 2000. Human intestinal es nucleoside transporter: molecular characterization and nucleoside inhibitory profiles. Cancer Chemother. Pharmacol. 45:273–278 [DOI] [PubMed] [Google Scholar]
- 35. Maag D., Castro C., Hong Z., Cameron C. E. 2001. Hepatitis C virus RNA-dependent RNA polymerase (NS5B) as a mediator of the antiviral activity of ribavirin. J. Biol. Chem. 276:46094–46098 [DOI] [PubMed] [Google Scholar]
- 36. Mangravite L. M., Badagnani I., Giacomini K. M. 2003. Nucleoside transporters in the disposition and targeting of nucleoside analogs in the kidney. Eur. J. Pharmacol. 479:269–281 [DOI] [PubMed] [Google Scholar]
- 37. McHutchison J. G., et al. 1998. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N. Engl. J. Med. 339:1485–1492 [DOI] [PubMed] [Google Scholar]
- 38. Morello J., et al. 2010. Influence of a single nucleotide polymorphism at the main ribavirin transporter gene on the rapid virological response to pegylated interferon-ribavirin therapy in patients with chronic hepatitis C virus infection. J. Infect. Dis. 202:1185–1191 [DOI] [PubMed] [Google Scholar]
- 39. Patil S. D., Ngo L. Y., Glue P., Unadkat J. D. 1998. Intestinal absorption of ribavirin is preferentially mediated by the Na+-nucleoside purine (N1) transporter. Pharm. Res. 15:950–952 [DOI] [PubMed] [Google Scholar]
- 40. Peavy D. L., Powers C. N., Knight V. 1981. Inhibition of murine plaque-forming cell responses in vivo by ribavirin. J. Immunol. 126:861–864 [PubMed] [Google Scholar]
- 41. Pfeiffer J. K., Kirkegaard K. 2005. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 79:2346–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poordad F., Reddy K. R., Martin P. 2008. Rapid virologic response: a new milestone in the management of chronic hepatitis C. Clin. Infect. Dis. 46:78–84 [DOI] [PubMed] [Google Scholar]
- 43. Shiffman M. L. 2009. What future for ribavirin? Liver Int. 29(Suppl. 1):68–73 [DOI] [PubMed] [Google Scholar]
- 44. Sintchak M. D., Nimmesgern E. 2000. The structure of inosine 5′-monophosphate dehydrogenase and the design of novel inhibitors. Immunopharmacology 47:163–184 [DOI] [PubMed] [Google Scholar]
- 45. Takagaki K., et al. 2004. Gene-expression profiling reveals down-regulation of equilibrative nucleoside transporter 1 (ENT1) in Ara-C-resistant CCRF-CEM-derived cells. J. Biochem. 136:733–740 [DOI] [PubMed] [Google Scholar]
- 46. Tam R. C., et al. 1999. Ribavirin polarizes human T cell responses towards a Type 1 cytokine profile. J. Hepatol. 30:376–382 [DOI] [PubMed] [Google Scholar]
- 47. Toan S. V., et al. 2003. Genomic organization and functional characterization of the human concentrative nucleoside transporter-3 isoform (hCNT3) expressed in mammalian cells. Pflugers Arch. 447:195–204 [DOI] [PubMed] [Google Scholar]
- 48. Vo N. V., Young K. C., Lai M. M. 2003. Mutagenic and inhibitory effects of ribavirin on hepatitis C virus RNA polymerase. Biochemistry 42:10462–10471 [DOI] [PubMed] [Google Scholar]
- 49. Wakita T., et al. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wohnsland A., Hofmann W. P., Sarrazin C. 2007. Viral determinants of resistance to treatment in patients with hepatitis C. Clin. Microbiol. Rev. 20:23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamamoto T., et al. 2007. Ribavirin uptake by cultured human choriocarcinoma (BeWo) cells and Xenopus laevis oocytes expressing recombinant plasma membrane human nucleoside transporters. Eur. J. Pharmacol. 557:1–8 [DOI] [PubMed] [Google Scholar]
- 52. Yao S. Y., et al. 1997. Molecular cloning and functional characterization of nitrobenzylthioinosine (NBMPR)-sensitive (es) and NBMPR-insensitive (ei) equilibrative nucleoside transporter proteins (rENT1 and rENT2) from rat tissues. J. Biol. Chem. 272:28423–28430 [DOI] [PubMed] [Google Scholar]
- 53. Zhang J., et al. 2007. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 26:85–110 [DOI] [PubMed] [Google Scholar]