

Abstract
The extracellular signal-regulated kinase 1/2 (ERK1/2) cascade is a central signaling pathway that regulates a wide variety of stimulated cellular processes, including mainly proliferation, differentiation, and survival, but apoptosis and stress response as well. The ability of this linear cascade to induce so many distinct and even opposing effects after various stimulations raises the question as to how the signaling specificity of the cascade is regulated. Over the past years, several specificity-mediating mechanisms have been elucidated, including temporal regulation, scaffolding interactions, crosstalks with other signaling components, substrate competition, and multiple components in each tier of the cascade. In addition, spatial regulation of various components of the cascade is probably one of the main ways by which signals can be directed to some downstream targets and not to others. In this review, we describe first the components of the ERK1/2 cascade and their mode of regulation by kinases, phosphatases, and scaffold proteins. In the second part, we focus on the role of MEK1/2 and ERK1/2 compartmentalization in the nucleus, mitochondria, endosomes, plasma membrane, cytoskeleton, and Golgi apparatus. We explain that this spatial distribution may direct ERK1/2 signals to regulate the organelles’ activities. However, it can also direct the activity of the cascade’s components to the outer surface of the organelles in order to bring them to close proximity to specific cytoplasmic targets. We conclude that the dynamic localization of the ERK1/2 cascade components is an important regulatory mechanism in determining the signaling specificity of the cascade, and its understanding should shed a new light on the understanding of many stimulus-dependent processes.
Keywords: MAPK, ERK, MEK, nucleus, mitochondria, Golgi
Introduction
The intracellular communication between membranal receptors and their nuclear or cytoplasmic targets upon stimulation is mediated by a limited number of signaling pathways, including a group of mitogen-activated protein kinase (MAPK) cascades. This group includes 4 distinct cascades, which are named after their MAPK tier component: 1) extracellular signal-regulated kinase 1 and 2 (ERK1/ 21-3); 2) c-Jun N-terminal kinase 1 to 3 (JNK1-34,5); 3) p38MAPK α, β, γ, and δ (p38α-δ6-9); and 4) ERK5 (also known as Big MAPK10,11). Additional MAPK-like molecules were termed ERK3/42,12 and ERK7/8,13,14 but at this stage, it is not clear whether these kinases are activated by phosphorylation cascades in a manner similar to the signaling via other MAPKs, and therefore, they are not considered as genuine MAPK cascades.15 The ERK1/2 cascade transmits mostly mitogenic signals, whereas the p38 and JNK cascades transmit mainly stress signals. ERK5 seems to play a role in both mitogenic and stress-response processes. Each MAPK cascade is activated by either a small GTP-binding protein (smGTP; Rap, Ras), or an adaptor protein, which transmits the signal directly, or through a mediator kinase (MAP4K) to the MAPK kinase kinase (MAP3K) level of the cascade. The signal is then transmitted to the MAPK kinase (MAPKK), MAPK, and eventually to MAPK-activated protein kinase (MAPKAPK). MAP3K, MAPKK, and MAPK are considered the core components, while MAP4K and MAPKAPK are required in specific conditions. While the MAP4K, MAP3K, and MAPKK levels are used mainly for the signal transmission, the MAPK and MAPKAPK components phosphorylate a large number of substrates that eventually regulate most stimulated cellular processes, including proliferation, differentiation, survival, apoptosis, and more.16-18
The first MAPK to be discovered is the ERK1/2 cascade that acts downstream of Ras and usually involves sequential phosphorylation and activation of the MAP3Ks Raf-1, B-Raf, and A-Raf (Rafs); MAPK/ERK kinases (MEK) 1/2; ERK1/2; and MAPKAPKs.3,19-20 In this review, we will describe the components of the cascade as well as the various aspects of their mode of regulation. In this sense, we will concentrate mainly on the subcellular distribution of MEK1/1b/2 and ERK1/1c/2 and their roles in the nucleus, plasma membranes, mitochondria, endosomes, cytoskeleton, and Golgi apparatus. Although Ras and Rafs may be localized in additional organelles, such as endoplasmic reticulum or lysosomes, these organelles do not seem to contain any MAPKK and MAPK components of the cascade and therefore will not be discussed here. Understanding the role of the ERK1/2 cascade in the above organelles should provide a wider view on their regulation as well as the determination of signaling specificity by the ERK1/2 cascade.
The Components of the ERK1/2 Cascade
The activation of the ERK1/2 cascade is mostly initiated at membrane receptors, such as receptor Tyr kinases (RTKs23), G protein–coupled receptors (GPCRs),24 ion channels,25 and others. These receptors transmit the signal by recruiting adaptor proteins (e.g., Grb2) and exchange factors (e.g., SOS) that, in turn, induce the activation of Ras at the plasma membranes, or membranes of other organelles. The activated, GTP-bound Ras then transmits the signal by activating the protein kinases Raf-1, B-Raf, and A-Raf (Rafs) within the MAP3K level of this cascade.26 This activation occurs by recruiting Rafs to the membranes, where they are then phosphorylated and activated by a mechanism that is not fully understood.27 Under specific conditions, other MAP3K components may participate in the activation of ERK1/2. Examples that depict this are c-Mos, which acts specifically in the reproductive system,28 the proto-oncogene TPL2, which seems to act in transformed cells,29 and MEKK1, which may act as a MAP3K in the ERK1/2 cascade under stress conditions.30
Upon activation, the Rafs transmit their signal by phosphorylating the MAPKKs, MEK1/2.31 The MEK1/2 were first identified as ERK1/2 activators,32,33 and their study revealed 2 main proteins with molecular masses of 45 kDa (MEK1) and 46 kDa (MEK2), which share a high degree (more than 85%) of homology.34-36 These proteins are composed of a large regulatory N-terminal domain containing a nuclear export signal (NES), followed by a catalytic kinase domain and a shorter C-terminal region. MEK1/2 are activated through serine phosphorylation at the MAPKK-typical Ser-Xaa-Ala-Xaa-Ser/Thr motif in their activation loop (residues 218-222 in human MEK137). In turn, MEK1/2 activate their only known substrates, native ERK1/2, which function as their sole downstream targets, suggesting that the MEK1/2 serve as the specificity-determining components of the ERK1/2 cascade. The MEK1/2 are the only kinases that can phosphorylate both regulatory Thr and Tyr residues of ERK1/2, and therefore, they belong to the small family of dual-specificity protein kinases.38
The next components of the cascade, namely ERK1/2, belong to its MAPK level and are important executers of the upstream signals. They are evolutionary conserved kinases that are gene products of ERK1 (MAPK3) and ERK2 (MAPK1). Each gene encodes one main product, which are the primary 44-kDa (ERK1) and 42-kDa (ERK2) proteins. In addition, these genes encode several splice variants, including ERK1b (46 kDa39), ERK1c (42 kDa40), and others that will be described below. The MEK1/2-mediated phosphorylation of ERK1/2 on the Thr and Tyr residues in their Thr-Xaa-Tyr signature motif (Thr202 and Tyr204 in human ERK141) is the mechanism that induces their full activation. Because of the high similarity between ERK1 and ERK2, their function and regulation are similar under most circumstances,42,43 although some differences between their functions have been recently elucidated under distinct conditions.44-46 The ERK1/2 are “Pro-directed” protein kinases, meaning that they phosphorylate Ser or Thr residues, neighbors to Pro residues. Pro-Xaa-Ser/Thr-Pro is the most common consensus sequence for substrate recognition by ERK1/2, although Ser/Thr-Pro can serve as a substrate as well.47
About 200 distinct substrates of ERK1/2 have been identified to date, which seem to be responsible for the induction and regulation of various ERK1/2-dependent processes.19,48 Among the first identified substrates/interactors of the ERK1/2 cascades are the cytoplasmic PLA2,26 cytoskeletal elements,49 and the intercellular domains of membranal receptors.50 However, not less important are the large number of nuclear ERK1/2 targets identified, including transcription factors such as Elk1,51 c-Fos,52 and c-Jun.53 These latter substrates require stimulation-dependent nuclear translocation of ERK1/2 for their phosphorylation, and this is regulated by the novel nuclear translocation signal (NTS) on ERK1/2, recently identified in our laboratory.54 Finally, among the large number of substrates, several MAPKAPKs may further extend the ERK1/2 cascade. The first identified MAPKAPK was RSK in which its phosphorylation by ERK1/2 was one of the main building blocks for the elucidation of the whole signaling cascade.1 MAPK-interacting protein kinase (MNK55,56) and mitogen- and stress-activated protein kinase (MSK57) were also identified as MAPKAPKs of the cascade. However, the activation of these kinases is not restricted to the ERK1/2 cascade, as they are also activated by the p38 cascade, mainly under stress conditions. Thus, the signals of the ERK1/2 cascade are well-disseminated, and it is estimated that a few minutes after extracellular stimulation, the cascade is responsible for no less than 500 phosphorylations throughout the cells.
Alternative Splicing within the ERK1/2 Cascade
As mentioned above, aside from the main components, each tier of the ERK1/2 cascade contains one or more alternatively spliced isoforms. These variants are much less abundant than the main isoforms described above, and their expression and activity are usually cell type or condition specific. However, the spliced forms have been reported to manifest unique important functions that are not shared by the main isoforms, and therefore, they may be considered as individual signaling components. At the MAP3K level of the cascade, B-Raf was reported to express up to 10 spliced variants in the mouse and human.58,59 Interestingly, some of the spliced isoforms demonstrate higher basal B-Raf activity, while in others, the activity is compromised. An example of such cases is the alternative splicing of exons 8b and 10 that are located just before the kinase domain.60 Thus, the insertion of the exon 10 sequence increases the affinity of B-Raf for MEK1/2, its basal kinase activity, and transforming properties. On the other hand, insertion of the exon 8b sequence seems to have the opposite, inhibitory effects. More recently, it was shown that aberrant B-Raf splicing can serve as an alternative mechanism for oncogenic B-Raf activation in vivo.61 Thus, novel B-Raf splice variants that lack the N-terminal autoinhibitory domain were detected in patients with thyroid carcinoma. These variants were constitutively active and significantly associated with the oncogenic V600E mutation and an advanced stage of the disease. All these studies indicate that splice variants of B-Raf may function differently from the most abundant isoform, either by activating or inhibiting downstream activities of the ERK1/2 cascade.
The MAP3K tier of the ERK1/2 cascade is not the only one to contain alternatively spliced isoforms. Thus, the MAPKK tier contains one alternatively spliced isoform, MEK1b, which lacks a 26–amino acid stretch in the kinase domain, as compared to the main transcribed form. This kinase was initially identified as an inactive form of MEK1, as it failed to phosphorylate ERK1/2,35,36 but was later shown to phosphorylate an alternative spliced isoform of ERK1 (ERK1c) and thereby to form an independent signaling cascade that differs from the main MEK1/2-ERK1/2 cascade.62 Thus, the elimination of 26 amino acids does not cause inactivation but rather modifies the substrate specificity of the kinase without modifying the remarkable selectivity of MEK family members to their respective ERKs.3 In the next tier of the cascade, the alternatively spliced isoform ERK1b was identified mainly in rodents as a 46-kDa protein that contains a 26–amino acids insert, just C-terminal to the kinase domain of ERK1.39 This kinase seems to be activated similar to the main ERK1/2, although under some circumstances, it is regulated differently because of distinct regulation of its altered CRS/CD domain.63 Interestingly, a similar splicing event also occurs in primates; in this case, however, the insert consists of 103 base pairs that are followed by a stop codon, which leads to the translation of a 42-kDa protein termed ERK1c.40 It was shown that ERK1c is expressed in most human cells, is activated primarily by MEK1b, and is unique in regulating mitotic Golgi fragmentation.62,64 Other ERK1/2 splice variants, such as ERK1d (unpublished) and ERK2b,12 seem to be generated as well, and extend the specificity of the ERK1/2 cascade, as described below.
Regulation of ERK1/2 by Kinases and Phosphatases
The activation of ERK1/2 is induced by MEK1/2 phosphorylation of both Thr and Tyr residues in the ERK1/2’s activation loop. This phosphorylation causes dramatic conformational changes, which enable full activation and interaction of ERK1/2 with their substrates. These global conformational changes elevate the catalytic rate of ERK1/2 to approximately 5 mM/min/mg, which is 5 to 6 orders of magnitude higher than the basal activity.65 Interestingly, aside from this activation loop phosphorylation, ERK1/2 were demonstrated to undergo regulatory phosphorylation on additional residues. For example, it was shown that 2 Ser residues (Ser244 and Ser246 of ERK2) in their kinase insert domain (KID) are phosphorylated upon stimulation.54 These phosphorylations, which are probably mediated by more than one kinase, are important for binding of ERK1/2 to importin7 and their nuclear translocation, as described below. In addition, autophosphorylation of a residue in the activation loop (Thr188 in ERK2) was shown to affect the subcellular localization of ERK1/2 as well.66 This phosphorylation seems to exhibit a unique role of ERK1/2 in integrating G protein–coupled receptor–initiated signals in order to induce cardiac hypertrophy. Importantly, other residues in close proximity to the activation loop of ERK1/2 are phosphorylated as well67 and thereby may affect this important region of ERK1/2.68 Notably, these Thr-Glu-Tyr–independent phosphorylations are usually not important merely for the activation mechanisms of ERK1/2 but rather play a role in the regulation of ERK1/2 localization or downregulation and thereby in the determination of their signaling specificity, as described below.
The activation of the ERK1/2 cascade upon extracellular stimulation lasts between 20 minutes (transient activation) up to 2 to 3 hours (sustained activation). Because the duration of the signal is necessary for proper ERK1/2 signaling (see below), the downregulation phase of the ERK1/2 cascade is not less important than its activation. Although degradation69 or distorting scaffolding interactions70 have been reported to participate in the regulation of ERK1/2, the main mechanism by which ERK1/2 are inactivated involves the removal of 1 or 2 of the phosphates from its activation loop.71 Since the phosphorylation of both Tyr and Thr residues is required to induce the full ERK1/2 activity, removal of phosphate from just one of them is sufficient for full inactivation. Thus, protein Ser/Thr phosphatases, protein Tyr phosphatases, and dual-specificity phosphatases (MKPs) all act to inactivate ERK1/2 under various conditions. MKPs are mostly products of inducible genes that are expressed only 30 to 60 minutes after stimulation, and therefore, the short-term inactivation of ERK1/2 is mediated mainly by protein Ser/Thr phosphatases or protein Tyr phosphatases that are responsible for ERK1/2’s transient activation.72 Escape from these 2 phosphatases often results in a prolonged ERK1/2 activation, which is finally downregulated by the induced, newly expressed MKPs.73 In summary, it is clear that ERK1/2 activity is heavily regulated by a set of MEK1/2-independent kinases and phosphatases that play an important role in regulating the substrate specificity of the ERK1/2 cascade.
Determination of Signaling Specificity
The ERK1/2 cascade is a linear signal transduction pathway that induces distinct and even opposing physiological processes. The above regulatory processes are not sufficient to allow the plethora of ERK1/2-induced effects, which raises the question as to the full scope of the mechanisms that are involved in the determination of the signaling specificity of this cascade. The current described mechanisms can be categorized into at least 5 distinct types,20 and these are as follows: 1) differences in the duration and strength of the signal; 2) interaction with scaffold proteins; 3) crosstalk of the MAPK components with other signaling pathways that are activated or inhibited simultaneously; 4) presence of multiple components in each level of the cascade, including substrate competition; and 5) compartmentalization of the MAPK cascade components and their targets in certain organelles or other cellular regions. The last mechanism is the main issue of this review and therefore will be covered in detail. A brief description of the first 4 mechanisms follows.
Differences in the duration and strength of the signal: This was the first mechanism elucidated for signaling specificity determination. In early experiments, it was shown that in PC12 cells, both EGF and NGF induce strong activation of ERK1/2, albeit with distinct outcomes. EGF stimulation caused a transient activation of ERK1/2 (peaking at 15 minutes and reduced back to basal levels after 40 minutes) and proliferation of PC12, while NGF stimulation led to a sustained activation (after 15-180 minutes), which resulted in differentiation of the cells.74,75 It was later demonstrated that these effects are mainly interpreted by immediate early genes that induce distinct cellular processes dependent on the signal length.76 As mentioned above, the duration of the signal is regulated mainly by protein phosphatases, which are therefore key regulators of ERK1/2 signaling outcomes.77
Scaffold proteins: The term “scaffold protein” is given to a protein that interacts with more than one protein in a specific cascade.78 In this way, scaffold proteins allow the formation of multicomponent complexes that may be important for the regulation of all MAPK cascades. For example, scaffold proteins may bring various components of the same cascade together and thereby facilitate the kinetics and duration of MAPK activation. Scaffold proteins may also direct the cascade to either specific upstream receptors or to unique downstream targets contributing to proper signal distribution. In addition, they may contribute to the stabilization of some components, determination of signaling thresholds, direction of the localization of the cascade components, or protecting signaling components from phosphatases. All these effects of scaffold proteins make them important regulators of the specificity of the ERK1/2 cascade.20 The regulation of ERK1/2 by scaffold proteins in different compartments is described below.
Crosstalk with other signaling cascades: Although the ERK1/2 cascade is a central signal transduction pathway in the cell, the same stimuli can also activate other cascades, such as PI3K-AKT, NF-κB, and others. These cascades may interact with each other and thus modulate the signaling output by cross-phosphorylation between the cascades, by combinatorial effects on downstream targets, or by modulation of activity.20 For example, several components of the PI3K-AKT pathway interact with and regulate the ERK1/2 cascade.79,80 Thus, MEK1/2 were suggested to be a focal point for cross-cascade regulation because they are affected by Rho family proteins and PAK1 downstream of PI3K.81 In addition, PI3K is thought to affect the ERK1/2 cascade via a direct interaction of its p110 catalytic subunit with Ras.82,83 Importantly, several improper interactions with other cascades are thought to induce pathologies, such as cancer.84
Multiple components in each level of the cascade: The presence of various components with distinct functions or regulation in each level of the cascade is, as yet, another important mechanism for the determination and extension of signaling specificity. Thus, different proteins in the MAP3K tier, including Rafs, MEKK1, Cot, and Mos, may be involved in the activation of the cascade under varying conditions. Although the activity of MEK1 and MEK2 in the MAPKK tier of the cascade is similar under most circumstances, the multiple phosphorylation sites in the Pro-rich domain of MEK1, but not MEK2, suggested different functions between these isoforms under some conditions. Indeed, it has been demonstrated that MEK1 and MEK2 have distinct functions during cell cycle progression85 and other processes.86 In addition, the alternative spliced isoform MEK1b is specifically instrumental in regulating mitotic Golgi fragmentation. In the next tier of the cascade, ERK1/2, which also share a high degree of similarity, were initially considered to be functionally redundant. However, several studies have demonstrated that differences between ERK1 and ERK2 do exist. For example, different outcomes were noticed with ERK1 or ERK2 depletion,85 whereas ERK2, and not ERK1, was shown to play a role in the induction of epithelial-to-mesenchymal transformation.46 Finally, the alternatively spliced isoforms described above, as well as substrate competition,87 might participate in the determination of signaling specificity as well.
5. Localization of Components of the ERK1/2 Cascade
Restriction of components of the ERK1/2 cascade to specific cellular compartments as well as the dynamic changes in their localization after stimulation is an important way of signaling specificity determination. In resting cells, the components of the ERK1/2 cascade are localized to the cytoplasm mainly because of interaction with specialized anchor/scaffold proteins.78 Upon stimulation, Rafs are recruited to the plasma membrane, and to membranes in other compartments, because of their interaction with activated Ras. Upon activation, MEK1/2, ERK1/2, and RSKs are usually released from their cytoplasmic anchors, and this allows translocation of a large portion of their molecules into the nucleus and other cellular organelles.88 It should be noted, however, that not all the molecules of the signaling kinases are released from their anchors, and it was shown that a significant portion of ERK1/2 molecules remain attached to specific cytoplasmic anchoring proteins (i.e., PEA1570), preventing their nuclear translocation while directing them to particular cytoplasmic targets. The ability of ERK1/2 molecules to be released from the anchoring proteins upon stimulation is dependent upon their mode of interaction. Thus, the interaction of ERK1/2 with many of their anchoring proteins is mediated through their CRS/CD domain.89,90 Activating phosphorylation of the ERK1/2’s Thr-Glu-Tyr domain induces a large conformational change, which forces a release of ERK1/2 from the CRS/CD-dependent interactions.91 Other docking sites of ERK1/2 that may mediate their cytoplasmic interaction are the DEJL as well as loop 6 of the kinase. The binding of ERK1/2 through these domains does not seem to be reversed upon stimulation, and thereby, it may fix the ERK1/2 molecules to a certain region in the cytoplasm and prevents their stimulated translocations.72 The hydrophobic DEJL domain is usually necessary for proper ERK1/2 substrate phosphorylation,92 and its irreversible binding may modulate ERK1/2 translocations and activity.93 The binding through loop 6 seems to mediate interaction of ERK1/2 with cytoskeletal elements.94,95 As for DEJL, this interaction is probably not affected by Thr-Glu-Tyr phosphorylation and seems to direct ERK1/2 to cytoskeletal elements and the proper site of action.
While most of the ERK1/2 molecules migrate to the nucleus, a smaller portion of these, as well as MEK1/2 and Raf molecules, migrates into distinct subcellular compartments/organelles, where they are conducting specific functions88 (Fig. 1). The various translocation events and distinct localizations, which have important physiological functions, can be categorized into 2 main functional groups. The first is the ability of the translocated ERK1/2 to regulate specific activities within certain organelles, such as the regulation of transcription in the nucleus and mitochondria22,96 or the regulation of mitotic Golgi fragmentation.64 The second function is to bring components of the ERK1/2 cascade into proper localization in the outer surface of the organelles, where the signaling components phosphorylate specialized substrates without significant nuclear translocation. These unique localizations often result in a distinct signaling fate under varying conditions, and thereby, it is an additional important method for the determination of signaling specificity.97,98 As described above, scaffold and other interacting proteins often regulate the targeting to the organelles. Examples of such directing proteins are MP1 that directs ERK1 to endosomes,99 VDAC to the mitochondria,96 and Sef1 to the Golgi.100 Cytoskeletal elements, as well as cytoskeleton-related proteins, may also participate in this effect.78 The distinct functions of ERK1/2 in various organelles are described below.
Figure 1.
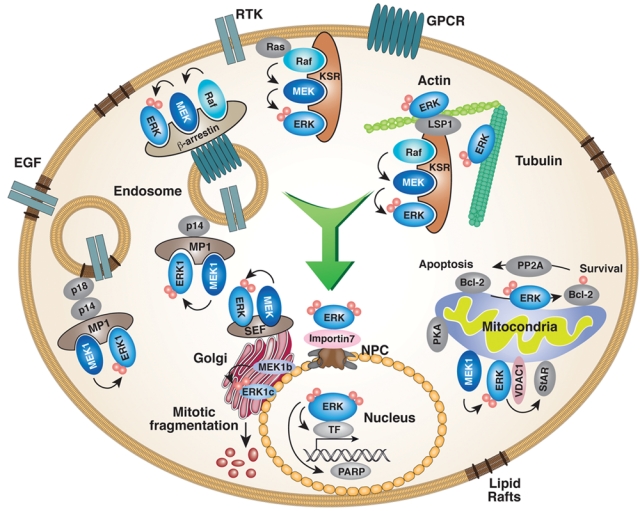
ERK1/2 distribution within the various compartments of the cell. The activation of the ERK1/2 cascade results in a significant translocation of the ERK1/2 molecules to the nucleus, which is mediated by interaction with importin7 to induce mainly proliferation and differentiation. In addition, ERK1/2 translocate into various cellular organelles usually because of interaction with specific scaffold proteins. In each of these organelles, ERK1/2 can either regulate intrinsic activities or direct ERK1/2 signals to nearby cytoplasmic substrates (for details, see text).
ERK1/2 in the Nucleus
As mentioned above, many ERK1/2 molecules detach from the anchor proteins upon stimulation and translocate to various organelles and other cellular compartments.88 Most notably, a large portion of the ERK1/2 molecules (50%-70%) can be found in the nucleus within 10 to 20 minutes after cellular stimulation.101 It was shown that specific abrogation of ERK1/2 translocation into the nucleus blocks growth factor–induced gene expression and other induced processes.70,102 Therefore, it is likely that understanding nuclear activities should shed light on the regulation of many growth factors or oncogene-dependent cellular processes including proliferation, differentiation, and oncogenic transformation. The nuclear effects of ERK1/2 are executed via a large number of substrates, interacting proteins, and direct DNA interactions in this location. Significantly, about half of the currently identified ERK1/2 substrates are nuclear proteins, and those participate in the regulation of many stimulated nuclear processes.19 These effects include mainly transcription (i.e., Elk1 activation103) as well as modulation of transcription suppression (i.e., Erf-1 suppression104), which are described below. Other mechanisms by which ERK1/2 affect the nuclear process include chromatin remodeling (i.e., PARP-1 regulation105) and nuclear translocation (i.e., phosphorylation of NUP50106). The direct effect of MEK1/2107 and RSKs108 translocation to the nucleus is less studied at this stage and therefore will not be covered here.
The main group of effectors identified is that of transcription factors, most notably those that regulate immediate early genes (IEG109). The rapid IEG transcription after stimulation requires activation of their transcription factors within minutes after stimulation, and this occurs mainly by the rapidly translocated ERK1/2. One of the best-studied ERK1/2-activated transcription factors is a nuclear ETS domain–containing Elk1.51,103 The rapid phosphorylation of Elk1 by ERK1/2 occurs on 6 to 9 sites110 and requires a direct docking interaction between the 2 proteins.111 One of the earliest transcriptional events regulated by Elk1, upon stimulation, is the induction of the IEG c-Fos, which is important for the proper progression of proliferation and differentiation.112 Thus, the expression of c-Fos, which is minimal in quiescent cells, is dramatically induced upon stimulation for durations that may vary from minutes to hours. These changes in duration are regulated by ERK1/2-dependent stabilization of c-Fos, which is achieved only when ERK1/2 activation is sustained, namely strong enough when the c-Fos is significantly expressed.113 Therefore, the differences in stability make c-Fos a good interpreter of differences in ERK1/2 kinetics of activation, which was shown to play a role in the determination of ERK1/2-dependent signaling specificity.75 Similar phosphorylation by ERK1/2 seems to be important for the stability and activity of additional IEGs, such as c-Myc and Fra1,76 which indicates that ERK1/2 serve as key regulators of transcription factors and IEGs upon various conditions.
Aside from the role of ERK1/2 in regulating IEGs, they can phosphorylate and modulate the activity of additional transcription factors that participate in the induction of intermediate or late genes. Notably, ERK1/2 were demonstrated to regulate several members in the subgroup of nuclear receptors, including estrogen receptor (ER), which under most conditions can be activated by this phosphorylation.114 In addition, components of the ERK1/2 cascade also affect the genomic activity of peroxisome proliferator–activated receptor gamma (PPARγ). However, unlike the activation of ER, the phosphorylation of this nuclear receptor by ERK1/2 actually inhibits the nuclear functions of this nuclear receptor.115 Attenuation of activity by ERK1/2-mediated phosphorylation was also reported for retinoic X receptor alpha (RXRα116), and some studies suggest that this might be the case for the glucocorticoid receptor (GR117) as well. Thus, the main function of ERK1/2-dependent phosphorylation of nuclear receptors is probably suppression of their activity or not activating them, as is the case for the IEG-related transcription factors. Interestingly, suppression of transcription by the ERK1/2 cascade was reported in the case of other transcription or repression factors as well. One of them is the Ets2 repressor factor (Erf1), which suppresses transcription in many resting cells.118 The phosphorylation by ERK1/2 induces CRM1-dependent nuclear export of Erf1, which thereby alleviates its suppression of transcription.104 Finally, the cascade was also implicated in suppressing gene activity by the direct interaction of ERK2 with DNA.119 This suggests that aside from its well-known regulation of transcription factors, ERK2 can regulate gene expression by activity-independent binding to promoter regions.
One of the questions that attracted much attention in the last few years is the mechanism by which ERK1/2 translocate into the nucleus.88,120 The nuclear envelope separates the nucleus from the rest of the cell and guarantees a selective transport into the nucleus. Proteins and other molecules shuttle into the nucleus via specialized nuclear pores (NPCs) that coordinate nucleocytoplasmic exchange.121 These NPCs allow free diffusion of small molecules and proteins (up to 40 kDa), whereby bigger proteins penetrate through the pore by using energy-dependent transport machinery. In many cases, this machinery includes the binding of a basic region in the transported protein (cargo), which serves as a nuclear localization signal (NLS) to the trafficking proteins, importins α and β. These importins then serve as shuttle for the NLS-containing cargo proteins and facilitate their transport through the NPCs.122 Other less-frequent and less-understood mechanisms of translocation seem to involve either nonconventional NLS,123 binding of the cargo to the NPC,124 or binding to non-α/β importins.125 Interestingly, ERK1/2 translocation seems to be mediated by the latter mechanism, namely by binding to importin7.126 Our recent findings54 suggest that upon stimulation, the activatory Thr and Tyr residues of ERK1/2 are phosphorylated by MEK1/2 to induce their activation and detachment from cytoplasmic anchoring proteins. This dissociation then allows the phosphorylation of 2 Ser residues within the kinase insert domain of ERK1/2 (Ser244 and Ser 246 in human ERK2, termed nuclear translocation signal [NTS] domain), which further induces interaction with importin7. NTS-like sequences were also found in other proteins like MEK1/2 (TPT) and SMAD3. In the nucleus, ERK1/2 are downregulated and are eventually exported from the nucleus, probably by coupling to MEK1/2 that induce the export by their binding to exportins.120 However, more studies are required in order to better understand the full mechanism by which ERK1/2 shuttling is regulated in various cell lines and processes.
ERK1/2 in the Mitochondria
The mitochondrion is a point of integration of signaling cascades because of its pivotal role in cellular metabolism, redox biochemistry, and survival/death decisions.127 Regulation of the number of the mitochondrial organelles in relation to environmental/cellular conditions would require coordinated transcription of nuclear and mitochondrial genes and the genesis of, or mitochondria trafficking to, appropriate regions with high-energy utilization. Various signaling components and cascades are involved in this type of cell fate processing via the mitochondria, including ERK1/2, PKA, PKC, PI3K-Akt, JNK, and p38 MAPK.128 In the following section, we describe the role of the ERK1/2 cascade in mitochondria regulation and activity. Unlike other organelles (see below), at this stage, there is no convincing evidence for ERK1/2 signaling from the outer surface of the mitochondria towards cytosolic targets.
Many studies, using pharmacological inhibitors of MEK1/2, have indicated that ERK1/2 can modulate mitochondrial functions, particularly those associated with cell death. While some of these studies demonstrated an antiapoptotic effect of ERK1/2,129,130 others showed that ERK1/2 have either proapoptotic effects or even induce nonapoptotic cell death.131,132 Although it is clear from these studies that ERK1/2 are important regulators of mitochondria activities, the reason for the different effects in specific cell types and conditions is not fully understood. These studies have also raised the question as to how the ERK1/2 signals are transmitted into the mitochondrial proteins and activities. Although it is conceivable that ERK1/2 may affect the mitochondrial activities by regulating the expression of mitochondrial proteins in the nucleus,133 it appears that these kinases may have intrinsic mitochondrial activities as well. One of the first evidence supporting mitochondrial localization and activity of ERK1/2 was derived from biochemical studies in renal tubular cells. In that study, it was found that upon cisplatin treatment, activated ERK1/2 as well as PKCα are present in enriched mitochondrial fractions. This localization was suggested to contribute to increased mitochondrial membrane potential, decreased oxidative phosphorylation, and increased caspase-3 activation and apoptosis in these cisplatin-treated cells.134 In addition, it was shown that ERK1/2 colocalize with Bcl2 in the mitochondria, where the latter is phosphorylated by ERK1/2 on Ser87 to exert its antiapoptotic effect. This effect of ERK1/2 on Bcl2 is reversed by mitochondrial PP2A.135,136 Additional cells in which ERK1/2 were identified in the mitochondria include murine cardiac myocytes, where the kinases are associated with enhanced Bad phosphorylation and cardioprotection,137 and human alveolar macrophages, where ERK1/2 maintain mitochondrial membrane potential and ATP production.138 Finally, detailed immunoelectron microscopy studies have established the presence of phosphorylated ERK1/2 in the outer membrane and intermembrane space of brain mitochondria,139,140 suggesting their role in multiple mitochondria functions.
The accumulation of ERK1/2 in the mitochondria can vary in different cellular conditions, suggesting that these kinases may translocate into this compartment upon stimulations. Thus, it was shown that in the brain of developing mice, the levels of mitochondrial ERK1/2 peak at stages E19 to P2, decreasing from P3 to adulthood. The decrease in mitochondrial ERK1/2 correlates with their increased nuclear translocation, providing information about mitochondrial energetic and redox status to the proliferating/differentiating cells.140 In addition, phosphorylated ERK1/2 were found within a subset of mitochondria in degenerating neurons from patients with Parkinson disease and Lewy body dementia.139 In the transformed Leydig-derived MA-10 cell line, presence of ERK1/2 in the mitochondria and their activation are obligatory for PKA-mediated steroidogenesis and thereby are probably associated with their oncogenic potential.141,142 Despite the clear accumulation and activation of ERK1/2 in the mitochondria, little is known about the mechanisms that allow these processes, especially because none of the components of the ERK1/2 cascade seems to contain a mitochondrial localization signal. In this regard, it was shown that ERK1/2, as well as p38, JNK, and their respective MAPKKs, are present in the mitochondria of a murine tumor cell line, and the traffic of these MAPKs in and out of the organelle is regulated by hydrogen peroxide.143 Another important step in understanding the mitochondrial localization, regulation, and role in mitochondrial activities was the recent proteomic study on ERK1/2-interacting proteins in the mitochondria.96 In this study, it was demonstrated that ERK1 physically associates with structural, signaling, transport, and metabolic proteins in the mitochondria of HeLa cells. Among the new interactors identified were the voltage-dependent anion channel 1 (VDAC1) that may facilitate the mitochondrial transport of ERK1/2. These interactions suggest that aside from their role in mitochondria-dependent survival/apoptosis, ERK1/2 participate in several unexpected intrinsic mitochondrial processes. These functions include the regulation of various proteins that govern the ATP source in the outer mitochondrial membrane, as well as the activation of mitochondrial transcription factors and transcriptional machinery. However, the exact mechanisms by which ERK1/2 are translocated and regulated in the mitochondria, the mechanism of MAPKKs activation, and the full scope of ERK1/2 functions in this location are still obscure.
ERK1/2 in the Endosomes
Formation of endosomes is crucial not only for signal termination of receptors but also for the activation of various cellular functions, such as nutrient intake/digestion, membrane protein cycling, cell migration, and intracellular signaling.144 Early endosomes, which are usually formed as clathrin-coated vesicles, are initiated by the internalization of either GPCRs or RTKs. Upon their detachment from the plasma membrane, the early endosomes either recycle back to the plasma membrane or mature into late endosomes, which often migrate into lysosomes, where the receptors are degraded.145,146 In many cases, the endocytosis of activated receptors is necessary not only for their downregulation but also for the induction of additional signaling outputs. Indeed, several early studies showed that endocytosis and endosomes are required for proper receptor signaling through the ERK1/2 cascade. For example, it was shown that inhibition of internalization attenuates ERK1/2 activation by lysophosphatidic acid (LPA), thrombin, and bombesin receptors in Rat-1 fibroblasts.147 Moreover, endocytosis of the GPCR β2 adrenergic receptor was shown necessary for the activation of the ERK1/2 cascade,148 and the internalization of PAR2 is required for the interaction of the receptor with its downstream effectors, Rafs, and ERK1/2 in the endocytic compartment.99
Since these initial findings, much information was accumulated on the role of endosomes in ERK1/2 signaling.149 The mechanism that allows the dissemination of the signals to downstream signaling components is primarily related to specialized scaffold proteins that bring these components to close proximity to each other and facilitate their activation. One such scaffold protein is β-arrestin, which was initially shown to enhance angiotensin II–induced Raf and MEK-dependent activation and endosomal targeting of ERK2.150,151 This scaffold directs ERK1/2 mainly to clathrin-coated endosomes that are formed together with receptor internalization, indicating a relatively rapid effect upon stimulation. Interestingly, the interaction of β-arrestin with activated ERK1/2 is irreversible and commits the ERK1/2 to phosphorylate cytoplasmic substrates without many effects on nuclear targets.152 Another scaffold protein that mainly binds ERK1 and directs it to endosomes is MEK1 partner 1 (MP1). MP1 was first identified in a yeast 2-hybrid screen as a binding partner of MEK1.153 MP1 binds MEK1 and ERK1 but not MEK2 and ERK2, and unlike β-arrestin that directs ERK1/2 to early endosomes, the recruitment of MP1 seems to be confined to late endosomes. Interestingly, MP1 seems to operate in conjunction with another binding protein, termed p14,99 which associates with the cytoplasmic face of the endosomes in a variety of cell types. ERK1 activation on endosomes regulates late endosomal traffic and cellular proliferation. While the MP1-p14 complex is required for ERK1 endosomal activation, this complex is irrelevant for ERK1 activation at the plasma membrane.154
Another type of ERK1/2 interaction with endosomes is through the adaptor protein p18 that directs the cascade components to lipid rafts on late endosomes and, consequently, to RTKs’ recycling.155 Lipid rafts are dynamic cholesterol-enriched microdomains in plasma and endomembranes and are proposed to serve as signaling platforms by facilitating protein-protein interactions.156 Importantly, the internalization of RTKs, like EGFR by clathrin-dependent endocytosis, usually results in a rapid recycling of the RTKs back to the plasma membranes. On the other hand, RTKs, but probably not GPCRs, that are internalized together with lipid rafts/caveolae are sorted from early endosomes to late endosomes, leading to lysosomal degradation.157,158 It was shown that p18 is anchored to lipid rafts of late endosomes and serves as a lipid raft anchor for p14-MP1-MEK1 signaling components.155 This complex can also recruit ERK1, while the p18-MEK1-ERK1 complex takes part in controlling intracellular membrane dynamics potentially by regulating organelle interactions and/or transports along cytoskeletal elements. Taken together, ERK1/2 do not seem to participate in the regulation of trafficking or other intrinsic endosomal activities. Rather, the endosomes seem to facilitate the activation of the ERK1/2 cascade downstream of internalizing RTKs and GPCRs and direct them to their proper targets in the cytoplasm. In these processes, RTKs and GPCRs are using a different set of scaffold/anchor proteins to execute their functions.
ERK1/2 in the Plasma Membrane and Cytoskeletal Elements
The plasma membrane is the origin of most intracellular signaling, as most of them are initiated by the membranal receptors. In addition, many signaling components are associated with the plasma membrane either by direct or indirect interactions. One of the main regulators of the ERK1/2 cascade in the plasma membrane is kinase suppressor of Ras (KSR), first identified by genetic screens in Drosophila and Caenorhabditis elegans as a positive regulator of the ERK1/2 cascade.159 Studies on the nature of KSR action led to the conclusion that it acts as a scaffold protein by facilitating ERK1/2 signaling, and as such, KSR was the first scaffold protein identified for the cascade. Interestingly, mammalian KSR1 (and probably also KSR2) is a central component of complex signaling machinery that initiates ERK1/2 signals from a close vicinity to the plasma membranes. Thus, in resting cells, KSR1 interacts with inactive MEK1/2160 but not with ERK1/2 or Rafs. For its regulation, KSR1 also interacts with c-Tak1, which constitutively phosphorylates its Ser392,161 as well as with the adaptor protein 14-3-3,162 inactive PP2A,163 and the inhibitory E3 ubiquitin ligase, IMP1.164 These components form a big protein complex that is localized primarily in the cytoplasm. Upon stimulation, IMP1 is recruited by Ras-GTP, which further induces its polyubiquitination and degradation. This induces a big change in the structure of the complex, allowing the associated PP2A to dephosphorylate Ser392 in KSR1, leading to dissociation of the KSR1 from the 14-3-3 protein and translocation to the plasma membrane. In this location, active Raf1 joins the complex and activates the pre-existing MEK1/2 that further recruit and activate ERK1/2 molecules.160 Finally, the activated ERK1/2 detach from the complex and shuttle to various cellular compartments, mainly the nucleus, to induce most ERK1/2-dependent cellular functions.20,159 Another membranal protein that may participate in the regulation of the ERK1/2 cascade is caveolin,165 localized mainly in caveolae. However, this interaction seems to mostly inhibit ERK1/2 activation and is probably specific to particular cell lines and conditions.
Yet another way to secure explicit localization of components of the ERK1/2 cascade and their proper regulation is achieved by interactions with cytoskeletal components.88,166 Several cytoskeletal elements have been reported to directly interact with ERK1/2 and other components of the cascade. Thus, it was initially shown that ERK1/2 associate with the microtubule and actin filaments both before and after cellular stimulation.49,167 This interaction may be induced by either a direct binding to microtubules or actin, or indirectly by adaptor proteins like calponin, which is an actin-binding protein. One of the main purposes of this interaction is to direct the ERK1/2 to their right localization and as such to restrict nuclear entry of activated ERK1/2. One example for the latter effect was demonstrated for retinoic acid–induced differentiation, which is accompanied by a reduction in cell proliferation. This reduced proliferation is mediated by restricting nuclear entry of ERK1/2, which requires intact actin and microtubule cytoskeleton.168 The association of ERK1/2 with the cytoskeleton was also suggested to be involved in the transport of phosphorylated ERK1/2 over long distances within the cell, using the cytoskeletal motors. For example, in lesioned nerves, the binding of vimentin to phosphorylated ERK1/2 enables spatial translocation of the kinases by importins and dynein.169 Aside from the direct interaction, and the interaction through cytoskeletal adaptors that recruit only ERK1/2 molecules, it was shown that scaffold proteins that recruit additional components of the ERK cascade might direct cytoskeletal localization of ERK1/2 as well. Thus, it was shown that IQGAP1 interacts with B-Raf, MEK1/2, and ERK1/2; recruits them to actin filaments; and thereby influences mitogenic, morphological, and migratory cell behavior.170,171 Another ERK1/2 scaffold protein that is also an actin-binding protein is LSP1, which associates with MEK1, ERK2, and KSR and targets them to peripheral actin filaments and thereby regulates signals to proliferation.172,173 Finally, not only are ERK1/2 regulated by cytoskeletal elements, they can also regulate cytoskeletal reorganization and thereby downstream cellular processes. An example of such an effect is the influence of ERK1/2 on cell motility, which is mediated in part by phosphorylation of myosin light chain kinase (MLCK) to enhance its activity and facilitate cell motility.174
ERK1/2 and ERK1c in the Golgi Apparatus
The Golgi apparatus of mammalian cells is organized into stacks of cisternae, which are anchored in the perinuclear region.175 Most of the processes within the Golgi, including glycosylation and trafficking, are probably not regulated by phosphorylation. However, the process of Golgi fragmentation may be different. Thus, once the cell enters mitosis, the perinuclear stacks of Golgi cisternae undergo extensive fragmentation, and the fragments are dispersed throughout the cytoplasm, later dividing between the separating cells. Golgi fragmentation is considered to be a “mitotic check point,” as once it starts, mitosis must be completed.15 Interestingly, it was previously shown that the fragmentation is inhibited by MEK1/2 inhibitors, indicating that the ERK1/2 cascade might be involved in its regulation.176 Moreover, it was shown that the fragmentation is associated with the accumulation of monophosphorylated ERK1/2 in the Golgi.177 However, although activated, no ERK1/2, and probably no MEK1/2, could be detected in the Golgi, either in the G2 or M phases of the cell cycle. Therefore, it became important to elucidate the mechanism by which MEK1/2 or ERK1/2 operate in order to induce the Golgi fragmentation. In this regard, our group has shown that the ERK1 splice variant, ERK1c, but not ERK1/2, accumulates in the Golgi in the relevant cell cycle stages.40,64 In addition, it was shown that ERK1c activity is elevated towards mitosis because of phosphorylation by MEK1b,62 and knockdown of MEK1b or ERK1c reduces mitotic Golgi fragmentation and attenuates mitotic progression. These results indicate that in late G2, Golgi-resident MEK1b activates ERK1c, which consequently regulates the Golgi fragmentation during mitosis (Fig. 2). One of the important questions that remained open in this study is what could be the downstream substrate of ERK1c that mediates its fragmentation effect? Interestingly, the Golgi protein GRASP55, which may be the protein that converts Golgi stacks into small fragments, was shown to be phosphorylated during early mitosis in a MEK-dependent manner.178 This and other Golgi proteins containing ERK1/2 consensus phosphorylation sites are likely candidates to be the MEK/ERK-dependent effectors during Golgi fragmentation, but the exact mechanism of action involving such proteins needs further clarification. A better understanding of the molecular mechanism by which ERK1c acts in the Golgi will shed light on a crucial step in mitosis and will expand our understanding of the signal specificity of the ERK1/2 cascade.
Figure 2.
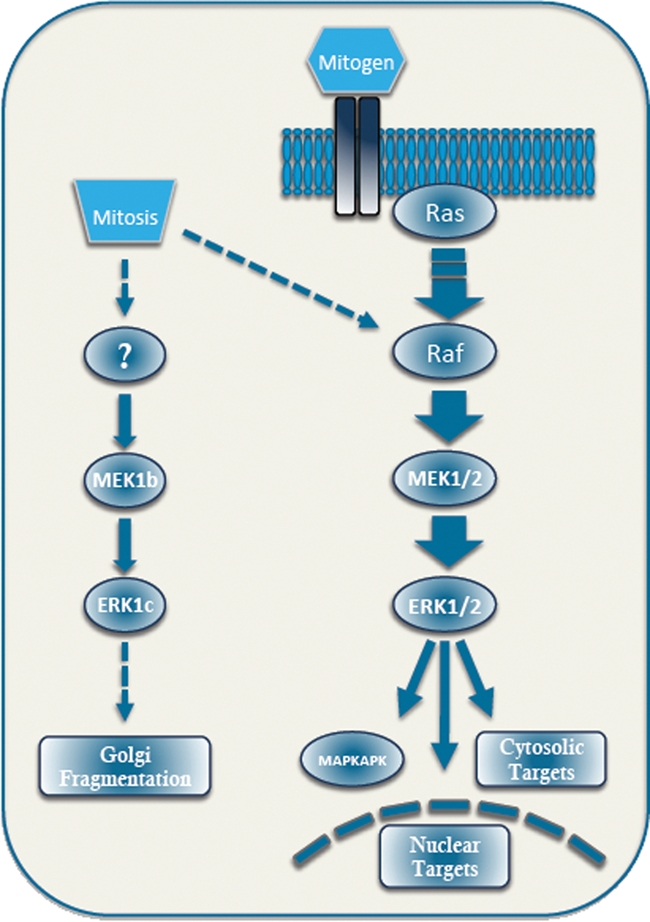
Schematic representation of the different components of the ERK1/2 cascade in resting and mitotic cells. In stimulated cells, most of the ERK cascade outcomes are facilitated by MEK1/2 and ERK1/2. During G2/M phases, some signals are transmitted by MEK1/2 and ERK1/2. In addition, the expression of MEK1 and ERK1 splice variants, MEK1b and ERK1c, is increased. At this stage, the activation of MEK1b and ERK1c is essential for mitotic Golgi fragmentation, while MEK1/2 and ERK1/2 activation induces other mitotic processes.
Aside from the role of MEK1b/ERK1c in the regulation of Golgi fragmentation, the outer surface of this organelle was suggested to serve as an anchoring platform for MEK1/2-ERK1/2–generated processes that lead to Golgi-independent cellular effects. This anchoring is mediated by at least one Golgi-specific scaffold protein, termed Sef1.100 This is a transmembranal Golgi protein that, in resting cells, seems to bind inactive ERK1/2 on the outer part of the Golgi apparatus. Upon stimulation, Sef1 binds irreversibly to activated MEK1/2 and thereby facilitates the activation of the prebound ERK1/2 molecules. At this stage, most of the Sef1-MEK1/2-ERK1/2 complexes remain bound to the Golgi, where they can initiate particular cytoplasmic signals. The irreversible nature of the ERK1/2-MEK1/2 interactions, induced by their interaction with Sef1, prevents the translocation of active ERK1/2 to the nucleus and thereby exerts an inhibitory effect on ERK1/2-dependent transcription. Indeed, knockdown of Sef1 results in enhanced ERK1/2 translocation to the nucleus and upregulation of ERK1/2-dependent genes, such as c-FOS, Egr1, and JunB.100 In addition, a small amount of the complexes translocate after stimulation to the plasma membranes and activate a particular set of targets at this vicinity as well. Another Golgi protein that may influence the ERK1/2 cascade is RKTG, which binds only to Raf1 and not MEK1/2 and ERK1/2.179 This protein seems to inhibit the ERK1/2 cascade by sequestering Raf1 from the other downstream signaling components. Thus, both Golgi scaffolds may inhibit rather than facilitate the rate of proliferation upon certain types of stimulations. In summary, it appears that although components of the ERK1/1c/2 cascade may influence Golgi function, the outer surface of this organelle may have a general inhibitory effect on part of the signal propagation downstream of the ERK1/2 cascade.
Summary
The ERK1/2 cascade is a central signaling pathway that participates in the regulation of many distinct and even opposing cellular processes. In order to execute all the diverse functions, the ERK1/2 cascade utilizes several mechanisms that determine its signaling specificity. These include differential temporal activation, scaffold proteins, interaction with different cascades, multiple components in each tier of the cascade, and differential spatial regulation. Interestingly, the relatively large number of ERK1/2 molecules in each cell (~107 molecules per cell) suggests that distinct pools of molecules mediate distinct functions. Such distinct subgroups of molecules can be maintained by the compartmentalization of the ERK1/2 molecules in different organelles or by interaction with specialized scaffold/anchoring proteins that prevent the translocation of the activated molecules to undesired compartments and bring them to close vicinity to their proper signaling partners under distinct conditions. These allow proper and specific signaling circuits that eventually result in the proper activation of the wanted cellular processes upon distinct activations.
Dysregulation of the ERK1/2 cascade is known to result in various pathologies, inducing neurodegenerative diseases,180 developmental diseases,181 diabetes,182 and cancer.183 The involvement of the ERK1/2 cascade in cancer is of particular interest, as it was shown that activating mutations of upstream components of ERK1/2 are responsible for more than half of all cancers.184 Notably, activated ERK1/2 were also found in cancers in which components of the cascade were not mutated, indicating that the ERK1/2 cascade plays a role even in carcinogenesis induced by apparently nonrelated oncogenes. It is also clear that the translocation of ERK1/2 into the nucleus, and probably other organelles, is essential for the proliferation of cancer cells as well as metastasis. Therefore, interference with the localization of certain components, and in particular with the translocation of ERK1/2 to the nucleus, may serve as potential therapeutic targets for some diseases. Further studies of the compartmentalization of the ERK1/2 cascade will broaden our knowledge of signal specificity determination and ERK1/2 involvement in diseases and may eventually lead to the development of new therapeutic strategies in combating diabetes, developmental disorders, and cancer.
Acknowledgments
The authors thank Dr. Efrat Glick-Saar and Ms. Martie Spiegel for their help in writing this article.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by grants from the Israeli Science Foundation, ICRF, and MINERVA. R.S. is an incumbent of the Yale Lewine and Ella Miller Lewine professorial chair for cancer research.
References
- 1. Sturgill TW, Ray LB, Erikson E, Maller JL. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature. 1988;334(6184):715-8 [DOI] [PubMed] [Google Scholar]
- 2. Boulton TG, Nye SH, Robbins DJ, et al. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65(4):663-75 [DOI] [PubMed] [Google Scholar]
- 3. Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9(9):726-35 [PubMed] [Google Scholar]
- 4. Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7(11):2135-48 [DOI] [PubMed] [Google Scholar]
- 5. Kyriakis JM, Banerjee P, Nikolakaki E, et al. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369(6476):156-60 [DOI] [PubMed] [Google Scholar]
- 6. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265(5173):808-11 [DOI] [PubMed] [Google Scholar]
- 7. Freshney NW, Rawlinson L, Guesdon F, et al. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78(6):1039-49 [DOI] [PubMed] [Google Scholar]
- 8. Rouse J, Cohen P, Trigon S, et al. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78(6):1027-37 [DOI] [PubMed] [Google Scholar]
- 9. Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372(6508):739-46 [DOI] [PubMed] [Google Scholar]
- 10. Zhou G, Bao ZQ, Dixon JE. Components of a new human protein kinase signal transduction pathway. J Biol Chem. 1995;270(21):12665-9 [DOI] [PubMed] [Google Scholar]
- 11. Lee JD, Ulevitch RJ, Han J. Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995;213(2):715-24 [DOI] [PubMed] [Google Scholar]
- 12. Gonzalez FA, Raden DL, Rigby MR, Davis RJ. Heterogeneous expression of four MAP kinase isoforms in human tissues. FEBS Lett. 1992;304(2-3):170-8 [DOI] [PubMed] [Google Scholar]
- 13. Abe MK, Kuo WL, Hershenson MB, Rosner MR. Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol. 1999;19(2):1301-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abe MK, Saelzler MP, Espinosa R, 3rd, et al. ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem. 2002;277(19):16733-43 [DOI] [PubMed] [Google Scholar]
- 15. Persico A, Cervigni RI, Barretta ML, Corda D, Colanzi A. Golgi partitioning controls mitotic entry through Aurora-A kinase. Mol Biol Cell. 2010;21(21):3708-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31(2):151-74 [DOI] [PubMed] [Google Scholar]
- 17. Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26(22):3100-12 [DOI] [PubMed] [Google Scholar]
- 18. Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3-38 [DOI] [PubMed] [Google Scholar]
- 19. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors. 2006;24(1):21-44 [DOI] [PubMed] [Google Scholar]
- 20. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213-26 [DOI] [PubMed] [Google Scholar]
- 21. Meloche S, Pouyssegur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227-39 [DOI] [PubMed] [Google Scholar]
- 22. Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: is subcellular localization the answer? Cell Cycle. 2009;8(8):1168-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marmor MD, Skaria KB, Yarden Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int J Radiat Oncol Biol Phys. 2004;58(3):903-13 [DOI] [PubMed] [Google Scholar]
- 24. Naor Z, Benard O, Seger R. Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab. 2000;11(3):91-9 [DOI] [PubMed] [Google Scholar]
- 25. Rane SG. Ion channels as physiological effectors for growth factor receptor and Ras/ERK signaling pathways. Adv Second Messenger Phosphoprotein Res. 1999;33:107-27 [DOI] [PubMed] [Google Scholar]
- 26. Kyriakis JM, Force TL, Rapp UR, Bonventre JV, Avruch J. Mitogen regulation of c-Raf-1 protein kinase activity toward mitogen-activated protein kinase-kinase. J Biol Chem. 1993;268(21):16009-19 [PubMed] [Google Scholar]
- 27. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5(11):875-85 [DOI] [PubMed] [Google Scholar]
- 28. Gotoh Y, Nishida E. Activation mechanism and function of the MAP kinase cascade. Mol Reprod Dev. 1995;42(4):486-92 [DOI] [PubMed] [Google Scholar]
- 29. Salmeron A, Ahmad TB, Carlile GW, Pappin D, Narsimhan RP, Ley SC. Activation of MEK-1 and SEK-1 by Tpl-2 proto-oncoprotein, a novel MAP kinase kinase kinase. EMBO J. 1996;15(4):817-26 [PMC free article] [PubMed] [Google Scholar]
- 30. Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL. A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science. 1993;260(5106):315-9 [DOI] [PubMed] [Google Scholar]
- 31. Kyriakis JM, App H, Zhang XF, et al. Raf-1 activates MAP kinase-kinase. Nature. 1992;358(6385):417-21 [DOI] [PubMed] [Google Scholar]
- 32. Ahn NG, Seger R, Bratlien RL, Diltz CD, Tonks NK, Krebs EG. Multiple components in an epidermal growth factor-stimulated protein kinase cascade: in vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J Biol Chem. 1991;266(7):4220-7 [PubMed] [Google Scholar]
- 33. Gomez N, Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353(6340):170-3 [DOI] [PubMed] [Google Scholar]
- 34. Crews CM, Alessandrini A, Erikson RL. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258(5081):478-80 [DOI] [PubMed] [Google Scholar]
- 35. Seger R, Seger D, Lozeman FJ, et al. Human T-cell mitogen-activated protein kinase kinases are related to yeast signal transduction kinases. J Biol Chem. 1992;267(36):25628-31 [PubMed] [Google Scholar]
- 36. Zheng CF, Guan KL. Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J Biol Chem. 1993;268(32):23933-9 [PubMed] [Google Scholar]
- 37. Alessi DR, Saito Y, Campbell DG, et al. Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J. 1994;13(7):1610-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dhanasekaran N, Premkumar Reddy E. Signaling by dual specificity kinases. Oncogene. 1998;17(11 Rev):1447-55 [DOI] [PubMed] [Google Scholar]
- 39. Yung Y, Yao Z, Hanoch T, Seger R. ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J Biol Chem. 2000;275(21):15799-808 [DOI] [PubMed] [Google Scholar]
- 40. Aebersold DM, Shaul YD, Yung Y, et al. Extracellular signal-regulated kinase 1c (ERK1c), a novel 42-kilodalton ERK, demonstrates unique modes of regulation, localization, and function. Mol Cell Biol. 2004;24(22):10000-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Payne DM, Rossomando AJ, Martino P, et al. Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J. 1991;10(4):885-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Voisin L, Saba-El-Leil MK, Julien C, Fremin C, Meloche S. Genetic demonstration of a redundant role of extracellular signal-regulated kinase 1 (ERK1) and ERK2 mitogen-activated protein kinases in promoting fibroblast proliferation. Mol Cell Biol. 2010;30(12):2918-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lefloch R, Pouyssegur J, Lenormand P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol. 2008;28(1):511-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity. 2005;23(4):431-43 [DOI] [PubMed] [Google Scholar]
- 45. Vantaggiato C, Formentini I, Bondanza A, Bonini C, Naldini L, Brambilla R. ERK1 and ERK2 mitogen-activated protein kinases affect Ras-dependent cell signaling differentially. J Biol. 2006;5(5):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shin S, Dimitri CA, Yoon SO, Dowdle W, Blenis J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell. 2010;38(1):114-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gonzalez FA, Raden DL, Davis RJ. Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J Biol Chem. 1991;266(33):22159-63 [PubMed] [Google Scholar]
- 48. von Kriegsheim A, Baiocchi D, Birtwistle M, et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol. 2009;11(12):1458-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci U S A. 1995;92(19):8881-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Northwood IC, Gonzalez FA, Wartmann M, Raden DL, Davis RJ. Isolation and characterization of two growth factor-stimulated protein kinases that phosphorylate the epidermal growth factor receptor at threonine 669. J Biol Chem. 1991;266(23):15266-76 [PubMed] [Google Scholar]
- 51. Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73(2):381-93 [DOI] [PubMed] [Google Scholar]
- 52. Chen RH, Abate C, Blenis J. Phosphorylation of the c-Fos transrepression domain by mitogen-activated protein kinase and 90-kDa ribosomal S6 kinase. Proc Natl Acad Sci U S A. 1993;90(23):10952-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Morton S, Davis RJ, McLaren A, Cohen P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003;22(15):3876-86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chuderland D, Konson A, Seger R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol Cell. 2008;31(6):850-61 [DOI] [PubMed] [Google Scholar]
- 55. Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16(8):1921-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997;16(8):1909-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17(15):4426-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Barnier JV, Papin C, Eychene A, Lecoq O, Calothy G. The mouse B-raf gene encodes multiple protein isoforms with tissue-specific expression. J Biol Chem. 1995;270(40):23381-9 [DOI] [PubMed] [Google Scholar]
- 59. Eychene A, Dusanter-Fourt I, Barnier JV, et al. Expression and activation of B-Raf kinase isoforms in human and murine leukemia cell lines. Oncogene. 1995;10(6):1159-65 [PubMed] [Google Scholar]
- 60. Papin C, Denouel-Galy A, Laugier D, Calothy G, Eychene A. Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J Biol Chem. 1998;273(38):24939-47 [DOI] [PubMed] [Google Scholar]
- 61. Baitei EY, Zou M, Al-Mohanna F, et al. Aberrant BRAF splicing as an alternative mechanism for oncogenic B-Raf activation in thyroid carcinoma. J Pathol. 2009;217(5):707-15 [DOI] [PubMed] [Google Scholar]
- 62. Shaul YD, Gibor G, Plotnikov A, Seger R. Specific phosphorylation and activation of ERK1c by MEK1b: a unique route in the ERK cascade. Genes Dev. 2009;23(15):1779-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yung Y, Yao Z, Aebersold DM, Hanoch T, Seger R. Altered regulation of ERK1b by MEK1 and PTP-SL and modified Elk1 phosphorylation by ERK1b are caused by abrogation of the regulatory C-terminal sequence of ERKs. J Biol Chem. 2001;276(38):35280-9 [DOI] [PubMed] [Google Scholar]
- 64. Shaul YD, Seger R. ERK1c regulates Golgi fragmentation during mitosis. J Cell Biol. 2006;172(6):885-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367(6465):704-11 [DOI] [PubMed] [Google Scholar]
- 66. Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15(1):75-83 [DOI] [PubMed] [Google Scholar]
- 67. Oppermann FS, Gnad F, Olsen JV, et al. Large-scale proteomics analysis of the human kinome. Mol Cell Proteomics. 2009;8(7):1751-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bendetz-Nezer S, Seger R. Role of non-phosphorylated activation loop residues in determining ERK2 dephosphorylation, activity, and subcellular localization. J Biol Chem. 2007;282(34):25114-22 [DOI] [PubMed] [Google Scholar]
- 69. Lu Z, Xu S, Joazeiro C, Cobb MH, Hunter T. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol Cell. 2002;9(5):945-56 [DOI] [PubMed] [Google Scholar]
- 70. Formstecher E, Ramos JW, Fauquet M, et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell. 2001;1(2):239-50 [DOI] [PubMed] [Google Scholar]
- 71. Yao Z, Seger R. The molecular mechanism of MAPK/ERK inactivation. Curr Genomics. 2004;5:385-93 [Google Scholar]
- 72. Yao Z, Dolginov Y, Hanoch T, et al. Detection of partially phosphorylated forms of ERK by monoclonal antibodies reveals spatial regulation of ERK activity by phosphatases. FEBS Lett. 2000;468(1):37-42 [DOI] [PubMed] [Google Scholar]
- 73. Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75(3):487-93 [DOI] [PubMed] [Google Scholar]
- 74. Nguyen TT, Scimeca JC, Filloux C, Peraldi P, Carpentier JL, Van Obberghen E. Co-regulation of the mitogen-activated protein kinase, extracellular signal-regulated kinase 1, and the 90-kDa ribosomal S6 kinase in PC12 cells: distinct effects of the neurotrophic factor, nerve growth factor, and the mitogenic factor, epidermal growth factor. J Biol Chem. 1993;268(13):9803-10 [PubMed] [Google Scholar]
- 75. Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80(2):179-85 [DOI] [PubMed] [Google Scholar]
- 76. Murphy LO, MacKeigan JP, Blenis J. A network of immediate early gene products propagates subtle differences in mitogen-activated protein kinase signal amplitude and duration. Mol Cell Biol. 2004;24(1):144-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev. 2008;27(2):253-61 [DOI] [PubMed] [Google Scholar]
- 78. Chuderland D, Seger R. Protein-protein interactions in the regulation of the extracellular signal-regulated kinase. Mol Biotechnol. 2005;29(1):57-74 [DOI] [PubMed] [Google Scholar]
- 79. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286(5445):1741-4 [DOI] [PubMed] [Google Scholar]
- 80. Rommel C, Clarke BA, Zimmermann S, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286(5445):1738-41 [DOI] [PubMed] [Google Scholar]
- 81. Frost JA, Steen H, Shapiro P, et al. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997;16(21):6426-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hu Q, Klippel A, Muslin AJ, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science. 1995;268(5207):100-2 [DOI] [PubMed] [Google Scholar]
- 83. Wennstrom S, Downward J. Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol. 1999;19(6):4279-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Halilovic E, She QB, Ye Q, et al. PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res. 2010;70(17):6804-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu X, Yan S, Zhou T, Terada Y, Erikson RL. The MAP kinase pathway is required for entry into mitosis and cell survival. Oncogene. 2004;23(3):763-76 [DOI] [PubMed] [Google Scholar]
- 86. Catalanotti F, Reyes G, Jesenberger V, et al. A Mek1-Mek2 heterodimer determines the strength and duration of the Erk signal. Nat Struct Mol Biol. 2009;16(3):294-303 [DOI] [PubMed] [Google Scholar]
- 87. Kim Y, Coppey M, Grossman R, et al. MAPK substrate competition integrates patterning signals in the Drosophila embryo. Curr Biol. 2010;20(5):446-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Yao Z, Seger R. The ERK signaling cascade: views from different subcellular compartments. Biofactors. 2009;35(5):407-16 [DOI] [PubMed] [Google Scholar]
- 89. Rubinfeld H, Hanoch T, Seger R. Identification of a cytoplasmic-retention sequence in ERK2. J Biol Chem. 1999;274(43):30349-52 [DOI] [PubMed] [Google Scholar]
- 90. Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2(2):110-6 [DOI] [PubMed] [Google Scholar]
- 91. Wolf I, Rubinfeld H, Yoon S, Marmor G, Hanoch T, Seger R. Involvement of the activation loop of ERK in the detachment from cytosolic anchoring. J Biol Chem. 2001;276(27):24490-7 [DOI] [PubMed] [Google Scholar]
- 92. Lee T, Hoofnagle AN, Kabuyama Y, et al. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14(1):43-55 [DOI] [PubMed] [Google Scholar]
- 93. Callaway K, Rainey MA, Dalby KN. Quantifying ERK2-protein interactions by fluorescence anisotropy: PEA-15 inhibits ERK2 by blocking the binding of DEJL domains. Biochim Biophys Acta. 2005;1754(1-2):316-23 [DOI] [PubMed] [Google Scholar]
- 94. Reszka AA, Bulinski JC, Krebs EG, Fischer EH. Mitogen-activated protein kinase/extracellular signal-regulated kinase 2 regulates cytoskeletal organization and chemotaxis via catalytic and microtubule-specific interactions. Mol Biol Cell. 1997;8(7):1219-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Perlson E, Michaelevski I, Kowalsman N, et al. Vimentin binding to phosphorylated Erk sterically hinders enzymatic dephosphorylation of the kinase. J Mol Biol. 2006;364(5):938-44 [DOI] [PubMed] [Google Scholar]
- 96. Galli S, Jahn O, Hitt R, et al. A new paradigm for MAPK: structural interactions of hERK1 with mitochondria in HeLa cells. PLoS One. 2009;4(10):e7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Casar B, Arozarena I, Sanz-Moreno V, et al. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol. 2009;29(5):1338-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Casar B, Pinto A, Crespo P. ERK dimers and scaffold proteins: unexpected partners for a forgotten (cytoplasmic) task. Cell Cycle. 2009;8(7):1007-13 [DOI] [PubMed] [Google Scholar]
- 99. Wunderlich W, Fialka I, Teis D, et al. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold mp1 on a late endosomal/lysosomal compartment. J Cell Biol. 2001;152(4):765-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Torii S, Kusakabe M, Yamamoto T, Maekawa M, Nishida E. Sef is a spatial regulator for Ras/MAP kinase signaling. Dev Cell. 2004;7(1):33-44 [DOI] [PubMed] [Google Scholar]
- 101. Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12(3):915-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brunet A, Roux D, Lenormand P, Dowd S, Keyse S, Pouyssegur J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999;18(3):664-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358(6385):414-7 [DOI] [PubMed] [Google Scholar]
- 104. Sgouras DN, Athanasiou MA, Beal GJ, Jr., Fisher RJ, Blair DG, Mavrothalassitis GJ. ERF: an ETS domain protein with strong transcriptional repressor activity, can suppress ets-associated tumorigenesis and is regulated by phosphorylation during cell cycle and mitogenic stimulation. EMBO J. 1995;14(19):4781-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cohen-Armon M, Visochek L, Rozensal D, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell. 2007;25(2):297-308 [DOI] [PubMed] [Google Scholar]
- 106. Kosako H, Yamaguchi N, Aranami C, et al. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat Struct Mol Biol. 2009;16(10):1026-35 [DOI] [PubMed] [Google Scholar]
- 107. Burgermeister E, Chuderland D, Hanoch T, Meyer M, Liscovitch M, Seger R. Interaction with MEK causes nuclear export and downregulation of peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 2007;27(3):803-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol. 2008;9(10):747-58 [DOI] [PubMed] [Google Scholar]
- 109. Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8(2):205-15 [DOI] [PubMed] [Google Scholar]
- 110. Cruzalegui FH, Cano E, Treisman R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene. 1999;18(56):7948-57 [DOI] [PubMed] [Google Scholar]
- 111. Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci. 2000;25(9):448-53 [DOI] [PubMed] [Google Scholar]
- 112. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3(11):859-68 [DOI] [PubMed] [Google Scholar]
- 113. Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4(8):556-64 [DOI] [PubMed] [Google Scholar]
- 114. Kato S, Endoh H, Masuhiro Y, et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491-4 [DOI] [PubMed] [Google Scholar]
- 115. Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100-3 [DOI] [PubMed] [Google Scholar]
- 116. Solomon C, White JH, Kremer R. Mitogen-activated protein kinase inhibits 1,25-dihydroxyvitamin D3-dependent signal transduction by phosphorylating human retinoid X receptor alpha. J Clin Invest. 1999;103(12):1729-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17(7):3947-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mavrothalassitis G, Ghysdael J. Proteins of the ETS family with transcriptional repressor activity. Oncogene. 2000;19(55):6524-32 [DOI] [PubMed] [Google Scholar]
- 119. Hu S, Xie Z, Onishi A, et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell. 2009;139(3):610-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Zehorai E, Yao Z, Plotnikov A, Seger R. The subcellular localization of MEK and ERK: a novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol Cell Endocrinol. 2010;314(2):213-20 [DOI] [PubMed] [Google Scholar]
- 121. Nigg EA. Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature. 1997;386(6627):779-87 [DOI] [PubMed] [Google Scholar]
- 122. Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell. 2006;125(6):1041-53 [DOI] [PubMed] [Google Scholar]
- 123. Christophe D, Christophe-Hobertus C, Pichon B. Nuclear targeting of proteins: how many different signals? Cell Signal. 2000;12(5):337-41 [DOI] [PubMed] [Google Scholar]
- 124. Fagotto F, Gluck U, Gumbiner BM. Nuclear localization signal-independent and importin/karyopherin-independent nuclear import of beta-catenin. Curr Biol. 1998;8(4):181-90 [DOI] [PubMed] [Google Scholar]
- 125. Waldmann I, Walde S, Kehlenbach RH. Nuclear import of c-Jun is mediated by multiple transport receptors. J Biol Chem. 2007;282(38):27685-92 [DOI] [PubMed] [Google Scholar]
- 126. Lorenzen JA, Baker SE, Denhez F, Melnick MB, Brower DL, Perkins LA. Nuclear import of activated D-ERK by DIM-7, an importin family member encoded by the gene moleskin. Development. 2001;128(8):1403-14 [DOI] [PubMed] [Google Scholar]
- 127. Smith DJ, Ng H, Kluck RM, Nagley P. The mitochondrial gateway to cell death. IUBMB Life. 2008;60(6):383-9 [DOI] [PubMed] [Google Scholar]
- 128. Horbinski C, Chu CT. Kinase signaling cascades in the mitochondrion: a matter of life or death. Free Radic Biol Med. 2005;38(1):2-11 [DOI] [PubMed] [Google Scholar]
- 129. Lee HJ, Bach JH, Chae HS, et al. Mitogen-activated protein kinase/extracellular signal-regulated kinase attenuates 3-hydroxykynurenine-induced neuronal cell death. J Neurochem. 2004;88(3):647-56 [DOI] [PubMed] [Google Scholar]
- 130. Jin K, Mao XO, Zhu Y, Greenberg DA. MEK and ERK protect hypoxic cortical neurons via phosphorylation of Bad. J Neurochem. 2002;80(1):119-25 [DOI] [PubMed] [Google Scholar]
- 131. Ishikawa Y, Kusaka E, Enokido Y, Ikeuchi T, Hatanaka H. Regulation of Bax translocation through phosphorylation at Ser-70 of Bcl-2 by MAP kinase in NO-induced neuronal apoptosis. Mol Cell Neurosci. 2003;24(2):451-9 [DOI] [PubMed] [Google Scholar]
- 132. Sperandio S, Poksay K, de Belle I, et al. Paraptosis: mediation by MAP kinases and inhibition by AIP-1/Alix. Cell Death Differ. 2004;11(10):1066-75 [DOI] [PubMed] [Google Scholar]
- 133. Seger R, Hanoch T, Rosenberg R, et al. The ERK signaling cascade inhibits gonadotropin-stimulated steroidogenesis. J Biol Chem. 2001;276(17):13957-64 [DOI] [PubMed] [Google Scholar]
- 134. Nowak G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J Biol Chem. 2002;277(45):43377-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Deng X, Ruvolo P, Carr B, May WS., Jr. Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc Natl Acad Sci U S A. 2000;97(4):1578-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tamura Y, Simizu S, Osada H. The phosphorylation status and anti-apoptotic activity of Bcl-2 are regulated by ERK and protein phosphatase 2A on the mitochondria. FEBS Lett. 2004;569(1-3):249-55 [DOI] [PubMed] [Google Scholar]
- 137. Baines CP, Zhang J, Wang GW, et al. Mitochondrial PKCepsilon and MAPK form signaling modules in the murine heart: enhanced mitochondrial PKCepsilon-MAPK interactions and differential MAPK activation in PKCepsilon-induced cardioprotection. Circ Res. 2002;90(4):390-7 [DOI] [PubMed] [Google Scholar]
- 138. Monick MM, Powers LS, Barrett CW, et al. Constitutive ERK MAPK activity regulates macrophage ATP production and mitochondrial integrity. J Immunol. 2008;180(11):7485-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 2003;13(4):473-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Alonso M, Melani M, Converso D, et al. Mitochondrial extracellular signal-regulated kinases 1/2 (ERK1/2) are modulated during brain development. J Neurochem. 2004;89(1):248-56 [DOI] [PubMed] [Google Scholar]
- 141. Poderoso C, Converso DP, Maloberti P, et al. A mitochondrial kinase complex is essential to mediate an ERK1/2-dependent phosphorylation of a key regulatory protein in steroid biosynthesis. PLoS One. 2008;3(1):e1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Poderoso C, Maloberti P, Duarte A, et al. Hormonal activation of a kinase cascade localized at the mitochondria is required for StAR protein activity. Mol Cell Endocrinol. 2009;300(1-2):37-42 [DOI] [PubMed] [Google Scholar]
- 143. Galli S, Antico Arciuch VG, Poderoso C, et al. Tumor cell phenotype is sustained by selective MAPK oxidation in mitochondria. PLoS One. 2008;3(6):e2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Miaczynska M, Bar-Sagi D. Signaling endosomes: seeing is believing. Curr Opin Cell Biol. 2010;22(4):535-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wiley HS, Burke PM. Regulation of receptor tyrosine kinase signaling by endocytic trafficking. Traffic. 2001;2(1):12-8 [DOI] [PubMed] [Google Scholar]
- 147. Luttrell LM, Daaka Y, Della Rocca GJ, Lefkowitz RJ. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in rat 1a fibroblasts: Shc phosphorylation and receptor endocytosis correlate with activation of Erk kinases. J Biol Chem. 1997;272(50):31648-56 [DOI] [PubMed] [Google Scholar]
- 148. Daaka Y, Luttrell LM, Ahn S, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273(2):685-8 [DOI] [PubMed] [Google Scholar]
- 149. Xu ZQ, Zhang X, Scott L. Regulation of G protein-coupled receptor trafficking. Acta Physiol (Oxf). 2007;190(1):39-45 [DOI] [PubMed] [Google Scholar]
- 150. Luttrell LM, Roudabush FL, Choy EW, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci U S A. 2001;98(5):2449-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483-510 [DOI] [PubMed] [Google Scholar]
- 152. Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem. 2002;277(11):9429-36 [DOI] [PubMed] [Google Scholar]
- 153. Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281(5383):1668-71 [DOI] [PubMed] [Google Scholar]
- 154. Teis D, Taub N, Kurzbauer R, et al. p14-MP1-MEK1 signaling regulates endosomal traffic and cellular proliferation during tissue homeostasis. J Cell Biol. 2006;175(6):861-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Nada S, Hondo A, Kasai A, et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 2009;28(5):477-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Patra SK. Dissecting lipid raft facilitated cell signaling pathways in cancer. Biochim Biophys Acta. 2008;1785(2):182-206 [DOI] [PubMed] [Google Scholar]
- 157. Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4(5):394-8 [DOI] [PubMed] [Google Scholar]
- 158. Sachse M, Urbe S, Oorschot V, Strous GJ, Klumperman J. Bilayered clathrin coats on endosomal vacuoles are involved in protein sorting toward lysosomes. Mol Biol Cell. 2002;13(4):1313-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91-118 [DOI] [PubMed] [Google Scholar]
- 160. Yu W, Fantl WJ, Harrowe G, Williams LT. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr Biol. 1998;8(1):56-64 [DOI] [PubMed] [Google Scholar]
- 161. Muller J, Ory S, Copeland T, Piwnica-Worms H, Morrison DK. C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold, KSR1. Mol Cell. 2001;8(5):983-93 [DOI] [PubMed] [Google Scholar]
- 162. Xing H, Kornfeld K, Muslin AJ. The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr Biol. 1997;7(5):294-300 [DOI] [PubMed] [Google Scholar]
- 163. Ory S, Zhou M, Conrads TP, Veenstra TD, Morrison DK. Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr Biol. 2003;13(16):1356-64 [DOI] [PubMed] [Google Scholar]
- 164. Matheny SA, Chen C, Kortum RL, Razidlo GL, Lewis RE, White MA. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature. 2004;427(6971):256-60 [DOI] [PubMed] [Google Scholar]
- 165. Krajewska WM, Maslowska I. Caveolins: structure and function in signal transduction. Cell Mol Biol Lett. 2004;9(2):195-220 [PubMed] [Google Scholar]
- 166. Pullikuth AK, Catling AD. Scaffold mediated regulation of MAPK signaling and cytoskeletal dynamics: a perspective. Cell Signal. 2007;19(8):1621-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Leinweber BD, Leavis PC, Grabarek Z, Wang CL, Morgan KG. Extracellular regulated kinase (ERK) interaction with actin and the calponin homology (CH) domain of actin-binding proteins. Biochem J. 1999;344(Pt 1):117-23 [PMC free article] [PubMed] [Google Scholar]
- 168. Smith ER, Smedberg JL, Rula ME, Xu XX. Regulation of Ras-MAPK pathway mitogenic activity by restricting nuclear entry of activated MAPK in endoderm differentiation of embryonic carcinoma and stem cells. J Cell Biol. 2004;164(5):689-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45(5):715-26 [DOI] [PubMed] [Google Scholar]
- 170. Ren JG, Li Z, Sacks DB. IQGAP1 modulates activation of B-Raf. Proc Natl Acad Sci U S A. 2007;104(25):10465-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. White CD, Brown MD, Sacks DB. IQGAPs in cancer: a family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009;583(12):1817-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Harrison RE, Sikorski BA, Jongstra J. Leukocyte-specific protein 1 targets the ERK/MAP kinase scaffold protein KSR and MEK1 and ERK2 to the actin cytoskeleton. J Cell Sci. 2004;117(Pt 10):2151-7 [DOI] [PubMed] [Google Scholar]
- 173. Jongstra-Bilen J, Jongstra J. Leukocyte-specific protein 1 (LSP1): a regulator of leukocyte emigration in inflammation. Immunol Res. 2006;35(1-2):65-74 [DOI] [PubMed] [Google Scholar]
- 174. Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137(2):481-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Lowe M. Structural organization of the Golgi apparatus. Curr Opin Cell Biol. 2011;23(1):85-93 [DOI] [PubMed] [Google Scholar]
- 176. Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell. 1998;92(2):183-92 [DOI] [PubMed] [Google Scholar]
- 177. Cha H, Shapiro P. Tyrosine-phosphorylated extracellular signal--regulated kinase associates with the Golgi complex during G2/M phase of the cell cycle: evidence for regulation of Golgi structure. J Cell Biol. 2001;153(7):1355-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Jesch SA, Lewis TS, Ahn NG, Linstedt AD. Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol Biol Cell. 2001;12(6):1811-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Feng L, Xie X, Ding Q, et al. Spatial regulation of Raf kinase signaling by RKTG. Proc Natl Acad Sci U S A. 2007;104(36):14348-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802(4):396-405 [DOI] [PubMed] [Google Scholar]
- 181. Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19(3):230-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Tanti JF, Jager J. Cellular mechanisms of insulin resistance: role of stress-regulated serine kinases and insulin receptor substrates (IRS) serine phosphorylation. Curr Opin Pharmacol. 2009;9(6):753-62 [DOI] [PubMed] [Google Scholar]
- 183. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26(22):3279-90 [DOI] [PubMed] [Google Scholar]
- 184. Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett. 2009;283(2):125-34 [DOI] [PubMed] [Google Scholar]