

Abstract
Perinatal hypoxia/ischemia (HI) is a common cause of neurological deficits in children. Our goal was to elucidate the underlying mechanisms that contribute to the neurological sequelae of HI-induced brain injury. HI was induced by permanent ligation of the left carotid artery followed by 90 minutes of hypoxia (7.8% O2) in female P7 rats. A two-dimensional differential proteome analysis was used to assess changes in protein expression in cortex 2h after HI. 17 proteins reflecting a 2 fold or higher perturbation of expression after HI as compared to sham-treated pups were identified by Mass Spectrometry. Of the altered proteins, 14-3-3ε and TUC-2, both playing an important role in the development of the central nervous system, decrease after HI, consistent with an early disturbance of cortical development. Also affected, DARPP-32 and alpha synuclein, two proteins important for dopamine neurotransmission, increased more than two fold 2h after HI injury. The differential expression of these proteins was validated by individual western blot assays. The expression of several metabolic enzymes and translational factors was also perturbed early after HI brain injury. These findings provide initial insights into the mechanisms underlying neurodegenerative events after HI, and may allow for the rational design of therapeutic strategies that enhance neuronal adaptation and compensation after HI.
Keywords: hypoxia/ischemia, cortex, proteomics
INTRODUCTION
Perinatal hypoxia-ischemia (HI) brain injury is a leading cause of neonatal morbidity and mortality. There is a significant loss of cortical neurons after HI brain injury and resultant progressive brain function impairment. Advances in obstetrical care have increased the number of infants surviving perinatal HI with moderate or severe brain injury. It is estimated that 20 to 30% of survivors of HI brain injury have severe long-term neurological sequelae, such as cerebral palsy;1 uncounted others have milder injuries that result in mental retardation, seizures, learning and behavioral disorders.2,3
HI results in prompt changes in the P7 neonatal brain during the first seven days after HI.4–6 Neuroimaging studies have shown that the parietal cortex is HI-sensitive. Diffusion-weighted magnetic resonance imaging shows a decrease in the apparent diffusion coefficient values in cortex on the affected side as early as 0.5h after HI.7,8 Maximal size of the lesion was reported at 3 to 6 hours after HI and declined thereafter. Furthermore, animals with MRI-detected lesions showed long-term functional deficits and abnormal histopathology at near-adult ages.7 Therefore, the elucidation of the mechanisms underlying the observed changes early after HI is important.
The mechanisms of HI-induced brain damage are heterogeneous. Biophysical and biochemical changes, such as blood flow deregulation, brain edema, hemorrhage, thrombosis, energy failure, accumulation of toxic metabolites, impaired intracellular calcium turnover, increase of neurotransmitter release and inhibition of uptake, release of interleukins and prostaglandins, and increased levels of free-radicals, are triggered by HI and may lead to brain dysfunction and neuronal cell death.9,10 However, the data are still incomplete regarding the biological processes responsible for the overall distribution and progression of neuronal injury and subsequent permanent brain damage. Understanding these mechanisms, especially at early time points after injury, is necessary to thoroughly explain the long term effects of HI-brain injury and find practical protective therapies.
The application of proteomic approaches could identify those proteins that are important in HI-induced brain injury and provide insights into potential mechanisms underlying the neuronal dysfunction and cell losses associated with HI injury. Here, application of proteomics showed differential expression of several proteins in the HI-injured brain. Our results indicate an early disturbance of cerebral cortical development and dopamine neurotransmission after HI injury, with long term neurological consequences.
MATERIALS AND METHODS
Animals
All animal procedures were approved by the institutional Animal Care and Use Committee. Dated, pregnant Wistar rats were purchased from a commercial breeder (Charles River), housed in individual cages, and fed a standard laboratory chow ad libidum. Offspring, delivered spontaneously, were maintained with their dams until 7 days of postnatal age. Litter size was reduced on P3 to 10 pups/litter to minimize variation in body and brain growth. Female pups were used in this study. Pups that show signs of respiratory or other infection are excluded from further study. The average body weight at P7 was 15 ± 3 g.
The Induction of Ischemia and Hypoxia
HI was induced as described in our previous publication by us.6 Briefly, the P7 rat pups were given Isoflurane in air by mask inhalation throughout surgery. The left common carotid artery was dually ligated and transected between the ligatures. The incision site was closed and cleaned. The operated pups recovered from surgery for approximately 30 minutes in a 37°C chamber with bedding from the cage. The rats were returned to the dam for 1–2 hours. Rats were transferred to a humidified hypoxia chamber where systemic hypoxia was induced by inhalation of 7.8% oxygen balanced with nitrogen for 90 minutes. The sham groups were treated with room air in the cage. The temperature in the hypoxia chamber was maintained at 37°C. The rats were sacrificed 2 hours after hypoxia. At sacrifice, animals were briefly anesthetized with Halothane, decapitated, the brains removed and cortex was dissected from the rest of the brain, frozen, and stored at −80°C.
Tissue Extracts
Dissected tissues were homogenized in ice-cold Buffer M, pH 7.4, containing sucrose (250mM), Hepes (20mM), EDTA (1mM), EGTA (1mM), dithiotheitol (DTT; 1mM), PMSF (1mM), leupeptin (5 mg/ml) and aprotinin (25 mg/ml) using a Wheaton Tenbroeck® manual homogenizer. The homogenate was centrifuged three times at 800g for 20 min to spin down nuclei and cell debris. The crude cytosolic extract containing cytosolic proteins, mitochondria and endoplasmic reticulum was centrifuged at 28000 g for 30 min to pellet down mitochondria and ER. All procedures are performed at 4°C. 150U of endonuclease Benzonase was added to 1ml of sample and incubate in room temperature for 30 min. After the incubation period, samples were vortexed again and spun in a microcentrifuge at 11,400rpm (12,000g) for 10 min. The supernatant containing the cytoplasmic extracts was frozen, and stored at −80°C. Protein concentration was determined by Biorad RC DC Protein Assay.
Two-dimensional electrophoresis
200 μg samples were prepared and the final volumes were adjusted to 200μl with rehydration buffer. 1μl IPG buffer was added to give a final concentration of 0.5%. The mixtures were spun in a microcentrifuge at 2,000rpm for 2 minutes. Paper wicks were placed over each electrode of the ceramic strip holders (Amersham-Biosciences). Each wick was wet with 8 μl of Milli-Q H2O just prior to adding the sample/rehydration buffer mix. An 11 cm IPG dry strip was added to each sample solution with strip gel side facing down. Strip was covered with mineral oil and placed on ceramic strip holder. Strip holder was placed in IPGhor focusing instrument (Amersham-Biosciences) and focused as follows: 50 V for 11 hrs (active rehydration); 250 V gradient 1 hr; 500 V gradient 1 hr; 1,000 V gradient 1 hr; 8,000 V gradient 2 hr; 8,000 V 48,000 Vhr. IPG strips were then removed and carefully blotted with damp filter paper to remove oil. At the end of the first dimensional separation the strips were equilibrated for 15 min in equilibrating buffer (50mM Tris-HCl, pH 8.8, 6M urea, 2% SDS, 20% glycerol, 10 mg DTT in 1ml buffer) with 10μl/ml TCEP, followed by 20 min equilibration in the above buffer containing 25 mg iodoacetamide per ml buffer. Strips were then rinsed in 1X Tris-Glycine-SDS running buffer and placed in IPG well of gels with the positive end of the strip on the left side of the gels. Strips were covered with molten 0.5% overlay agarose. Gels were placed in electrophoresis apparatus and run at 150 V for approximately 2.25 hr at 4°C in prechilled 1X running buffer.
Gel Fixation and SYPRO Ruby Staining
Gels were removed from plates, rinsed in dIH2O and fixed in fixative/destain solution (10% methanol, 7% acetic acid prepare with Milli-Q H2O) for 30 minutes with shaking at RT. Gels were rinsed again in dI H2O and stained with SYPRO® Ruby overnight on a shaker at room temperature. The gel was covered with foil to protect from light. The gels were then rinse with dIH2O, destained in 10% methanol/7% acetic acid for 1 h and were imaged on ProExpress Proteomic Imaging System (Perkin Elmer) with 480mm excitation and 620mm emission filter.
Gel Analysis
Total of six 2D gel images (3 from sham-treated animals and 3 from HI-injured animals) were analyzed using Progenesis Discovery software v2006 (Nonlinear Dynamics, United Kingdom). This software automatically detects individual protein spot within each image and matches identical protein spots across all images. It also removes noise from measurements of spot volumes using a proprietary algorithm for noise determination and correction. After automatic matching, manual review and adjustment were done to confirm proper spot detection and matching. The intensity of each protein spot was normalized based on the total volume of each gel, i.e. the pixel intensity of each spot area is divided by the sum of all spots in the gel. Spots present on less than 2 gels or with normalized volumes less than 150 were filtered out. A ±2-fold change was used as a cut off point for determining useful spots. In addition to these spots we also include missing and new spots as spots of interest.
Mass spectrometry
Protein gel spots were excised and prepared for Matrix-Assisted Laser Desorption Ionization Time-of-Flight/Time-of-Flight Mass Spectrometry (MALDI-TOF/TOF MS) analysis. Gel samples were cut into 1mm size pieces or smaller and incubated in 100 μl of ammonium bicarbonate buffer at 37°C for 30 min. The buffer was removed and 100 l of water was added to each tube. The samples were then incubated again at 37°C for 30 min. Gels were washed twice in 100% acetonitrile with vortex for 5 min to dehydrate the gels. Acetonitrile was removed and gels were dried in a Speed-Vac for 30 min. Trypsin (10 μg/mL) (sequencing grade modified T; Promega) in 25 mM ammonium bicarbonate, pH 8.0, was used for enzymatic digestion. Samples were incubated at 37°C for 6h. After digestion, the samples were removed from the incubator and 1μl of sample solution was spotted directly onto a MALDI target plate and allowed to dry. 1 uL of matrix (alpha-cyano-4-hydroxycinnamic acid; Aldrich Chemical Co.) was then applied on the sample spot and allowed to dry. MALDI-TOF/TOF MS was performed using an Applied Biosystems model 4700 Proteomics Analyzer for peptide mass fingerprinting. Following MALDI MS analysis, MALDI MS/MS was performed on several ions from each sample spot. Applied Biosystems software was used in conjunction with MASCOT to search databases for protein identification. Protein match probabilities were determined using expectation values and/or MASCOT protein scores.
Western Blot Analysis
Proteins were separated on 10% SDS-PAGE gels and electroblotted onto polyvinylidene difluoride membranes (Millipore). Proteins were analyzed by Western blot assays performed as previously described.11 14-3-3ε monoclonal antibody was purchased from Chemicon (Temecula, CA). TUC-2 monoclonal antibody was purchased from Immunobiological Lab (Gunma, Japan). DARPP-32 and alpha-synuclein polyclonal antibodies were purchased from BD pharmingen (San Diego, CA).
Immunofluorescence Staining
The rats were euthanized with an intraperitoneal injection (i.p.) of 120-mg/kg pentobarbital (Nembutal) and then perfused through the left ventricle with saline followed by 4% paraformaldehyde. Brains were removed and postfixed in 4% paraformaldehyde overnight at 4°C. The brains were blocked in the coronal plane and the right sides were notched. Brains were put into 30% sucrose/PBS solution at 4°C until the brain section sunk to the bottom of the container. Brain were then embedded in Optimal Cutting Temperature compound (OCT), immediately frozen with dry ice for 1–2 minutes then stored at −80°C. Coronal sections were cut at a thickness of 10 μm. Slides were washed 3 times in 0.1M TBS/Triton 100, 7 minutes each time, and rinse with TBS. Sections were blocked with signal enhancer (Molecular probes, Eugene, OR) for 30 minutes at room temperature. Sections were then incubated separately with primary antibodies in diluent buffer (TBS+ Triton-x-100 +1% NGS) at room temperature overnight inside moisture chamber. After washing with TBS 3 times (7 minutes each time), sections were incubated with fluorescent labeled secondary antibody (1:1000 Alexa Fluor 568 (Red) Goat-anti Rabbit IgG, Molecular probes, Eugene, OR) for 2 hours at room temperature in dark. Sections were washed with TBS 5 times, each for 12 minutes, coverslipped using fluorescence mounting medium with DAPI, and sealed with clear nail polish. Stained sections were scanned with a confocal laser scanning microscope (Olympus BX50 equipped with a Flouview Krypton-Argon-Ion Laser scanning unit). Controls omitted primary antibody and it was replaced by an irrelevant antibody of the same IgG subclass. Images were captured from a FXA photomicroscope (Nikon) with a DEI-470 digitally enhanced color microscope video camera (Optronics) with a built-in Digital Image Processor.
Statistical analysis
For 2D gel analysis, finding genes with significantly changed expression levels was done by using Statistical Analysis of Microarrays (SAM). The complete version of SAM software is available for downloading from: http://www-stat.stanford.edu/~tibs/SAM/index.html. This method computes the false discovery rate (FDR) by averaging the number of significance over the appropriately chosen permutations of data among experimental groups. With the single tuning parameter (called Δ in the original work) the FDR for the dataset can be reduced, at the expense of a reduction in the number of significant change. In our analysis, we selected the adjustable parameter Δ such that FDR was <5% for the list of significant proteins. For statistical analysis of western blot results, mean values (±SEM) for each experiment were determined, and values statistically different from shams calculated using student t test.
RESULTS
Identification of differentially expressed proteins after HI injury
In the present study, comparative protein profiling was performed using a well characterized 7-day old rat HI model to determine changes in the proteome of cortex subjected to HI injury. Cytoplasmic protein was extracted from sham-treated or HI-treated cortical samples (n=3 for each group), and separated by two-dimensional electrophoresis. Figure 1 shows representative 2-D images, spanning the molecular weights between 250kd and 10kd and pIs between 3 and 10, of sham-treated and HI-treated animals. Image analysis with Progenesis software (Nonlinear) showed approximately 900 spots on each gel after spot filtering. Spots present on less than 2 gels or with normalized volumes less than 150 were filtered out. After quantification of spot signal intensities, the fold change (ratio of average normalized spot volume of HI-treated versus sham-treated) and a two tailed t test incorporating FDR correction were calculated. We focused on identifying proteins that were found to be either statistically significantly altered more than 2 fold after HI injury or newly show/miss in HI-treated pups. 17 proteins were successfully identified by MALDI-TOF/TOF Mass Spectrometry (Table 1). Four proteins were under-expressed while three proteins were over-expressed. Ten spots were found to be present only in the HI-treated animals. The apparent observed molecular mass and isoelectric point for all identified proteins matched very well with their calculated values. We classified the altered proteins based on their published biological functions (Table 2). The majority of the proteins whose levels were perturbed by HI could be classified into four general functional groups: cortical development, metabolism enzymes, translation factors and neurotransmission.
Figure 1.
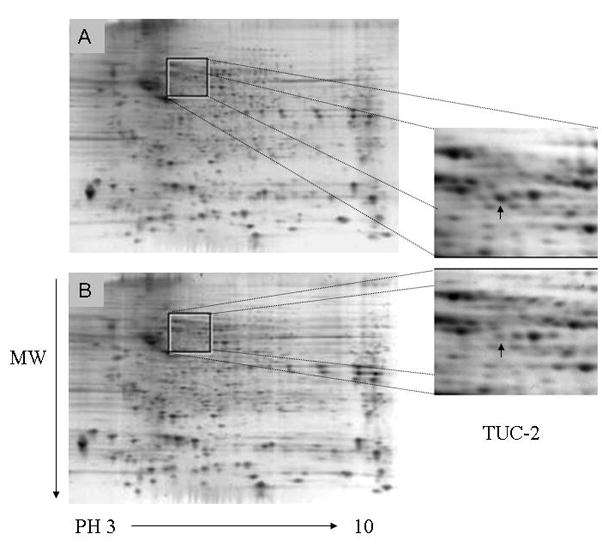
Sypro-ruby stained two-dimensional gels. The cytoplasmic extracts from sham (A) or HI injured (B) P7 rat cortexes were separated by two-dimensional electrophoresis. Differential expression of spots was evaluated with Progenesis. These changed spots were identified by MALDI-TOF-TOF-MS. Identified protein spots are listed in Table 1. Magnified image shows changes in spot intensity of TUC-2 (arrow).
Table I.
Proteins identified to change 2 hour after hypoxia/ischemia injury
| Spot# | Protein Name | Accession Number | pI | MW | Expect Value | Fold Change |
|---|---|---|---|---|---|---|
| 1 | Tyrosine 3- monooxygenase/tryptophan 5- monooxygenase activation protein (14-3-3 ε) | 13928824 | 4.55 | 29.27 | 8.5e−4 | −2.12* |
| 2 | TUC-2: Dihydropyrimidinase related protein-2 (DRP-2) Turned on after division, 64kDa protein(TOAD-64) Collapsin response mediator protein 2 (CRMP-2) | 40254595 | 5.95 | 62.64 | 7.0e−55 | −2.57* |
| 3 | Phosphoglycerate kinase-1 | 40254752 | 8.02 | 44.92 | 2.0e−5 | 2.59* |
| 4 | Isocitrate dehydrogenase1 (NADP+) | 13928690 | 6.53 | 47.05 | 3.5e−16 | + |
| 5 | Glyceraldehyde-3-phosphate dehydrogenase | 8393418 | 8.14 | 36.09 | 2.8e−24 | + |
| 6 | Eukaryotic translation enlongation factor 2 | 8393296 | 6.41 | 96.19 | 3.5e−21 | + |
| 7 | Dopamine- and cAMP-regulated phosphoprotein | 47155821 | 4.54 | 22.96 | 8.8e−13 | 2.12* |
| 8 | Synuclein, alpha | 9507125 | 4.74 | 14.51 | 1.8e−7 | + |
| 9 | Preproapolipoprotein A-1 | 55747 | 5.52 | 30.13 | 1.1e−8 | + |
| 10 | Cofilin 1 | 8393101 | 8.22 | 18.75 | 1.1e−33 | 4.99* |
| 11 | Chain F, Crystal Structure Of The Adenylyl Cyclase Domain Of Anthrax Edema Factor (Ef) In Complex With Calmodulin, 3′,5′ Cyclic Amp (Camp), And Pyrophosphate | 49259042 | 4.09 | 16.70 | 3.5e−12 | + |
| 12 | Unamed protein | 56929 | 6.63 | 58.29 | 4.4e-26 | + |
| 13 | Predicted: similar to 2010012F05Rik protein | 62648463 | 5.9 | 33.76 | 4.4e−7 | + |
| 14 | Predicted: similar to esterase D/formyglutathione hydrolase | 62661724 | 6.45 | 37.32 | 2.8e−17 | + |
| 15 | Unknown protein MGC108615 | 60551773 | 8.56 | 23.00 | 2.8e-23 | −2.39* |
| 16 | Predicted: similar to tumor protein, translational controlled 1 | 34879492 | 5.2 | 19.10 | 3.5e−19 | + |
| 17 | Glyoxylase 1 | 38181954 | 5.12 | 20.98 | 1.1e−16 | −2.59* |
Proteins were extracted from cortexes of sham or HI treated P7 rats, separated by two-dimensional electrophoresis, and identified by MALDI-TOF mass spectrometry following in gel digestion with trypsin. The expectation value is the number of matches expected to occur by chance only. Identifications were filtered for expectation score lower than 0.001. The accession numbers indicated are from National Center for Biotechnology Information (NCBI).
spots detected in HI treated animals but not in sham treated animals.
indicating significant change when comparing HI-treated versus sham-treated cortices using two tailed t test with false discovery rate correction.
Table 2.
Functional category of significantly changed proteins after HI injury
| Protein Name | Change | Function |
|---|---|---|
| Cortical Development | ||
| Tyrosine 3-monooxygenase/tryptophan 5- monooxygenase activation protein (14-3-3 ε) | Decrease | essential for normal brain development and neuronal migration |
| TUC-2: Dihydropyrimidinase related protein-2 (DRP-2) Turned on after division, 64kDa protein(TOAD-64) Collapsin response mediator protein 2 (CRMP-2) | Decrease | axon guidance and outgrowth |
| Metabolism Enzymes | ||
| Phosphoglycerate kinase-1 | Increase | enzyme involved in the glycolytic pathway |
| Isocitrate dehydrogenase 1 (NADP+) | Increase | enzyme in Kreb circle |
| Glyceraldehyde-3-phosphate dehydrogenase | Increase | enzyme involved in the glycolytic pathway |
| Translation Factor | ||
| Eukaryotic translation enlongation factor 2 | Increase | Protein translation |
| Neurotransmission | ||
| Dopamine- and cAMP-regulated phosphoprotein | Increase | Major target for dopamine activated adenylyl cyclase; crucial mediator of the biochemical, electrophysiological, transcriptional, and behavioral effects of dopamine |
| Synuclein, alpha | Increase | Involved in multiple steps of dopamine metabolism, including synthesis, storage, release, and uptake. |
The expression levels of several metabolic enzymes were increased 2h after HI brain injury. Isocitrate dehydrogenase 1 (Table 1, spot 4) that catalyzes the oxidative decarboxylation of isocitrate and the production of alpha-ketoglutarate was not detectable in sham-treated pups but was significantly expressed in the HI-treated animals. Two enzymes involved in the glycolytic pathway, Phosphoglycerate kinase-1 (PK-1, Table 1, spot 3), and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Table 1, spot 5), were upregulated 2h after HI injury. Both PK-1 and GAPDH are target genes of hypoxia inducible factor-1 (HIF-1), a transcription factor activated under hypoxia stress.12 The upregulation of these two enzymes is consistent with the hyperglycolysis13–15 and activation of HIF-1 after HI. We also found that two proteins associated with cortical development, 14-3-3ε (Table 1, spot 1) and TUC-2 (Table 1, spot 2), were down regulated early after HI injury. There was also upregulation of Dopamine- and cAMP-regulated phosphoprotein-32 (DARPP-32, Table 1, spot 7) and alpha Synuclein (alpha-Syn, Table 1, spot 8), two proteins important for dopamine neurotransmission, in the HI-treated pups. The expression of an important translation factor, translation elongation factor 2 (Table 1, spot 6), also increased after HI injury.
Validation of changes of 14-3-3ε and TUC-2 after HI injury
We validated the expression changes of 14-3-3ε and TUC-2 in HI-treated P7 cortex with western blot assays. The enlarged representative illustrations in Figure 2A and Figure 3A showed decreased 14-3-3ε and TUC-2 in cortex 2h after HI injury by 2D gel electrophoresis. The 14-3-3ε, an essential protein for neuronal developmental migration, decreased about 50% in HI-treated compared to sham-treated cortices. The TUC-2, a protein important for axonal guidance, was also found to decrease to 39% of sham levels 2h after HI injury. Since the distribution of 14-3-3ε in the neonatal brain is unknown, we did immunofluorecent staining for 14-3-3ε (Figure 2B) in P7 rat cortex. Immunofluorescent staining was also performed to confirm the expression in P7 cortex of TUC-2 (Figure 3B), a protein present early in development in most cortical neurons and gradually downregulated in most brain areas after birth.16 Immunoreactivity of 14-3-3ε and TUC-2 was widespread in P7 cortecal areas. We observed that 14-3-3ε and TUC-2 surrounded DAPI-counterstained nuclei with no overlap, consistent with the cytoplasmic distribution of these proteins. There were no immunoreactive products in the negative control sections in which the primary antibody was omitted (Data do not shown). The increased levels in 14-3-3ε and TUC-2 were further confirmed by western blot analysis using specific antibodies to these proteins. In agreement with the proteomics results, the expression of 14-3-3ε (Figure 2C) and TUC-2 (Figure 3C) decreased significantly 2h after HI.
Figure 2.

14-3-3ε decreased after HI. (A) Enlarged images of 2D protein profile representing decreases in protein expression of 14-3-3ε after HI. (B) Immunolabeling (×60) of sham-treated P7 rat cortex shows distribution of 14-3-3ε (Red) and co-localization of 14-3-3ε with nuclear DAPI (blue) stain. (C) Representative western blot assays and graphic representation of densitometry analyses of 14-3-3ε protein expression in cytoplasmic extracts of P7 rat cortices. Lane 1–3: Sham-treated; lane 4–6: HI-treated. β-actin was used as loading control. Values are mean ± SEM. N=7 in each group. *: p<0.05 compared to sham-treated group.
Figure 3.

TUC-2 decreased after HI. (A) Enlarged images of 2D protein profile representing decreases in protein expression of TUC-2 after HI. (B) Immunolabeling (×60) of sham-treated P7 rat cortex shows distribution of TUC-2 (Red) and co-localization of TUC-2 with nuclear DAPI (blue) stain. (C) Representative western blot assays and graphic representation of densitometry analyses of TUC-2 protein expression in cytoplasmic extracts of P7 rat cortices. Lane 1–3: Sham-treated; lane 4–6: HI-treated. β-actin was used as loading control. Values are mean ± SEM. N=7 in each group. *: p<0.05 compared to sham-treated group.
Validation of changes of alpha Synuclein and DARPP-32 after HI injury
Our proteome analyses showed better than a two-fold increase in DARPP-32 after HI injury. The expression of alpha-Syn was not detectable by 2D gel analysis in the sham-treated pups but was present 2h after HI injury. The enlarged illustrations show increases in alpha-Syn (Figure 4A) and DARPP-32 (Figure 5A) in cortex of P7 rat subjected to HI. Western blots were performed to further ensure the validity of our proteomics-based observations of changes in the expression of alpha-Syn and DARPP-32. We found significant increases in both alpha-Syn (Figure 4B) and DARPP-32 (Figure 5B) in P7 rat cortex 2h after HI injury.
Figure 4.

Alpha Synuclein increased after HI. (A) Enlarged images of 2D protein profile representing increases in alpha Synuclein after HI. (B) Representative western blot assays and graphic representation of densitometry analyses of alpha Synuclein protein expression in cytoplasmic extracts of P7 rat cortices. Lane 1–3: Sham-treated; lane 4–6: HI-treated. β-actin was used as loading control. Values are mean ± SEM. N=7 in each group. *: p<0.05 compared to sham-treated group.
Figure 5.

DARPP-32 increased after HI. (A) Enlarged images of 2D protein profile representing decreases in protein expression of DARPP-32 after HI. (B) Representative western blot assays and graphic representation of densitometry analyses of DARPP-32 protein expression in cytoplasmic extracts of P7 rat cortices. Lane 1–3: Sham-treated; lane 4–6: HI-treated. β-actin was used as loading control. Values are mean ± SEM. N=7 in each group. *: p<0.05 compared to sham-treated group.
DISCUSSION
This is the first 2-dimentional proteome profiling of protein expression in the P7 rat brain after HI. To exclude the possibility of differences due to small variations in experimental setup or operation, we arbitrarily set a high cutoff in our evaluation of proteome expression levels. We accepted only those protein values with 2 fold up or down regulation. T test with FDR correction was used to analyze the significance of over- or under-expression. Among the 17 proteins identified by Mass Spectrometry, two proteins are associated with neuronal migration and differentiation, consistent with a trauma-dependant early disturbance of cortical development. Cerebral cortical development occurs in a precisely-timed manner and can be divided into three major phases: neurogenesis, neuronal migration and neuronal differentiation.17 Neocortical neurons are generated mainly in the cortical ventricular zone between embryonic (E) days 14 and 20 in rats17 or during the early and middle parts of gestation in primates.18 Postmitotic neurons then migrate to their final positions in the cortex, forming an inside-out layered pattern.19 In rodents, migration occurs generally 2–4 days after neuronal generation.17 Cortical neuronal differentiation includes cell maturation and dendrite formation, axon extension and the development of intercellular connections. The first 3–4 weeks postnatal are critical for the differentiation of the rodent brain because most synapses in the neocortex are formed and many intrinsic and extrinsic cortical connections are refined during this period.20,21 The organization of mature cerebral cortex and the establishment of cortical circuitry are determined by a variety of early developmental signals and processes. Although the mechanisms responsible for how nerve cells from different parts of the brain properly target and assemble to form functional circuits are not completely understood, a large number of experimental and clinical data would suggest that HI and other forms of brain injury during the pre- or perinatal period may disturb these mechanisms, cause neuronal disorder and result in irreversible structural modifications.22–24 These malformations have been associated with severe long-term neurological sequelae such as mental retardation, motor dysfunction and epilepsy.
Our proteomics and western blot results showed that 14-3-3 epsilon, which is essential for normal brain development and neuronal migration,25 decreased in the cortex 2h after HI injury. 14-3-3 proteins are major evolutionarily conserved dimeric regulators in eukaryotic cells that involve several different cellular events, including signal transduction, cell cycle regulation, apoptosis, stress responses, cytoskeletal organization, and neurotransmitter synthesis.26 Recent studies have shown that the 14-3-3 protein detected in the cerebrospinal fluid of some neurological diseases such as Creutzfeldt-Jacob disease,27 multiple sclerosis28 and transmissible spongiform encephalopathy,29 is a marker for extensive brain destruction. It has been reported that, in humans and mice, defects in brain development and neuronal migration occur when 14-3-3ε protein is defective or missing. The 14-3-3ε gene is always deleted on one chromosome in Miller-Dieker syndrome (MDS), patients with a severe developmental defect of the brain caused when neurons born deep within the brain are unable to migrate normally to areas such as the hippocampus and cortex.25 Our immunofluoresence staining showed that 14-3-3ε proteins are cytoplasmic with widespread distribution in the P7 cortex. Our results presented here show for the first time that there is a decrease in 14-3-3ε levels early after HI, which might impair the migration of cortical neurons and the proper development of the cortex in these pups. Definitive understanding of the mechanisms of action of 14-3-3ε proteins remain unknown. There are reports that 14-3-3ε binds to CDK5/p35-phosphorylated NUDEL and maintains its phosphorylated state.25 Phosphorylated NUDEL is a LIS1-binding protein that participates with LIS1 in the regulation of the dynein motor complex and of microtubular organizing centers,30 which are known to be essential for neuronal migration. There are also aspects of the anti-apoptotic functions of 14-3-3 proteins that have been implicated in signaling apoptotic commitment via pro-apoptotic molecules such as Bad, FKHRL1, ASK1, and Nur77.26, 31,32 The 14-3-3ε protein has been identified as one of the caspase-3 substrates using a modified yeast two-hybrid genetic system.33 The cleavage of 14-3-3ε proteins by caspase-3 during apoptosis might contribute to cell death by preventing the association of 14-3-3ε with pro-apoptotic molecules. The early decrease in 14-3-3ε after HI might cause the improper cell death of developing neurons in the P7 rat. For example, it has been documented that there are decreases in neuronal numbers, especially the transient neuronal populations such as Cajal–Retzius neurons and subplate neurons that are known to be critical for guiding migration of newly generated cortical neurons, with possible consequences being defective radial migration and aberrant laminar patterning.34–36
The early stages of neurogenesis and neuronal migration are followed by a period of cellular differentiation, including axonal pathfinding to near or distant target regions.37 The TUC (TOAD-64/Ulip/CRMP/DRP) family of proteins has been reported to be important in axonal guidance and outgrowth and thus contributing to neuronal connectivity.16 TUCs consist of four 64-kDa isoforms, known as TUC-1 (CRMP-1/Ulip-3), TUC-2 (CRMP-2/Ulip-2/DRP-2/Toad64), TUC-3 (CRMP-3/Ulip4), and TUC-4 (CRMP-4/Ulip-1). In our proteome analyses TUC-2 was found to decrease after HI. In rats, all TUC proteins are expressed exclusively in the nervous system primarily during development.38 TUC-2 is present early in development in a majority of neurons and in a selected group of adult neurons, such as pyramidal cells of the hippocampus and Purkinje cells of the cerebellum. It is down-regulated in the adult brain but is re-expressed in adult motor neurons that are regrowing axons following axotomy.39 We observed a strong immunoreactivity of TUC-2 protein in the P7 cortex. A role for TUC-2 protein in axon guidance was implicated by the observation that antibodies to CRMP-62, the chick TUC-2 protein, can inhibit growth cone collapsing activity of neurons induced by semaphorin-3A, which is an important repulsive axonal guidance cue.40 Mutation of UNC-33, the C. elegans homologue of TUC proteins, results in severely uncoordinated movements and error pathfinding in neurons.41,42 Furthermore, overexpression of TUC-2 in cultured neurons, or SH-SY5Y neuroblastoma cells, promotes axonal growth and branching.43–45 Therefore, the observed early decrease in TUC-2 after HI might reflect a disturbance of normal signaling required for axonal guidance and thus, result in abnormal pathfinding. Although the mechanism of TUC-2 action has not been completely elucidated, TUC-2 has been reported to be associated with tubulin heterodimers and microtubule bundles, and it has been proposed that it is involved in the regulation of microtubule assembly dynamics.44,46 Neurons with expression of truncated TUC-2 protein lacking the region for microtubule assembly exhibit less axonal growth in a dominant-negative manner.
Our results showed that some proteins associated with neuronal migration and differentiation decreased early after HI brain injury, consistent with an early impairment of cortical development after HI injury. These findings are complementary to reports of HI- stimulated cell proliferation and neurogenesis in the subventricular zone (SVZ) area and peri-infarct striatum.47 Thus, the early impairment of cortical development might be transient and followed by compensatory neurogenesis necessary for neuronal recovery after neonatal brain injury. However, considering that P7 is an important time for the refinement of intracortical connections and precisely timed cortical development;48 these early disturbances might result in long term deficits not overcome by the repair processes.
Our proteome analyses also showed increases in the expression of DARPP-32 and alpha Synuclein, two proteins important for dopamine neurotransmission, in HI-treated animals. Dopamine is a neurotransmitter with crucial biochemical, electrophysiological and behavioral effects in the nervous system. Abnormalities in the dopamine signaling pathway are found in several neurological and psychiatric diseases such as Parkinsonism, schizophrenia, Attention Deficit Hyperactivity Disorder and drug abuse.49 Alternatively, excessive cytosolic dopamine levels could lead to increased levels of dopamine-quinone and the subsequent generation of reactive oxygen species with ensuing damage to mitochondria, proteins and DNA and eventually dopamine neuronal cell death.50 Dopamine was the first neurotransmitter to be identified as having a role in ischemic damage.51 Several investigators had previously determined that levels of dopamine were reduced early after ischemia.52 It has also been shown that the depletion of dopamine prior to ischemic stroke not only protects dopaminergic neurons from ischemic damage but the surrounding neurons as well.53
Alpha-Syn, a presynaptic protein in central nervous system, has been shown to play a role in the pathogenesis of Parkinson’s disease. Although the normal functions of alpha-Syn are not completely elucidated, accumulating evidence shows that the molecule is involved in multiple steps of dopamine metabolism, including synthesis, storage, release, and uptake.54 Alpha-Syn can inhibit tyrosine hydroxylase,55 the rate-limiting enzyme in dopamine synthesis, and reduce cytoplasmic dopamine synthesis at nerve terminals. Our proteomics result showed a decrease of alpha-Syn after HI injury, which may cause early reduction of dopamine, thereby limiting its conversion to highly reactive oxidative molecules.
DARPP-32 serves an obligatory role in the dopamine signaling pathway. It becomes a potent inhibitor of multifunctional Serine/Threonine Phosphatase PP-1 after being phosphorylated at Thr34 by dopamine receptor 1-activated protein kinase A (PKA), and is therefore involved in the modulation of synaptic neurotransmission by regulating the phosphorylation status of neurotransmitter receptors, ion channels or ion pumps. The regulation of DARPP-32 phosphorylation provides a mechanism for integrating information arriving at dopaminoceptive neurons in multiple brain regions.56 Consistent with its function as a target of DA receptor 1 activated PKA, DARPP-32 is present in dopaminoceptive neurons and concentrated in a subpopulation containing D-1 receptors.57 We found that DARPP-32 expression increased 2h after HI, a time when dopamine synthesis is inhibited. This should compensate the reduction of dopamine production after HI injury and might be a useful bypassing mechanism to keep normal neurontransmission function without increasing the oxidative stress induced by dopamine.
We have shown the differential expression of proteins that play an important role in development in animals subjected to HI brain injury. The proteins identified suggest that HI affects brain development and neurotransmission, and thus contribute to the neuropathology associated with HI. While more thorough analyses focusing on specific brain regions and multiple time points will better define the sequelae of events, these initial results relevant to cortical development and dopaminergic function need further analyses.
Synopsis.
Hypoxia/ischemia (HI) was induced in female P7 rats. A two-dimensional differential proteome analysis was used to assess changes in protein expression in cortex 2h after HI. 14-3-3ε and TUC-2 decreased after HI, consistent with an early disturbance of cortical development. DARPP-32 and alpha-synuclein, two proteins important for dopamine neurotransmission, increased after HI injury. The differential expression of theses proteins was validated by individual western blot assays. These findings provide insights into the mechanisms underlying neurodegenerative events after HI.
Acknowledgments
This study is supported by Grant 8540 from the Shriners Burns Institute, NICHD Grant HD39833 and a Postdoctoral Fellowship from NIEHS (T32-07254).
References
- 1.Kuban KC, Leviton A. Cerebral palsy. N Engl J Med. 1994;330:188–195. doi: 10.1056/NEJM199401203300308. [DOI] [PubMed] [Google Scholar]
- 2.Krageloh-Mann I, Toft P, Lunding J, Andresen J, Pryds O, Lou HC. Brain lesions in preterms: origin, consequences and compensation. Acta Paediatr. 1999;88:897–908. doi: 10.1080/08035259950168856. [DOI] [PubMed] [Google Scholar]
- 3.Vexler ZS, Ferriero DM. Molecular and biochemical mechanisms of perinatal brain injury. Semin Neonatol. 2001;6:99–108. doi: 10.1053/siny.2001.0041. [DOI] [PubMed] [Google Scholar]
- 4.Northington FJ, Ferriero DM, Flock DL, Martin LJ. Delayed neurodegeneration in neonatal rat thalamus after hypoxia-ischemia is apoptosis. J Neurosci. 2001;21:1931–1938. doi: 10.1523/JNEUROSCI.21-06-01931.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puka-Sundvall M, Wallin C, Gilland E, Hallin U, Wang X, Sandberg M, Karlsson J, Blomgren K, Hagberg H. Impairment of mitochondrial respiration after cerebral hypoxia-ischemia in immature rats: relationship to activation of caspase-3 and neuronal injury. Brain Res Dev Brain Res. 2000;125:43–50. doi: 10.1016/s0165-3806(00)00111-5. [DOI] [PubMed] [Google Scholar]
- 6.Hu X, Nesic-Taylor O, Qiu J, Rea HC, Fabian R, Rassin DK, Perez-Polo JR. Activation of nuclear factor-kappaB signaling pathway by interleukin-1 after hypoxia/ischemia in neonatal rat hippocampus and cortex. J Neurochem. 2005;93:26–37. doi: 10.1111/j.1471-4159.2004.02968.x. [DOI] [PubMed] [Google Scholar]
- 7.Aden U, Dahlberg V, Fredholm BB, Lai LJ, Chen Z, Bjelke B. MRI evaluation and functional assessment of brain injury after hypoxic ischemia in neonatal mice. Stroke. 2002;33:1405–1410. doi: 10.1161/01.str.0000014608.78503.db. [DOI] [PubMed] [Google Scholar]
- 8.Rumpel H, Nedelcu J, Aguzzi A, Martin E. Late glial swelling after acute cerebral hypoxia-ischemia in the neonatal rat: a combined magnetic resonance and histochemical study. Pediatr Res. 1997;42:54–59. doi: 10.1203/00006450-199707000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Calvert J, Zhang J. Pathophysiology of an hypoxic-ischemic insult during the perinatal period. Neurol Res. 2005;27:246–260. doi: 10.1179/016164105X25216. [DOI] [PubMed] [Google Scholar]
- 10.Amato M, Donati F. Update on perinatal hypoxic insult: mechanism, diagnosis and interventions. Eur J Paediatr Neurol. 2000;4:203–209. doi: 10.1053/ejpn.2000.0307. [DOI] [PubMed] [Google Scholar]
- 11.Qiu J, Grafe MR, Schmura SM, Glasgow JN, Kent TA, Rassin DK, Perez-Polo JR. Differential NF-kappa B regulation of bcl-x gene expression in hippocampus and basal forebrain in response to hypoxia. J Neurosci Res. 2001;64:223–234. doi: 10.1002/jnr.1070. [DOI] [PubMed] [Google Scholar]
- 12.Sharp FR, Ran R, Lu A, Tang Y, Strauss KI, Glass T, Ardizzone T, Bernaudin M. Hypoxic preconditioning protects against ischemic brain injury. NeuroRx. 2004;1:26–35. doi: 10.1602/neurorx.1.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiraishi K, Sharp FR, Simon RP. Sequential metabolic changes in rat brain following middle cerebral artery occlusion: a 2-deoxyglucose study. J Cereb Blood Flow Metab. 1989;9:765–73. doi: 10.1038/jcbfm.1989.110. [DOI] [PubMed] [Google Scholar]
- 14.Yao H, Ginsberg MD, Eveleth DD, LaManna JC, Watson BD, Alonso OF, Loor JY, Foreman JH, Busto R. Local cerebral glucose utilization and cytoskeletal proteolysis as indices of evolving focal ischemic injury in core and penumbra. J Cereb Blood Flow Metab. 1995;15:398–408. doi: 10.1038/jcbfm.1995.50. [DOI] [PubMed] [Google Scholar]
- 15.Tohyama Y, Sako K, Yonemasu Y. Hypothermia attenuates hyperglycolysis in the periphery of ischemic core in rat brain. Exp Brain Res. 1998;122:333–338. doi: 10.1007/s002210050521. [DOI] [PubMed] [Google Scholar]
- 16.Quinn CC, Gray GE, Hockfield S. A family of proteins implicated in axon guidance and outgrowth. J Neurobiol. 1999;41:158–164. [PubMed] [Google Scholar]
- 17.Bayer S, Altman J, Russo R, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- 18.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 19.Angevine J, Sidman R. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 20.Sur M, Cowey A. Cerebral cortex: function and development. Neuron. 1995;15:497–505. doi: 10.1016/0896-6273(95)90139-6. [DOI] [PubMed] [Google Scholar]
- 21.Berger-Sweeney J, Hohmann Behavioral consequences of abnormal cortical development: insights into developmental disabilities. Behav Brain Res. 1997;86:121–142. doi: 10.1016/s0166-4328(96)02251-6. [DOI] [PubMed] [Google Scholar]
- 22.Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. I: Hemorrhagic lesions of neocortex. J Neuropathol Exp Neurol. 1996;55:758–773. doi: 10.1097/00005072-199607000-00002. [DOI] [PubMed] [Google Scholar]
- 23.Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. J Neuropathol Exp Neurol. 1999;58:407–429. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- 24.McQuillen PS, Ferriero DM. Perinatal subplate neuron injury: implications for cortical development and plasticity. Brain Pathol. 2005;15:250–260. doi: 10.1111/j.1750-3639.2005.tb00528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, Ayala R, Tsai LH, Dobyns W, Ledbetter D, Hirotsune S, Wynshaw-Boris A. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- 26.Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- 27.Zerr I, Bodemer M, Gefeller O, Otto M, Poser S, Wiltfang J, Windl O, Kretzschmar HA, Weber T. Detection of 14-3-3 protein in the cerebrospinal fluid supports the diagnosis of Creutzfeldt-Jakob disease. Ann Neurol. 1998;43:32–40. doi: 10.1002/ana.410430109. [DOI] [PubMed] [Google Scholar]
- 28.Satoh J, Yukitake M, Kurohara K, Takashima H, Kuroda Y. Detection of the 14-3-3 protein in the cerebrospinal fluid of Japanese multiple sclerosis patients presenting with severe myelitis. J Neurol Sci. 2003;212:11–20. doi: 10.1016/s0022-510x(03)00083-2. [DOI] [PubMed] [Google Scholar]
- 29.Hsich G, Kenney K, Gibbs CJ, Lee KH, Harrington MG. The 14-3-3 brain protein in cerebrospinal fluid as a marker for transmissible spongiform encephalopathies. N Engl J Med. 1996;335:924–930. doi: 10.1056/NEJM199609263351303. [DOI] [PubMed] [Google Scholar]
- 30.Niethammer M, Smith DS, Ayala R, Peng J, Ko J, Lee MS, Morabito M, Tsai LH. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 31.Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000;275:25865–25869. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 32.Meller R, Schindler CK, Chu XP, Xiong ZG, Cameron JA, Simon RP, Henshall DC. Seizure-like activity leads to the release of BAD from 14-3-3 protein and cell death in hippocampal neurons in vitro. Cell Death Differ. 2003;10:539–547. doi: 10.1038/sj.cdd.4401206. [DOI] [PubMed] [Google Scholar]
- 33.Won J, Kim DY, La M, Kim D, Meadows GG, Joe CO. Cleavage of 14-3-3 protein by caspase-3 facilitates bad interaction with Bcl-x(L) during apoptosis. J Biol Chem. 2003;278:19347–19351. doi: 10.1074/jbc.M213098200. [DOI] [PubMed] [Google Scholar]
- 34.Shinozaki K, Miyagi T, Yoshida M, Miyata T, Ogawa M, Aizawa S, Suda Y. Absence of Cajal-Retzius cells and subplate neurons associated with defects of tangential cell migration from ganglionic eminence in Emx1/2 double mutant cerebral cortex. Development. 2002;129:3479–3792. doi: 10.1242/dev.129.14.3479. [DOI] [PubMed] [Google Scholar]
- 35.Sarnat HB, Flores-Sarnat L. Role of Cajal-Retzius and subplate neurons in cerebral cortical development. Semin Pediatr Neurol. 2002;9:302–308. doi: 10.1053/spen.2002.32506. [DOI] [PubMed] [Google Scholar]
- 36.Ferguson KL, McClellan KA, Vanderluit JL, McIntosh WC, Schuurmans C, Polleux F, Slack RS. A cell-autonomous requirement for the cell cycle regulatory protein, Rb, in neuronal migration. EMBO J. 2005;24:4381–4391. doi: 10.1038/sj.emboj.7600887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luhmann HJ, Hanganu I, Kilb W. Cellular physiology of the neonatal rat cerebral cortex. Brain Res Bull. 2003;60:345–453. doi: 10.1016/s0361-9230(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 38.Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996;16:6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minturn JE, Fryer HJ, Geschwind DH, Hockfield S. TOAD-64, a gene expressed early in neuronal differentiation in the rat, is related to unc-33, a C. elegans gene involved in axon outgrowth. J Neurosci. 1995;15:6757–6766. doi: 10.1523/JNEUROSCI.15-10-06757.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- 41.Hedgecock EM, Culotti JG, Thomson JN, Perkins LA. Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev Biol. 1985;111:158–170. doi: 10.1016/0012-1606(85)90443-9. [DOI] [PubMed] [Google Scholar]
- 42.Li W, Herman RK, Shaw JE. Analysis of the Caenorhabditis elegans axonal guidance and outgrowth gene unc-33. Genetics. 1992;132:675–689. doi: 10.1093/genetics/132.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inagaki N, Chihara K, Arimura N, Menager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K. CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci. 2001;4:781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- 44.Fukata Y, Itoh T, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 45.Cole A, Knebel A, Morrice N, Robertson L, Irving A, Connolly C, Sutherland C. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu Y, Ihara Y. Evidence that collapsin response mediator protein-2 is involved in the dynamics of microtubules. J Biol Chem. 2000;275:17917–17920. doi: 10.1074/jbc.C000179200. [DOI] [PubMed] [Google Scholar]
- 47.Plane JM, Liu R, Wang TW, Silverstein FS, Parent JM. Neonatal hypoxic-ischemic injury increases forebrain subventricular zone neurogenesis in the mouse. Neurobiol Dis. 2004;16:585–595. doi: 10.1016/j.nbd.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 48.Lipska BK. Using animal models to test a neurodevelopmental hypothesis of schizophrenia. J Psychiatry Neurosci. 2004;29:282–286. [PMC free article] [PubMed] [Google Scholar]
- 49.Greengard P. The neurobiology of dopamine signaling. Biosci Rep. 2001;21(3):247–269. doi: 10.1023/a:1013205230142. [DOI] [PubMed] [Google Scholar]
- 50.Maker HS, Weiss C, Silides DJ, Cohen G. Coupling of dopamine oxidation (monoamine oxidase activity) to glutathione oxidation via the generation of hydrogen peroxide in rat brain homogenates. J Neurochem. 1981;36:589–593. doi: 10.1111/j.1471-4159.1981.tb01631.x. [DOI] [PubMed] [Google Scholar]
- 51.Weinberger J. The role of dopamine in cerebral ischemic damage: a review of studies with Gerald Cohen. Parkinsonism Relat Disord. 2002;8:413–416. doi: 10.1016/s1353-8020(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 52.Lust WD, Mrsulja BB, Mrsulja BJ, Passonneau JV, Klatzo I. Putative neurotransmitters and cyclic nucleotides in prolonged ischemia of the cerebral cortex. Brain Res. 1975;98:394–399. doi: 10.1016/0006-8993(75)90021-9. [DOI] [PubMed] [Google Scholar]
- 53.Weinberger J, Nieves-Rosa J, Cohen G. Nerve terminal damage in cerebral ischemia: protective effect of alpha-methyl-para-tyrosine. Stroke. 1985;16:864–870. doi: 10.1161/01.str.16.5.864. [DOI] [PubMed] [Google Scholar]
- 54.Yu S, Ueda K, Chan P. Alpha-synuclein and dopamine metabolism. Mol Neurobiol. 2005;31:243–254. doi: 10.1385/MN:31:1-3:243. [DOI] [PubMed] [Google Scholar]
- 55.Perez RG, Waymire JC, Lin E, Liu JJ, Guo F, Zigmond MJ. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J Neurosci. 2002;22:3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 57.Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3′:5′-monophosphate-regulated phosphoprotein enriched in dopamine-innervated brain regions. I. Regional and cellular distribution in the rat brain. J Neurosci. 1984;4:84–98. doi: 10.1523/JNEUROSCI.04-01-00084.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]