

Abstract
This perspective summarizes recent results, which demonstrate that stable carbenes can activate small molecules (CO, H2, NH3 and P4) and stabilize highly reactive intermediates (main group elements in the zero oxidation state and paramagnetic species). These two tasks were previously exclusive for transition metal complexes.
Introduction
At the end of the 19th century, Sabatier observed the formation of ethane in the addition of H2 to ethylene over thin slivers of reduced nickel. Since this pivotal discovery the activation of H2 and other small molecules has attracted considerable interest. Until recently, most of the chemical and biological systems that have been successful in activating small molecules, and in more general terms unreactive bonds, involve a transition metal. Even the so-called iron-sulfur cluster-free hydrogenase (Hmd) uses an iron center in combination with an adjacent methenyl-H4MPT+, which serves as a hydride acceptor.1
In 2006, our group discovered that acyclic2 and cyclic(alkyl)-(amino)carbenes3 react with carbon monoxide to afford the corresponding ketenes.4 Examination of the literature showed that organic molecules were not known to react with CO, except a few transient carbenes5 and silylenes,6 whereas transition metal centers classically do. This led to the question of whether singlet carbenes could mimic the chemical behavior of transition metals. This new paradigm appeared very reasonable since singlet carbenes possess both a lone pair of electrons and an accessible vacant orbital, and therefore resemble, at least to some extent, transition metal centers.
This perspective summarizes the results obtained during the last four years, which demonstrate that stable carbenes can do some of the tasks transition metal complexes are known for. The activation of small molecules such as CO, H2, NH3 and P4 will first be discussed. Then, as transition metals are also well known for stabilizing highly reactive species within their coordination sphere, examples showing that stable singlet carbenes can be equally stabilizing entities will be presented. This second aspect can appear paradoxical since it consists of using carbenes that were considered for a long time as prototypical reactive intermediates,7 for isolating otherwise unstable molecules.
Although this review focuses on carbenes, the reactivity of their heavier analogues (silylenes, germylenes and stannylenes) will be briefly presented. In order to facilitate the reading, the five families of stable carbenes, which will often be involved, have been abbreviated as shown in Fig. 1.
Fig. 1.
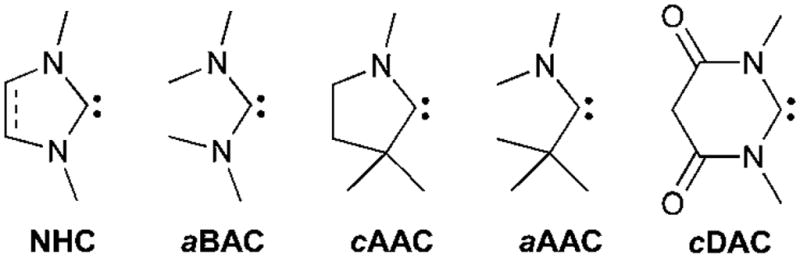
Stable carbenes frequently used in this perspective.
1 Activation of small molecules
1.1 Activation of carbon monoxide
In 1994 it was claimed that imidazol-2-ylidene NHC1 reacts with CO to give the corresponding stable ketene.8 However, a year later Arduengo et al.9 were not able to duplicate these experimental results. They found computationally that there is no stable structure associated with the combination of the parent imidazol-2-ylidene and CO, other than “a non-bonded weakly interacting (van der Waals) complex”; the ketene not even being found as a transition state.
According to calculations,10 the singlet–triplet gap and the HOMO for the parent acyclic and cyclic (alkyl)(amino)carbenes aAAC0 and cAAC0 are much smaller and higher in energy, respectively, than for the acyclic and cyclic bis(amino)carbenes aBAC0 and NHC0 (Fig. 2). Consequently, (alkyl)(amino)-carbenes are more nucleophilic but also more electrophilic than their bis(amino) counterparts, and are therefore better suited for metal-like behavior. Calculations predicted that the reaction of CO with the acyclic bis(amino)carbene aBAC0 was endergonic (ΔG = +4.6 kJ mol−1), whereas with the acyclic (alkyl)(amino)-carbene aAAC0 it was exergonic (ΔG = −111.6 kJ mol−1). Indeed, although aBAC1 is inert towards CO, cAAC1 and aAAC1 react instantaneously and quantitatively at room temperature, affording the corresponding ketenes 1 and 2. Note that ketene 1 is by far less thermally stable than 2, in agreement with the larger singlet–triplet gap and therefore weaker electrophilicity of cAACs compared to aAACs. Very interestingly, Bielawski et al.11 have recently prepared cyclic di(amido)carbenes, such as cDAC1, which are more electrophilic than classical NHCs and shown that they reversibly react with CO.
Fig. 2.

Activation of CO, H2 and NH3 by stable carbenes.
In 2009, Power et al.12 reported the first example of a room temperature reaction of CO with a heavier group 14 carbene analogue.13 Using a diaryl germylene, they observed formation of compound 3, most likely resulting from the rearrangement of the first formed germaketene (Scheme 1).
Scheme 1.
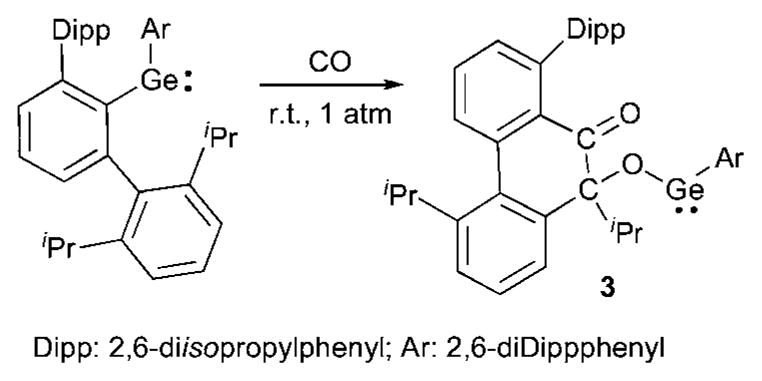
Activation of CO by a germylene.
1.2 Activation of dihydrogen
As mentioned in the introduction, for a long time, the splitting of H2 was almost exclusively the domain of metals. The only exceptions were compounds only stable in matrices at a few K such as subvalent group 13 species,14 triplet carbenes15 and nitrenes,16 and the highly electrophilic singlet difluorovinylidene.17 In the last few years considerable progress has been achieved in the metal-free activation of H2. Power et al.18 showed that room temperature stable digermynes and distannynes (ArE≡EAr, E = Ge, Sn) were able to split H2 under mild conditions. Li and Zu19 reported that fullerenes, fullerene anions, and their combination can activate hydrogen under UV irradiation at room temperature; even more striking, these species are able to catalyze the reduction of nitrobenzene into aniline. Stephan et al.20 have reported the reversible activation of hydrogen using intramolecular phosphine-borane pairs. The latter system, the so-called frustrated Lewis pairs (FLPs), has received considerable attention. Several intra-molecular and intermolecular combinations of Lewis acids/bases have been proven to efficiently split H2, reversibly or irreversibly, and even catalyze the hydrogenation of reactive unsaturated compounds.21
Although NHCs have been used as the Lewis base component of the FLPs,22 acyclic and cyclic bis(amino)carbenes, on their own, are inert towards H2.23 In marked contrast, cyclic and acyclic (alkyl)(amino)carbenes cAAC1 and aAAC1 can operate as single-site molecules for the activation of H2 under mild conditions (Fig. 2).10 The calculated energy changes (ΔE) show that although the reactions of all carbenes with H2 are exothermic, the reactions are significantly more favored in the case of mono(amino)carbenes (cAAC0 and aAAC0) than bis(amino)-carbenes (aBAC0 and NHC0).24 More importantly, the activation energy is at least 45 kJ mol−1 lower in the case of mono(amino)carbenes, which readily rationalizes the experimental results. Note that although di(amido)carbenes cDAC, react with CO and NH3 (vide infra), they are inert towards hydrogen even under forcing conditions (65 atm H2, 200 °C).25
Importantly, the mode of activation of H2 by mono-(amino)carbenes and transition metals is totally different. For the latter, H2-splitting results from the primary interaction of a vacant orbital at the metal and the σ-bonding orbital of H2.26 In the case of carbenes, the cleavage comes from the primary interaction of the carbene’s lone pair with an H2 antibonding orbital. In the transition state, the H–H bond is considerably elongated (1.071 Å versus 0.77 Å for H2 gas) and polarized with the positively charged H (+0.178) already bonded to the carbon (1.206 Å) (Fig. 3). In other words, in contrast to the electrophilic activation/proton transfer pathway favored by transition metal centers, carbenes act by initial nucleophilic activation followed by hydride transfer.
Fig. 3.
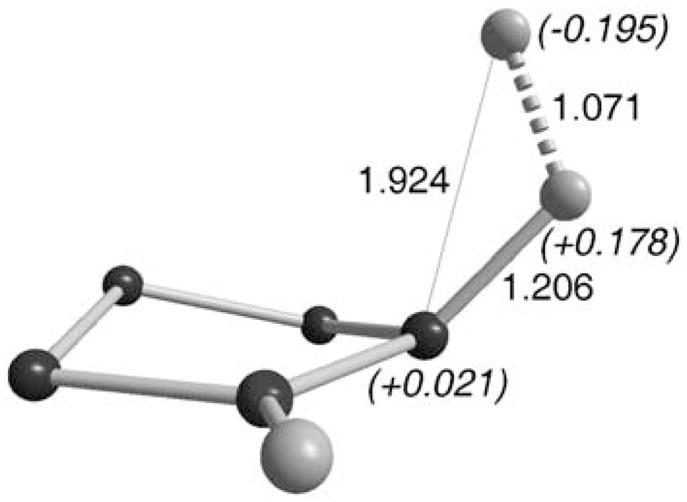
Transition state for H2 activation by cAACs.
There are also recent reports covering the activation of hydrogen by germylenes and stannylenes (Scheme 2).27,28 These reactions occurred under relatively mild conditions; however, depending on the nature of the aryl substituents, subsequent arene elimination sometimes occurs.
Scheme 2.
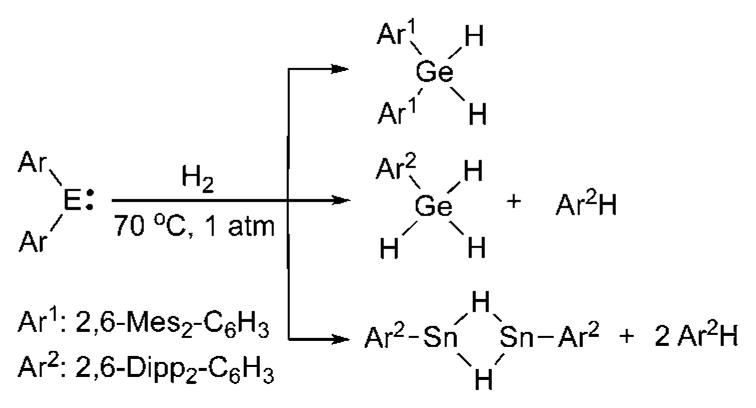
H2 activation by heavier carbene analogues.
1.3 Activation of ammonia
In contrast with H2, NH3 is reluctant to be split by transition metal centers.29 Since H–H and N–H bond dissociation energies are comparable, the main obstacle is the electrophilicity of the metal, which instead favors the formation of the so-called Werner complexes LnM-NH3. As mentioned above, the activation of H2 by mono(amino)carbenes relies on the primary interaction between the HOMO of the carbene (lone pair) and the LUMO of the reactant. This suggests that no Lewis acid–base adducts can be formed between a carbene and NH3. According to calculations, the reaction barriers for NH3 splitting by carbenes are comparable to those for H2 activation, although the former processes are less exothermic (Fig. 2).
Consistent with the computational results, NHCs are inert towards NH3,30 but cAAC1 and aAAC1 rapidly react with liquid ammonia, even at −40 °C, cleanly affording the corresponding adducts.10 Similarly, Bielawski et al.25 have found that the rather electrophilic N,N′-di(amido)carbene cDAC2 also activates NH3 under mild conditions.
The mode of approach of NH3 to cAACs was calculated to be very similar to that observed with H2. In the transition state, one of the N–H bonds elongates up to 1.50 Å, and becomes strongly polarized with the positively charged H (+0.316) bonded to the carbon (1.155 Å) (Fig. 4). Note that the nitrogen lone pair is pointing away from the carbene vacant orbital, supporting the nucleophilic character of the activation process.
Fig. 4.
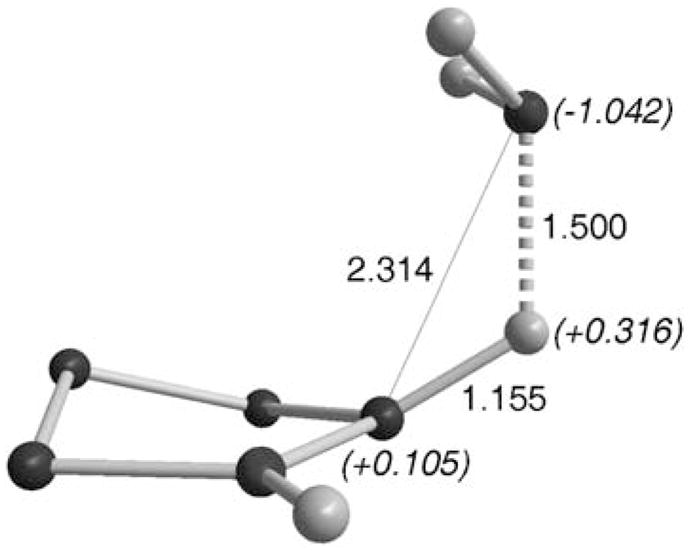
Transition state for NH3 activation by cAACs.
Heavier carbene analogues have also been used for the activation of ammonia, and depending on the heteroatom very different reactions were observed (Scheme 3). Silylene 431 reacts at room temperature with NH3 gas to give adduct 5, formally resulting from an oxidative addition at the silicon(II) center.32 Conversely, the analogous germylene 633 undergoes a 1,4-addition affording 7.34 The different behavior of 4 and 6 has not been rationalized. In the case of stannylenes 8, the addition of NH3 results in the formation of dimeric derivative 9 with concomitant elimination of an arene, as observed for the activation of H2 by this species.27
Scheme 3.
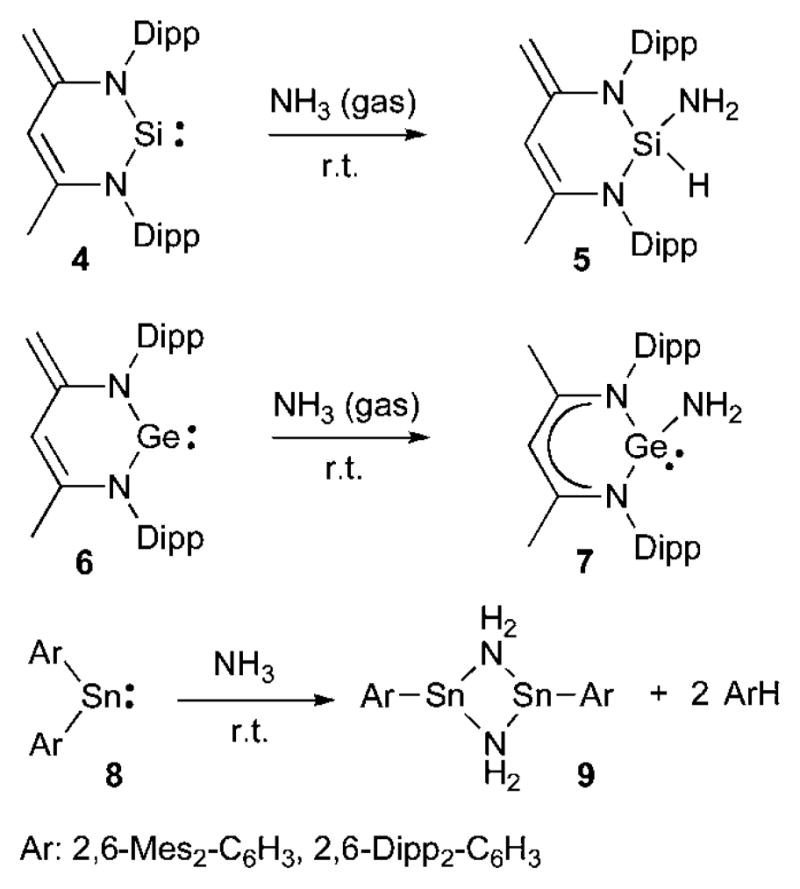
NH3 activation by heavier carbene analogues.
1.4 Activation of white phosphorus (P4)
White phosphorus, the most reactive allotrope of the element, is another small molecule of industrial importance. It is the precursor to PCl3 and PH3, and thus of most organophosphorus derivatives. To meet the increasingly stringent environmental regulations, new processes using P4, but avoiding chlorine, are highly desirable, which explains why its reactivity with organometallic compounds has been widely studied.35 Consequently, P4 is an excellent model to further explore the analogy between carbenes and transition metals.
At the end of the 1990s, West et al.36 reported that stable silylene 10 catalyzed the conversion of white phosphorus to the red allotrope (Scheme 4). More recently, Driess et al.37 reported the mono- and di-insertion of silylene 4 into P4, which leads to strained polycyclic silaphosphanes 11 and 12.38 Interestingly, the germylene analogue 6 is inert towards P4 even in boiling toluene, possibly due to the lower reduction potential of Ge(II) versus Si(II). It should also be mentioned that mono insertion products similar to 11 have been observed in the reaction of white phosphorus with aluminium(I) derivatives39 and phosphenium cations.40
Scheme 4.

P4 activation by silylenes.
In 2007, the clean formation of the bis(carbene) adduct 14a was observed in the addition of two equivalents of the bulky cAAC1 on P4 (Scheme 5).41 This was the first demonstration that stable singlet carbenes can activate white phosphorus. Compound 14a has to be regarded as a 2,3,4,5-tetraphosphatriene, and a neutral P4 chain as seen in 14a is unprecedented. This compound features highly reactive functional groups, a diphosphene and two phosphaalkene fragments, thus allowing for the challenging and desirable direct formation of organophosphorus derivatives from P4. For instance, 14a reacts diastereoselectively with 2,3- dimethylbutadiene to afford the [4 + 2] cycloaddition product 15.
Scheme 5.

Activation, degradation and aggregation of P4 by stable carbenes.
Calculations, using the parent cAACo predicted that the first step of the reaction is the nucleophilic attack of the carbene at one of the phosphorus atoms of P4, affording triphosphirene 13.41,42 A second carbene then reacts at one of the unsaturated phosphorus centers of the triphosphirene moiety, giving a transient bis(carbene) adduct; the latter undergoes a facile ring opening to afford the observed 2,3,4,5-tetraphosphatriene 14. Several complexes containing metal-stabilized mono-substituted triphosphirene fragments have been observed in the reaction of transition metal complexes with P4.43 The transient formation of 13 was demonstrated by reacting cAAC1 with P4 in the presence of a large excess of 2,3-dimethylbutadiene, which afforded the corresponding [4 + 2] cycloaddition product 19a. Further proof of the initial formation of triphosphirenes was found when acyclic (alkyl)(amino)carbene aAAC1 was used, instead of cAAC1. Indeed, the bicyclic compound 16 was obtained in 66% yield.44
Also similar to transition metals,45 cAACs can promote the degradation of P4 into P1 and P2 fragments (Scheme 5).44 Excess of the relatively small cAAC2, instead of the bulky cAAC1 or more electrophilic aAAC1, affords products 17a and 18, which were isolated in moderate yields. The formation of tris-carbene P4-adduct 18 implies that two cAACs react on the primarily formed adduct of type 13. The minor product 17a is the desired bis-carbene-P2 adduct, and the reaction leading to this compound represents the first example of fragmentation of P4 by a neutral organic species.
P2-adduct 17a most probably results from the attack of the carbene to the β-phosphorus centers of a P4 bis-carbene adduct similar to 14a; this fragmentation is made possible by the small size of cAAC2. When the least sterically demanding stable carbene known was used, bis(diisopropylamino)cyclopropenylidene CP1,46,47 the cationic bis(carbene)-P1 adduct 20 was obtained along with an anionic P3 fragment, which has not been fully identified.44 Clearly, 20 results from the attack of the carbene at a phosphorus center in a position α to the carbene carbon(s) of adducts of type 13 or 14. Notably, analogous (NHC)2P+ systems have previously been reported by Schmidpeter48a and Macdonald,48b but using (R3P)2P+, (RNPCl)2, or PCl3 as P1 sources.
Another task that transition metal centers are capable of doing with P4 is to induce its aggregation, although complexes with more than six phosphorus atoms are usually obtained as byproducts in low yields.49 NHC250 reacts at 70 °C overnight with white phosphorus, giving the P12 cluster 21 in high yield (81%).51 The architecture of the P12 core is very different from that found in the only known P12-transition metal complex.52 Trapping experiments with dimethylbutadiene demonstrated that mono-and bis(carbene) adducts of type 13 and 14b were involved. The different outcome of reactions with NHCs versus cAACs can be rationalized by the different electronic properties of these two carbenes. As already mentioned, cAACs are more electrophilic (π-acceptor) and thus strengthen the PC bonds of 14, while NHCs are less basic and therefore better leaving groups, favoring the formation of clusters such as 21.
2 Stabilization of highly reactive species
There are numerous examples of the use of NHCs for stabilizing reactive Lewis acids as well as transition metal centers with unusual coordination and/or oxidation states. This section is limited to two recent applications of stable carbenes in main group element chemistry: the isolation of compounds featuring group 14 and 15 elements in the zero oxidation state, and the synthesis of phosphorus and boron centered radicals.
2.1 Stabilization of non-transition metal elements in the zero oxidation state
The zero oxidation state is classically found in metals, when stabilized by ligands that donate electron pairs into their empty orbitals. Such a state has been difficult to realize for main group elements except in their allotropes.
The four main allotropes of phosphorus are: white, red, violet and black. Except white phosphorus, all these compounds are polymeric and therefore it is easy to recognize that the P12 cluster 21 (Scheme 5) can be viewed as a carbene-stabilized phosphorus allotrope.51,53 In 2008, Robinson et al.54 discussed the structure of compound 17b prepared by reduction of the (NHC)-PCl3 adduct 22 with potassium graphite (Scheme 6). Due to donation of the nitrogen’s lone pair of electrons into the P=C double bonds, 17b can be represented by the canonical forms 17b(I) and 17b(II), or can even be viewed as a bis-phosphinidene unit coordinated by two carbenes, as shown by representation 17b(III). X-ray crystal data and DFT computations showed that the PC bonds of 17b have a modest double bond character, while the PP bond is single. Moreover, each phosphorus has two lone pairs, supporting the carbene stabilized bis-phosphinidene structure 17b(III). Of course, the structure of this bis(carbene)-P2 adduct is very different from the highly unstable diphosphorus allotrope (P2), which exhibits a phosphorus-phosphorus triple bond. Both compounds, however, have phosphorus centers with formal zero oxidation states. Note that a bis(carbene)-phosphonitride analogue (NHC-NP-cAAC) has recently been isolated.55
Scheme 6.

Synthesis and different forms of P2-biscarbene adduct 17b.
Robinson et al. used the same synthetic strategy for preparing other diatomic molecules of group 15 (arsenic),56 and of group 14 elements (silicon),57 in the formal oxidation state of zero (Scheme 7). Compound 23 represents a landmark in low coordinate silicon chemistry since each silicon center is involved in a multiple bond and, at the same time, features a lone pair of electrons, two attributes usually associated with extreme instability.58 The zero oxidation state of silicon was clearly evidenced by a distinctly non-linear C–Si–Si–C backbone (C–Si–Si = 93.37°) and long C–Si bonds (1.927 Å). This geometry strikingly contrasts that expected for form 23(I), with silicon atoms formally in the +II oxidation state. The latter would exhibit a perfectly planar skeleton with a linear C–Si–Si–C arrangement and short carbon-silicon bonds. Thus, in compound 23, the carbene ligand is only serving as a two-electron donor and is non-oxidizing (not withdrawing electron density from silicon), leaving a non-bonding electron pair on each silicon center as shown in 23(II). With such a bonding description,59 compound 23 should be regarded as a diatomic silicon(0) unit coordinated by two L ligands; in other words it is a Lewis base-stabilized silicon allotrope. Note that a germanium analogue of 23 has also recently been isolated.60
Scheme 7.

Synthesis and different forms of Si2-biscarbene adduct 23.
What about carbon in the zero oxidation state? Several allotropes are known such as graphite, diamond, fullerenes, etc. and atomic carbon has been identified in the interstellar space.61 The latter is a short lived species with a triplet ground state, but most of the reactions reported so far involve a singlet excited state located 125 kJ mol−1 higher in energy. A hallmark of atomic carbon chemistry is the large amount of energy that it brings, which makes it so reactive that it inserts into very inert bonds. A simplistic view of singlet atomic carbon is a carbon atom with two lone pairs and two vacant orbitals. Therefore one could hope that electron donation from two Lewis bases could stabilize such a species while maintaining neutrality of the compound. Frenking et al.62 first tested this hypothesis computationally, using phosphines as Lewis bases. They showed that carbodiphosphoranes 24, which have been known for decades,63 feature a carbon atom with its four valence electrons in two orthogonal lone pairs that are not engaged in chemical bonding (24b) (Fig. 5). In other words, the commonly used allenic structure 24a is not relevant because of the weakness of back-donation of the carbon lone-pairs into the antibonding P–C orbitals.
Fig. 5.

Allenic structure a versus L2C(0) formulation b.
More surprisingly, Frenking et al.64,65 also proposed NHCs as alternative ligands for carbon(0), and suggested to name the corresponding complexes “carbodicarbenes”. At first glance, the prediction that the central carbon of 25 prefers to keep its four electrons as two lone pairs as shown in 25b seemed astonishing. Indeed, in contrast to the form 24a of carbodiphosphoranes, in which phosphorus is hypervalent, 25a is simply an allene with regular tetravalent carbon atoms.
Soon after the publication of this computational study, we reported the synthesis and isolation of the bis(benzimidazolin-2-ylidene)carbon(0) complex 26 (Fig. 6) that we classified as an acyclic bent-allene.66 We simply reasoned that strong electron-donating groups at the carbon termini of the allene would induce a strong polarization, and therefore a weakening of the π-bonds. As a consequence, the CCC framework should be more flexible, and ultimately a bending of the otherwise rigid and linear CCC skeleton should occur. A single crystal X-ray diffraction study revealed that the four amino groups do cause a dramatic effect on the geometry of 26. Although the bond lengths are only slightly longer (C1–C2 = 1.343 Å) than the standard C=C bond length of an allene (1.31 Å), the two NCN planes are not perpendicular but twisted by 69°. Remarkably, the allene framework is severely bent with a CCC angle of 134.8°. Clearly the allene π-system has been severely perturbed, and the central carbon atom is no longer sp-hybridized as in typical allenes, but likely approaching a configuration with two lone pairs. As a consequence of its peculiar electronic structure, and in contrast with “regular allenes”, an η1-coordination mode involving the central carbon was observed with metals.66,67
Fig. 6.

Solide state stucture of acyclic bent-allene 26.
Interestingly, it has even been suggested that tetrakis (dimethylamino)allene, displays “hidden” lone pairs, despite a classical linear geometry.68 Alcarazo, Fürstner et al. have shown that the reactivity of this compound is analogous to that of carbodiphosphorane 24 and carbodicarbene 26. They stated that the bonding in the tetrakis(dimethylamino)allene is still best described by the capto-dative formalism, i.e. σ-donation from the L ligands to the central carbon, and π-back donation of the lone pairs of the carbon(0) to L, exactly as in metal complexes. They concluded that “carbon is capable of serving as the central atom of a complex – just as a metal can do”.68b
Although the bent geometry is not a common feature for carbodicarbenes, all of them are highly flexible. Calculations have shown that widening the bond angle of the carbodicarbene 25 from the equilibrium geometry (131.8°) up to the “classical” linear structure requires only 15.5 kJ mol−1.64 As a consequence of its flexibility, the CCC framework of carbodicarbenes can be confined in rather small cyclic systems, as shown by the isolation of derivatives 2769 and 2870 (Fig. 7).
Fig. 7.

Cyclic carbodicarbenes.
Carbodicarbenes, as well as other carbon(0) derivatives (L:→ C←:L′), are predicted to be extremely basic. Computational studies showed that 26 has not only a very high first proton affinity (1230 versus 1100 kJ mol−1 for NHCs), but also a large second proton affinity (700 versus 300 kJ mol−1 for NHCs).71 Consequently, carbon(0) derivatives should be prone to double protonation, and should also be capable of bonding two transition metals at the same carbon site. This hypothesis is supported by the existence of dimetallated carbodiphosphoranes,72 and confirmed by recent results. Addition at room temperature of two equivalents of tetrafluoroboric acid to four-membered heterocycle 28 afforded dication 29,70 and Fürstner et al.,68b were able to prepare 30, a dinuclear gold complex of a carbon(0) derivative, featuring a phosphine and a dialkoxycarbene as “ligands” (Fig. 8).
Fig. 8.
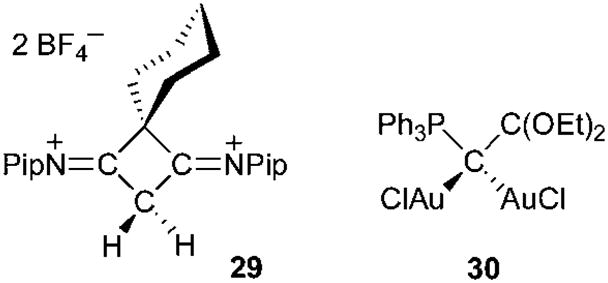
Double protonation and metalation of carbon(0) derivatives.
The concept of carbodicarbenes may be extended to heavier main group 14 elements (Fig. 9).73 The central atom can be silicon(0), germanium(0) or tin(0), whereas the L ligands are silylenes, germylenes or stannylenes.74 Several combinations are known, although surprisingly neither compounds featuring a heavier main group 14 element(0) ligated by two carbenes, nor a carbon(0) stabilized by heavier carbene analogues, have been prepared. Interestingly, Kira et al. noted that most of these compounds are highly flexible, the X-ray analysis showing dynamic disorders of the central atom even at low temperature.
Fig. 9.
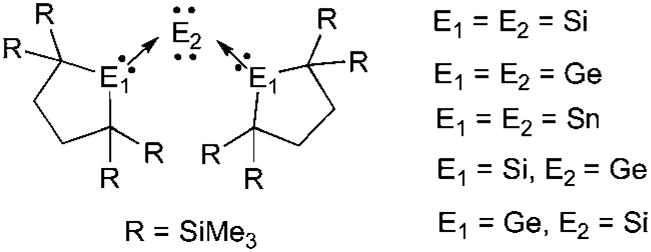
Heavier group 14 element(0) derivatives.
2.2 Stabilization of main-group element radicals
Recently, Malacria and Curran et al. used NHC-BH3 adducts as hydrogen-atom donors in the Barton–McCombie deoxygenation reaction,75 and as co-initiators for the radical photo-polymerization of acrylate.76 These results are particularly interesting since BH3 itself, and even amine- and phosphineboranes, are not suitable for these transformations. The higher reactivity of NHC-BH3 complexes was rationalized by a considerable reduction of the B–H bond dissociation energy. For BH3 and its amine and phosphine complexes, the calculated bond dissociation energies are in the 390–450 kJ mol−1 range, whereas for NHC-BH3 complexes the value dropped to 367 kJ mol−1.77 This value is smaller than that of triethylsilane (397 kJ mol−1), and only slightly higher than those calculated for standard radical hydrogen transfer reagents such as (Me3Si)3SiH (351 kJ mol−1) and tributylstannane (328 kJ mol−1). This trend has been confirmed by implementing kinetic experiments to measure rate constants for radical hydrogen abstraction from NHC-boranes by alkyl radicals.78 The lowering of the B–H bond dissociation energy points towards the enhanced stability of the NHC-BH2 radical 32, which is due to spin delocalization away from boron,79 as shown by EPR studies.77,80 Despite the presence of nitrogen centers in β-positions of the boron, larger a(14N) isotropic hyperfine coupling constants were observed for 32 than for R3N-boryl radicals 31. In addition, radicals 32 display a much smaller a(11B), showing that the boron center is in a planar environment and not pyramidalized as in 31 (Fig. 10). Stable boron-containing radicals are quite rare.81,82 Therefore, although NHC-BH2, or even NHC-BAr2 radicals,83 have so far not been isolated, their superior stability compared to other Lewis base stabilized analogues is a clear indication of the stabilizing effect of NHCs on adjacent radical centers. This conclusion has been corroborated by recent results in phosphorus chemistry.
Fig. 10.
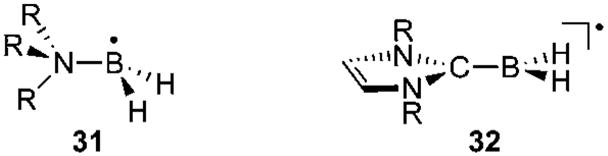
Planar versus pyramidalized boron radicals.
Until 2010, only resonance-stabilized phosphorus radicals, featuring a rather small spin density at phosphorus were structurally characterized by single crystal X-ray diffraction studies.84 The few phosphinyl radicals, which are stable in solution at room temperature, such as 33, dimerize in the solid state despite bulky substituents (Fig. 11).85 In contrast, the phosphinyl radical cation 35, readily available by oxidation of phosphaalkene 34 (derived from a cAAC), is indefinitely stable both in solution and in the solid state.86 The crystallographic study of 35 shows that both the PC (1.81 Å) and PN (1.68 Å) bond distances are comparable to those observed in the gas phase electron diffraction study of 33a (PC: 1.85; PN: 1.62 Å), 85 and collectively are in agreement with those expected for a phosphinyl radical bearing a cationic substituent. Similarly, the isotropic hyperfine coupling constant with P [a(31P) = 99 G] is comparable to those observed for phosphinyl radicals 33 [a(31P) = 96.3 G (33a) and 91.8 G (33b)], in which the odd electron resides predominantly in a 3p(P) valence orbital. Calculations and the EPR spectrum of 35 in a frozen fluorobenzene solution show that 67% of the spin density is localized at phosphorous, with only small contributions from the nitrogen atoms (Fig. 12). Therefore, the stability of phosphinyl radical 35 is partly due to steric factors, but more importantly to the presence of the cationic substituent. The latter prevents, by electrostatic repulsion, the dimerization observed for other phosphinyl radicals, such as 33.
Fig. 11.

Stable localized phosphinyl radicals.
Fig. 12.
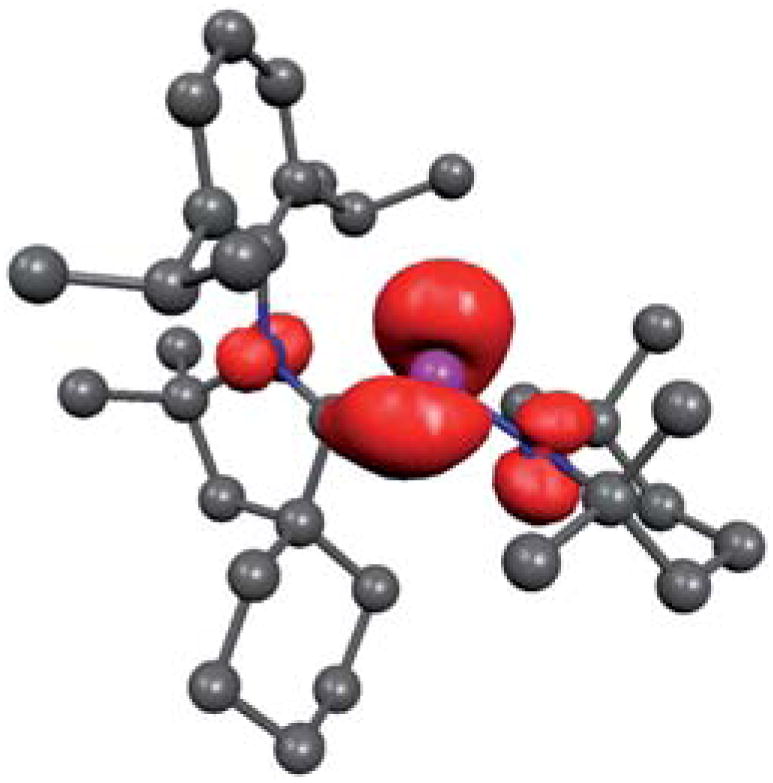
Calculated spin density for 35.
Radical cation 35 can also be regarded as a R2N–P+• unit stabilized by a carbene; in other words as a carbene–phospheniumyl adduct. Along this line, it has been shown that singlet carbenes are also capable of stabilizing P2+• fragments (Fig. 13). One-electron oxidation of bis(carbene)-P2 adducts 17a and 17b afforded the corresponding radical cations 17a+• and 17b+•, which have been fully characterized, including X-ray diffraction studies.87 According to calculations and EPR spectroscopy, the spin density of 17a+• is distributed between the phosphorous atoms (0.27 e at each P) and the nitrogen atoms (0.14 e at each N) of the cAAC ligands. In contrast, the unpaired electron in 17b+• is nearly exclusively localized at phosphorous (0.33 e and 0.44 e) with less than 0.07 e for any other atoms.
Fig. 13.

Cyclic voltammograms of 17a and 17b.
In this review, there are numerous examples, showing that although the donor properties of cAACs and NHCs are comparable, the former are by far more electron accepting. The comparison of the cyclic voltammograms of 17a and 17b provides another striking illustration (Fig. 13). For the cAAC adduct 17a, a reversible one-electron oxidation at E1/2 = −0.536 V vs. Fc+/Fc is observed. In contrast, for the NHC adduct 17b, two reversible one-electron oxidations can be seen. The first oxidation occurs at a much lower potential (E1/2 = −1.408 V vs. Fc+/Fc) than 17a, and the second at E1/2 = −0.178 V vs. Fc+/Fc; consequently, even the P22+ bisNHC adduct 36b can be prepared and isolated (Fig. 14).
Fig. 14.
P22+-bisNHC adduct.
Conclusions
Beginning as laboratory curiosities at the end of the 20th century,88 stable carbenes first found applications as ligands for transition metal based catalysts.89 It was also quickly discovered that stable carbenes are excellent catalysts in their own right due to their Lewis basicity.90 This perspective summarizes some novel applications of these fascinating species, and it is extremely likely that many others will soon be discovered. For example, there is already one publication demonstrating that stable carbenes can themselves induce organometallic transformations; in other words, a carbon-based catalyst can promote metal–metal bond formation.91 Will carbon centers compete with or even surpass transition metals for applications the latter are known for?
Acknowledgments
We are grateful to NSF (CHE-0808825 and -0924410), NIH (R01 GM 68825), DOE (DE-FG02-09ER16069), and RHODIA Inc. for financial support of our work. G. B. is grateful to his dedicated co-workers who are co-authors of the papers cited in this review.
Biographies

David Martin was born in 1976 at Clermont-Ferrand, France. After graduating from the Ecole Normale Supérieure (Lyon, France) in 2000, he joined Pr. G. Bertrand and Dr A. Baceiredo at the Université Paul Sabatier (Toulouse, France). He completed his PhD in 2004 and moved to Switzerland as a postdoctoral assistant in the group of Pr. A. Alexakis (University of Geneva). In 2006 he became CNRS research associate and joined Pr. G. Buono at the University of Marseille (France). He is currently a member of the UCR/CNRS Joint Research Laboratory at the University of California, Riverside.

Michele Soleilhavoup studied chemistry at the University Paul Sabatier in Toulouse and received her PhD in 1993 under the supervision of Guy Bertrand. From 1993 to 1995, she worked for BASF AG at Ludwigshafen. In 1995, she moved to the University Paris VI as a ≪Chargée de Recherche CNRS≫, and from 2000 to 2001, she worked in the Remi Chauvin’s group at the Laboratoire de Chimie de Coordination in Toulouse, before joining the UCR/CNRS Joint Research Laboratory at the University of California, Riverside. Her current research interests are focused on carbene chemistry and their application as tunable ligands for transition metal catalysts.

Guy Bertrand was an undergraduate in Montpellier, and obtained his PhD from the University of Toulouse. From 1988 to 1998 he was a “Director of Research” at the Laboratoire de Chimie de Coordination du CNRS, and from 1998 to 2005 the Director of the Laboratoire d’Hétérochimie Fondamentale et Appliquée at the University Paul Sabatier (Toulouse). Since 2001 he has been Distinguished Professor and Director of the UCR/CNRS Joint Research Chemistry Laboratory at the University of California at Riverside. His research spans a wide range of topics at the border between organic and inorganic chemistry. He is a member of the French Academy of Sciences.
Notes and references
- 1.(a) Lyon EJ, Shima S, Buurman G, Chowdhuri S, Batschauer A, Steinbach K, Thauer RK. Eur J Biochem. 2004;271:195–204. doi: 10.1046/j.1432-1033.2003.03920.x. [DOI] [PubMed] [Google Scholar]; (b) Pilak O, Mamat B, Vogt S, Hagemeier CH, Thauer RK, Shima S, Vonrhein C, Warkentin E, Ermler U. J Mol Biol. 2006;358:798–809. doi: 10.1016/j.jmb.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 2.Lavallo V, Mafhouz J, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. J Am Chem Soc. 2004;126:8670–8671. doi: 10.1021/ja047503f. [DOI] [PubMed] [Google Scholar]
- 3.(a) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2005;44:5705–5709. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jazzar R, Dewhurst RD, Bourg JB, Donnadieu B, Canac Y, Bertrand G. Angew Chem, Int Ed. 2007;46:2899–2902. doi: 10.1002/anie.200605083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zeng X, Frey GD, Kinjo R, Donnadieu B, Bertrand G. J Am Chem Soc. 2009;131:8690–8696. doi: 10.1021/ja902051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Angew Chem, Int Ed. 2006;45:3488–3491. doi: 10.1002/anie.200600987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Sander W, Bucher G, Wierlacher S. Chem Rev. 1993;93:1583–1621. [Google Scholar]; (b) Sander W, Hubert R, Kraka E, Grafenstein J, Cremer D. Chem–Eur J. 2000;6:4567–4579. doi: 10.1002/1521-3765(20001215)6:24<4567::aid-chem4567>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]; (c) Visser P, Zuhse R, Wong MW, Wentrup C. J Am Chem Soc. 1996;118:12598–12602. [Google Scholar]; (d) Ammann JR, Subramanian R, Sheridan RS. J Am Chem Soc. 1992;114:7592–7594. [Google Scholar]
- 6.(a) Arrington CA, Petty JT, Payne SE, Haskins WCK. J Am Chem Soc. 1988;110:6240–6241. doi: 10.1021/ja00226a046. [DOI] [PubMed] [Google Scholar]; (b) Bornemann H, Sander W. J Organomet Chem. 2002;641:156–164. [Google Scholar]
- 7.Moss RA, Platz MS, Jones M Jr, editors. Reactive Intermediate Chemistry. Wiley; New York: 2004. [Google Scholar]
- 8.Lyashchuk SN, Skrypnik YG. Tetrahedron Lett. 1994;35:5271–5274. [Google Scholar]
- 9.Dixon DA, Arduengo AJ, III, Dobbs KD, Khasnis DV. Tetrahedron Lett. 1995;36:645–648. [Google Scholar]
- 10.Frey GD, Lavallo V, Donnadieu B, Schoeller WW, Bertrand G. Science. 2007;316:439–441. doi: 10.1126/science.1141474. [DOI] [PubMed] [Google Scholar]
- 11.Hudnall TW, Bielawski CW. J Am Chem Soc. 2009;131:16039–16041. doi: 10.1021/ja907481w. [DOI] [PubMed] [Google Scholar]
- 12.Wang XP, Zhu ZL, Peng Y, Lei H, Fettinger JC, Power PP. J Am Chem Soc. 2009;131:6912–6913. doi: 10.1021/ja9017286. [DOI] [PubMed] [Google Scholar]
- 13.See also: Power PP. Nature. 2010;463:171–177. doi: 10.1038/nature08634.
- 14.(a) Himmel HJ. Dalton Trans. 2003:3639–3649. [Google Scholar]; (b) Zhu Z, Wang X, Peng Y, Lei H, Fettinger JC, Rivard E, Power PP. Angew Chem, Int Ed. 2009;48:2031–2034. doi: 10.1002/anie.200805982. [DOI] [PubMed] [Google Scholar]
- 15.Tomioka H. In: Reactive Intermediate Chemistry. Moss RA, Platz MS, Jones M Jr, editors. Wiley; New York: 2004. pp. 375–461. [Google Scholar]
- 16.Bettinger HF, Filthaus M, Neuhaus P. Chem Commun. 2009:2186–2188. doi: 10.1039/b817270f. [DOI] [PubMed] [Google Scholar]
- 17.Kotting C, Sander W. J Am Chem Soc. 1999;121:8891–8897. [Google Scholar]
- 18.(a) Spikes GH, Fettinger JC, Power PP. J Am Chem Soc. 2005;127:12232–12233. doi: 10.1021/ja053247a. [DOI] [PubMed] [Google Scholar]; (b) Peng Y, Brynda M, Ellis BD, Fettinger JC, Rivard E, Power PP. Chem Commun. 2008:6042–6044. doi: 10.1039/b813442a. [DOI] [PubMed] [Google Scholar]
- 19.Li BJ, Xu Z. J Am Chem Soc. 2009;131:16380–16382. doi: 10.1021/ja9061097. [DOI] [PubMed] [Google Scholar]
- 20.Welch GC, San Juan RR, Masuda JD, Stephan DW. Science. 2006;314:1124–1126. doi: 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- 21.Results related to FLP have recently been summarized: Stephan DW, Erker G. Angew Chem Int Ed. 2010;49:46–76. doi: 10.1002/anie.200903708.Stephan DW. Dalton Trans. 2009:3129–3136. doi: 10.1039/b819621d.
- 22.(a) Chase PA, Stephan DW. Angew Chem, Int Ed. 2008;47:7433–7437. doi: 10.1002/anie.200802596. [DOI] [PubMed] [Google Scholar]; (b) Chase PA, Gille AL, Gilbert TM, Stephan DW. Dalton Trans. 2009:7179–7188. doi: 10.1039/b908737k. [DOI] [PubMed] [Google Scholar]; (c) Holschumacher D, Taouss C, Bannenberg T, Hrib CG, Daniliuc CG, Jones PG, Tamm M. Dalton Trans. 2009:6927–6929. doi: 10.1039/b908074k. [DOI] [PubMed] [Google Scholar]; (d) Holschumacher D, Bannenberg T, Hrib CG, Jones PG, Tamm M. Angew Chem, Int Ed. 2008;47:7428–7432. doi: 10.1002/anie.200802705. [DOI] [PubMed] [Google Scholar]
- 23.Denk MK, Rodezno JM, Gupta S, Lough AJ. J Organomet Chem. 2001;617:242–253. [Google Scholar]
- 24.Rullich M, Tonner R, Frenking G. New J Chem. 2010;34:1760–1773. [Google Scholar]
- 25.Hudnall TW, Moerdyk JP, Bielawski CW. Chem Commun. 2010;46:4288–4290. doi: 10.1039/c0cc00638f. [DOI] [PubMed] [Google Scholar]
- 26.(a) Kubas GJ, Ryan RR, Swanson BI, Vergamini PJ, Wasserman HJ. J Am Chem Soc. 1984;106:451–452. [Google Scholar]; (b) Kubas GJ. Adv Inorg Chem. 2004;56:127–177. [Google Scholar]; (c) McGrady GS, Guilera G. Chem Soc Rev. 2003;32:383–392. doi: 10.1039/b207999m. [DOI] [PubMed] [Google Scholar]; (d) Crabtree RH. Acc Chem Res. 1990;23:95–101. [Google Scholar]
- 27.(a) Peng Y, Guo JD, Ellis BD, Zhu Z, Fettinger JC, Nagase S, Power PP. J Am Chem Soc. 2009;131:16272–16282. doi: 10.1021/ja9068408. [DOI] [PubMed] [Google Scholar]; (b) Peng Y, Ellis BD, Wang X, Power PP. J Am Chem Soc. 2008;130:12268–12269. doi: 10.1021/ja805358u. [DOI] [PubMed] [Google Scholar]
- 28.Possible mechanisms of activation reactions of H2 with a variety of acyclic and cyclic silylenes and germylenes have been investigated using the density functional theory: Wang Y, Ma J. J Organomet Chem. 2009;694:2567–2575.
- 29.van der Vlugt JI. Chem Soc Rev. 2010;39:2302–2322. doi: 10.1039/b925794m. [DOI] [PubMed] [Google Scholar]
- 30.Herrmann WA, Elison M, Fischer J, Köcher C, Artus GRJ. Chem–Eur J. 1996;2:772–780. [Google Scholar]
- 31.Driess M, Yao S, Brym M, van Wullen C, Lentz D. J Am Chem Soc. 2006;128:9628–9629. doi: 10.1021/ja062928i. [DOI] [PubMed] [Google Scholar]
- 32.(a) Jana A, Schulzke C, Roesky HW. J Am Chem Soc. 2009;131:4600–4601. doi: 10.1021/ja900880z. [DOI] [PubMed] [Google Scholar]; (b) Meltzer A, Inoue S, Prasang C, Driess M. J Am Chem Soc. 2010;132:3038–3046. doi: 10.1021/ja910305p. [DOI] [PubMed] [Google Scholar]
- 33.Driess M, Yao S, Brym M, van Wullen C. Angew Chem, Int Ed. 2006;45:4349–4352. doi: 10.1002/anie.200600237. [DOI] [PubMed] [Google Scholar]
- 34.Jana A, Objartel I, Roesky HW, Stalke D. Inorg Chem. 2009;48:798–800. doi: 10.1021/ic801964u. [DOI] [PubMed] [Google Scholar]
- 35.For reviews on P4 activation see for example: Scheer M, Balazs G, Seitz A. Chem Rev. 2010;110:4236–4256. doi: 10.1021/cr100010e.Cossairt BM, Piro NA, Cummins CC. Chem Rev. 2010;110:4164–4177. doi: 10.1021/cr9003709.Caporali M, Gonsalvi L, Rossin A, Peruzzini M. Chem Rev. 2010;110:4178–4235. doi: 10.1021/cr900349u.Peruzzini M, Gonsalvi L, Romerosa A. Chem Soc Rev. 2005;34:1038–1047. doi: 10.1039/b510917e.Cummins CC. Angew Chem, Int Ed. 2006;45:862–870. doi: 10.1002/anie.200503327.
- 36.Haaf M, Schmiedl A, Schmedake TA, Powell DR, Millevolte AJ, Denk M, West R. J Am Chem Soc. 1998;120:12714–12719. [Google Scholar]
- 37.Xiong Y, Yao S, Brym M, Driess M. Angew Chem, Int Ed. 2007;46:4511–4513. doi: 10.1002/anie.200701203. [DOI] [PubMed] [Google Scholar]
- 38.The reaction of silylenes with P4 has also bee studied computationally: Schoeller WW. Phys Chem Chem Phys. 2009;11:5273–5280. doi: 10.1039/b818396a.Damrauer R, Pusede SE. Organometallics. 2009;28:1289–1294.
- 39.Peng Y, Fan H, Zhu H, Roesky HW, Magull J, Hughes CE. Angew Chem, Int Ed. 2004;43:3443–3445. doi: 10.1002/anie.200353406. [DOI] [PubMed] [Google Scholar]
- 40.Holthausen MH, Weigand JJ. J Am Chem Soc. 2009;131:14210–14211. doi: 10.1021/ja906878q. [DOI] [PubMed] [Google Scholar]
- 41.Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2007;46:7052–7055. doi: 10.1002/anie.200703055. [DOI] [PubMed] [Google Scholar]
- 42.The reaction of P4 with singlet and triplet methylene has also been computationally studied: Damrauer R, Pusede SE, Staton GM. Organometallics. 2008;27:3399–3402.
- 43.See for examples: Yakhvarov D, Barbaro P, Gonsalvi L, Carpio SM, Midollini S, Orlandini A, Peruzzini M, Sinyashin O, Zanobini F. Angew Chem, Int Ed. 2006;45:4182–4185. doi: 10.1002/anie.200601048.Barbaro P, Ienco A, Mealli C, Peruzzini M, Scherer OJ, Schmitt G, Vizza F, Wolmershaeuser G. Chem–Eur J. 2003;9:5195–5210. doi: 10.1002/chem.200305091.
- 44.Back O, Kuchenbeiser G, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2009;48:5530–5533. doi: 10.1002/anie.200902344. [DOI] [PubMed] [Google Scholar]
- 45.It has even been shown that P1- and P2-niobium complexes prepared from P4 can be used as phosphorus transfer agents. Cossairt BM, Diawara MC, Cummins CC. Science. 2009;323:602–602. doi: 10.1126/science.1168260.Cossairt BM, Cummins CC. Angew Chem, Int Ed. 2008;47:8863–8866. doi: 10.1002/anie.200803971.Piro NA, Cummins J Am Chem Soc. 2008;130:9524–9535. doi: 10.1021/ja802080m.Fox AR, Clough CR, Piro NA, Cummins CC. Angew Chem, Int Ed. 2007;46:973–976. doi: 10.1002/anie.200604736.Piro NA, Figueroa JS, McKellar JT, Cummins CC. Science. 2006;313:1276–1279. doi: 10.1126/science.1129630.
- 46.(a) Lavallo V, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. Science. 2006;312:722–724. doi: 10.1126/science.1126675. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lavallo V, Ishida Y, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2006;45:6652–6655. doi: 10.1002/anie.200602701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Another stable cyclopropenylidene is known: Holschumacher D, Hrib CG, Jones PG, Tamm M. Chem Commun. 2007:3661–3663. doi: 10.1039/b706708a.
- 48.(a) Schmidpeter A, Lochschmidt S, Willhalm A. Angew Chem, Int Ed Engl. 1983;22:545–546. [Google Scholar]; (b) Ellis BD, Dyker CA, Decken A, Macdonald CLB. Chem Commun. 2005:1965–1967. doi: 10.1039/b500692a. [DOI] [PubMed] [Google Scholar]
- 49.(a) Ehses M, Romerosa A, Peruzzini M. Topics in Current Chemistry. Vol. 220. Springer-Verlag; Berlin Heidelberg: 2002. pp. 107–140. [Google Scholar]; (b) Peruzzini M, Abdreimova RR, Budnikova Y, Romerosa A, Scherer OJ, Sitzmann H. J Organomet Chem. 2004;689:4319–4331. [Google Scholar]
- 50.Arduengo AJ, III, Krafczyk R, Schmutzler R, Craig HA, Goerlich JR, Marshall WJ, Unverzagt M. Tetrahedron. 1999;55:14523–14534. [Google Scholar]
- 51.Masuda JD, Schoeller WW, Donnadieu B, Bertrand G. J Am Chem Soc. 2007;129:14180–14181. doi: 10.1021/ja077296u. [DOI] [PubMed] [Google Scholar]
- 52.Scherer OJ, Berg G, Wolmershauser G. Chem Ber. 1996;129:53–58. [Google Scholar]
- 53.Dyker CA, Bertrand G. Science. 2008;321:1050–1051. doi: 10.1126/science.1162926. [DOI] [PubMed] [Google Scholar]
- 54.Wang Y, Xie Y, Wei P, King RB, Schaefer HF, III, Schleyer PvR, Robinson GH. J Am Chem Soc. 2008;130:14970–14971. doi: 10.1021/ja807828t. [DOI] [PubMed] [Google Scholar]
- 55.(a) Kinjo R, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2010;49:5930–5933. doi: 10.1002/anie.201002889. [DOI] [PubMed] [Google Scholar]; (b) Weber L. Angew Chem, Int Ed. 2010;49:5829–5830. doi: 10.1002/anie.201003479. [DOI] [PubMed] [Google Scholar]
- 56.Abraham MY, Wang Y, Xie Y, Wei P, Schaefer HF, III, Schleyer PvR, Robinson GH. Chem–Eur J. 2010;16:432–435. doi: 10.1002/chem.200902840. [DOI] [PubMed] [Google Scholar]
- 57.Wang Y, Xie Y, Wei P, King RB, Schaefer HF, Schleyer PvR, Robinson GH. Science. 2008;321:1069–1071. doi: 10.1126/science.1160768. [DOI] [PubMed] [Google Scholar]
- 58.Fischer RC, Power PP. Chem Rev. 2010;110:3877–3923. doi: 10.1021/cr100133q. [DOI] [PubMed] [Google Scholar]
- 59.For a more detailed computational study, see: Liu Z. J Phys Chem A. 2009;113:6410–6414. doi: 10.1021/jp902308d.
- 60.Sidiropoulos A, Jones C, Stasch A, Klein S, Frenking G. Angew Chem Int Ed. 2009;48:9701–9704. doi: 10.1002/anie.200905495. [DOI] [PubMed] [Google Scholar]
- 61.Shevlin PB. In: Reactive Intermediate Chemistry. Moss RA, Platz MS, Jones M Jr , editors. Wiley; New York: 2004. pp. 463–500. [Google Scholar]
- 62.Tonner R, Öxler F, Neumüller B, Petz W, Frenking G. Angew Chem, Int Ed. 2006;45:8038–8042. doi: 10.1002/anie.200602552. [DOI] [PubMed] [Google Scholar]
- 63.(a) Ramirez F, Desai NB, Hansen B, McKelvie N. J Am Chem Soc. 1961;83:3539–3540. [Google Scholar]; (b) Schmidbaur H. Angew Chem, Int Ed Engl. 1983;22:907–927. [Google Scholar]
- 64.Tonner R, Frenking G. Angew Chem, Int Ed. 2007;46:8695–8698. doi: 10.1002/anie.200701632. [DOI] [PubMed] [Google Scholar]
- 65.(a) Tonner R, Frenking G. Chem–Eur J. 2008;14:3260–3272. doi: 10.1002/chem.200701390. [DOI] [PubMed] [Google Scholar]; (b) Tonner R, Frenking G. Pure Appl Chem. 2009;81:597–614. [Google Scholar]; (c) Kaufhold O, Hahn FE. Angew Chem, Int Ed. 2008;47:4057–4061. doi: 10.1002/anie.200800846. [DOI] [PubMed] [Google Scholar]
- 66.Dyker CA, Lavallo V, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2008;47:3206–3209. doi: 10.1002/anie.200705620. [DOI] [PubMed] [Google Scholar]
- 67.Tonner R, Frenking G. Organometallics. 2009;28:3901–3905. [Google Scholar]
- 68.(a) Fürstner A, Alcarazo M, Goddard R, Lehmann CW. Angew Chem, Int Ed. 2008;47:3210–3214. doi: 10.1002/anie.200705798. [DOI] [PubMed] [Google Scholar]; (b) Alcarazo M, Lehmann CW, Anoop A, Thiel W, Fürstner A. Nat Chem. 2009;1:295–301. doi: 10.1038/nchem.248. [DOI] [PubMed] [Google Scholar]; (c) Dyker A, Bertrand G. Nat Chem. 2009;1:265–266. doi: 10.1038/nchem.265. [DOI] [PubMed] [Google Scholar]
- 69.(a) Lavallo V, Dyker CA, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2008;47:5411–5414. doi: 10.1002/anie.200801176. [DOI] [PubMed] [Google Scholar]; (b) Lavallo V, Dyker CA, Donnadieu B, Bertrand G. Angew Chem, Int Ed. 2009;48:1540–1542. [Google Scholar]; (c) Fernández I, Dyker CA, DeHope A, Donnadieu B, Frenking G, Bertrand G. J Am Chem Soc. 2009;131:11875–11881. doi: 10.1021/ja903396e. [DOI] [PubMed] [Google Scholar]; (d) Hänninen MM, Peuronen A, Tuononen HM. Chem–Eur J. 2009;15:7287–7291. doi: 10.1002/chem.200900928. [DOI] [PubMed] [Google Scholar]
- 70.Melaimi M, Parameswaran P, Donnadieu B, Frenking G, Bertrand G. Angew Chem, Int Ed. 2009;48:4792–4795. doi: 10.1002/anie.200901117. [DOI] [PubMed] [Google Scholar]
- 71.(a) Tonner R, Heydenrych G, Frenking G. Chem Phys Chem. 2008;9:1474–1481. doi: 10.1002/cphc.200800208. [DOI] [PubMed] [Google Scholar]; (b) Tonner R, Frenking G. Chem–Eur J. 2008;14:3273–3289. doi: 10.1002/chem.200701392. [DOI] [PubMed] [Google Scholar]
- 72.(a) Schmidbaur H, Sherbaum F, Huber B, Müller G. Angew Chem, Int Ed Engl. 1988;27:419–421. [Google Scholar]; (b) Schmidbaur H, Gasser O. Angew Chem, Int Ed Engl. 1976;15:502–503. [Google Scholar]; (c) Vicente J, Singhal AR, Jones PG. Organometallics. 2002;21:5887–590. [Google Scholar]; (d) Romeo I, Bardaji M, Gimeno MC, Laguna M. Polyhedron. 2000;19:1837–1841. [Google Scholar]; (e) Petz W, Oxler F, Neumuller B, Tonner R, Frenking G. Eur J Inorg Chem. 2009:4507–4517. [Google Scholar]
- 73.(a) Takagi N, Shimizu T, Frenking G. Chem–Eur J. 2009;15:8593–8604. doi: 10.1002/chem.200901401. [DOI] [PubMed] [Google Scholar]; (b) Takagi N, Shimizu T, Frenking G. Chem–Eur J. 2009;15:3448–3456. doi: 10.1002/chem.200802739. [DOI] [PubMed] [Google Scholar]; (c) Kosa M, Karni M, Apeloig Y. J Chem Theory Comput. 2006;2:956–964. doi: 10.1021/ct050154a. [DOI] [PubMed] [Google Scholar]; (d) Pinter B, Olasz A, Petrov K, Veszpremi T. Organometallics. 2007;26:3677–3683. [Google Scholar]
- 74.(a) Wiberg N, Lerner HW, Vasicht SK, Wagner S, Karaghiosoff K, Noth H, Ponikwar W. Eur J Inorg Chem. 1999:1211–1218. [Google Scholar]; (b) Ishida S, Iwamoto T, Kabuto C, Kira M. Nature. 2003;421:725–727. doi: 10.1038/nature01380. [DOI] [PubMed] [Google Scholar]; (c) Iwamoto T, Masuda H, Kabuto C, Kira M. Organometallics. 2005;24:197–199. [Google Scholar]; (d) Iwamoto T, Abe T, Kabuto C, Kira M. Chem Commun. 2005:5190–5192. doi: 10.1039/b509878e. [DOI] [PubMed] [Google Scholar]; (e) Iwamoto T, Abe T, Ishida S, Kabuto C, Kira M. J Organomet Chem. 2007;692:263–270. [Google Scholar]; (f) Kira M, Iwamoto T, Ishida S, Masuda H, Abe T, Kabuto C. J Am Chem Soc. 2009;131:17135–17144. doi: 10.1021/ja904525a. [DOI] [PubMed] [Google Scholar]; (g) Kira M. Chem Commun. 2010;46:2893–2903. doi: 10.1039/c002806a. [DOI] [PubMed] [Google Scholar]
- 75.(a) Ueng SH, Makhlouf Brahmi M, Derat E, Fensterbank L, Lacôte E, Malacria M, Curran DP. J Am Chem Soc. 2008;130:10082–10083. doi: 10.1021/ja804150k. [DOI] [PubMed] [Google Scholar]; (b) Ueng SH, Fensterbank L, Lacôte E, Malacria M, Curran DP. Org Lett. 2010;12:3002–3005. doi: 10.1021/ol101015m. [DOI] [PubMed] [Google Scholar]
- 76.(a) Tehfe MA, Makhlouf Brahmi M, Fouassier JP, Curran DP, Malacria M, Fensterbank L, Lacote E, Lalevée J. Macromolecules. 2010;43:2261–2267. [Google Scholar]; (b) Lalevée J, Blanchard N, Chany AC, Tehfe MA, Allonas X, Fouassier JP. J Phys Org Chem. 2009;10:986–993. [Google Scholar]
- 77.Ueng S-H, Solovyev A, Yuan X, Geib SJ, Fensterbank L, Lacôte E, Malacria M, Newcomb M, Walton JC, Curran DP. J Am Chem Soc. 2009;131:11256–11262. doi: 10.1021/ja904103x. [DOI] [PubMed] [Google Scholar]
- 78.Solovyev A, Ueng S-H, Monot J, Fensterbank L, Malacria M, Lacôte E, Curran DP. Org Lett. 2010;12:2998–3001. doi: 10.1021/ol101014q. [DOI] [PubMed] [Google Scholar]
- 79.Rablen PR. J Am Chem Soc. 1997;119:8350–8360. [Google Scholar]
- 80.Walton JC, Makhlouf Brahmi M, Fensterbank L, Lacôte E, Malacria M, Chu Q, Ueng S-H, Solovyev A, Curran DP. J Am Chem Soc. 2010;132:2350–2358. doi: 10.1021/ja909502q. [DOI] [PubMed] [Google Scholar]
- 81.For reviews on boron-containing radicals, see: Power PP. Chem Rev. 2003;103:789–809. doi: 10.1021/cr020406p.Lee VY, Nakamoto M, Sekiguchi A. Chem Lett. 2008;37:128–133.
- 82.For recent examples of stable boron-containing radicals, see: Wood TK, Piers WE, Keay BA, Parvez M. Chem Commun. 2009:5147–5149. doi: 10.1039/b911982e.Chiu CW, Gabbai FP. Angew Chem, Int Ed. 2007;46:1723–1725. doi: 10.1002/anie.200604119.Tuononen HM, Chivers T, Armstrong A, Fedorchuk C, Boere RT. J Organomet Chem. 2007;692:2705–2715.Venkatasubbaiah K, Zakharov LN, Kassel WS, Rheingold AL, Jakle F. Angew Chem, Int Ed. 2005;44:5428–5433. doi: 10.1002/anie.200502148.Emslie DJH, Piers WE, Parvez M. Angew Chem, Int Ed. 2003;42:1252–1255. doi: 10.1002/anie.200390320.Scheschkewitz D, Amii H, Gornitzka H, Schoeller WW, Bourissou D, Bertrand G. Science. 2002;295:1880–1881. doi: 10.1126/science.1068167.
- 83.Matsumoto T, Gabbai FP. Organometallics. 2009;28:4252–4253. [Google Scholar]
- 84.(a) Scheer M, Kuntz C, Stubenhofer M, Linseis M, Winter RF, Sierka M. Angew Chem, Int Ed. 2009;48:2600–2604. doi: 10.1002/anie.200805892. [DOI] [PubMed] [Google Scholar]; (b) Ito S, Kikuchi M, Yoshifuji M, Arduengo AJ, III, Konovalova TA, Kispert LD. Angew Chem, Int Ed. 2006;45:4341–4345. doi: 10.1002/anie.200600950. [DOI] [PubMed] [Google Scholar]; (c) Agarwal P, Piro NA, Meyer K, Muller P, Cummins CC. Angew Chem, Int Ed. 2007;46:3111–3114. doi: 10.1002/anie.200700059. [DOI] [PubMed] [Google Scholar]; (d) Armstrong A, Chivers T, Parvez M, Boere RT. Angew Chem, Int Ed. 2004;43:502–505. doi: 10.1002/anie.200353108. [DOI] [PubMed] [Google Scholar]
- 85.(a) Hinchley SL, Morrison CA, Rankin DWH, Macdonald CLB, Wiacek RJ, Cowley AH, Lappert MF, Gundersen G, Clyburne JAC, Power PP. Chem Commun. 2000:2045–2046. doi: 10.1021/ja010615b. [DOI] [PubMed] [Google Scholar]; (b) Hinchley SL, Morrison CA, Rankin DWH, Macdonald CLB, Wiacek RJ, Voigt A, Cowley AH, Lappert MF, Gundersen G, Clyburne JAC, Power PP. J Am Chem Soc. 2001;123:9045–9053. doi: 10.1021/ja010615b. [DOI] [PubMed] [Google Scholar]; (c) Bezombes JP, Borisenko KB, Hitchcock PB, Lappert MF, Nycz JE, Rankin DWH, Robertson HE. Dalton Trans. 2004:1980–1988. doi: 10.1039/b402926g. [DOI] [PubMed] [Google Scholar]
- 86.Back O, Celik MA, Frenking G, Melaimi M, Donnadieu B, Bertrand G. J Am Chem Soc. 2010;132:10262–10263. doi: 10.1021/ja1046846. [DOI] [PubMed] [Google Scholar]
- 87.Back O, Donnadieu B, Parameswaran P, Frenking G, Bertrand G. Nat Chem. 2010;2:369–373. doi: 10.1038/nchem.617. [DOI] [PubMed] [Google Scholar]
- 88.For the discovery of the first stable carbenes, see: Igau A, Grützmacher H, Baceiredo A, Bertrand G. J Am Chem Soc. 1988;110:6463–6466.Igau A, Baceiredo A, Trinquier G, Bertrand G. Angew Chem, Int Ed Engl. 1989;28:621–622.Arduengo AJ, III, Harlow RL, Kline M. J Am Chem Soc. 1991;113:361–363.Arduengo AJ, III, Goerlich JR, Marshall WJ. J Am Chem Soc. 1995;117:11027–11028.Enders D, Breuer K, Raabe G, Runsink J, Teles JH, Melder JP, Ebel K, Brode S. Angew Chem, Int Ed Engl. 1995;34:1021–1023.Hahn FE, Wittenbecher L, Boese R, Bläser D. Chem–Eur J. 1999;5:1931–1935.
- 89.Arduengo AJ, III, Bertrand G. Chem Rev. 2009;109:3209–3884. doi: 10.1021/cr900241h. Special issue dedicated to carbenes. [DOI] [PubMed] [Google Scholar]
- 90.(a) Enders D, Niemeier O, Henseler A. Chem Rev. 2007;107:5606–5655. doi: 10.1021/cr068372z. [DOI] [PubMed] [Google Scholar]; (b) Marion N, Diez-Gonzalez S, Nolan SP. Angew Chem, Int Ed. 2007;46:2988–3000. doi: 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]; (c) Kamber NE, Jeong W, Waymouth RM, Pratt RC, Lohmeijer BGG, Hedrick JL. Chem Rev. 2007;107:5813–5840. doi: 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- 91.Lavallo V, Grubbs RH. Science. 2009;326:559–562. doi: 10.1126/science.1178919. [DOI] [PMC free article] [PubMed] [Google Scholar]