

Abstract
5-thyminyl-5,6-dihydrothymine (commonly called spore photoproduct or SP) is the exclusive DNA photo-damage product in bacterial endospores. It is generated in the bacterial sporulation phase and repaired by a radical SAM enzyme, spore photoproduct lyase (SPL), at the early germination phase. SPL utilizes a special [4Fe-4S] cluster to reductively cleave S-adenosylmethionine (SAM) to generate a reactive 5′-dA radical. The 5′-dA radical is proposed to abstract one of the two H atoms at the C6 carbon of SP to initiate the repair process. Via organic synthesis and DNA photochemistry, we selectively labeled the 6-HproS or 6-HproR position with a deuterium in a dinucleotide SP TpT substrate. Monitoring the deuterium migration in enzyme catalysis (employing Bacillus subtilis SPL) revealed that it is the 6-HproR atom of SP that is abstracted by the 5′-dA radical. Surprisingly, the abstracted deuterium was not returned to the resulting TpT after enzymatic catalysis, an H atom from the aqueous buffer was incorporated into TpT instead. This result questions the currently hypothesized SPL mechanism which excludes the involvement of protein residue(s) in SPL reaction, suggesting that some protein residue(s), which is capable of exchanging a proton with the aqueous buffer, is involved in the enzyme catalysis. Moreover, evidence has been obtained for a possible SAM regeneration after each catalytic cycle; however, such a regeneration process is more complex than currently thought, with one or even more protein residues involved as well. These observations have enabled us to propose a modified reaction mechanism for this intriguing DNA repair enzyme.

Introduction
Spore-forming bacteria are responsible for a number of serious diseases in humans, including botulism (Clostridium botulinum), gas gangrene and food poisoning (Clostridium perfringens), tetanus (Clostridium tetani), and anthrax (Bacillus anthracis).1 At the sporulation phase, these bacteria are extremely resistant to normal sterilization means such as heat, oxidizing chemicals, and UV or gamma irradiation. For instance, some Bacillus spores can be 100-fold more resistant to UV light than the corresponding vegetative cells.2,3 As spores contain a minimum amount of proteins and a complete genomic DNA, the ability to protect their DNA against external damage is suggested to be the key for their survival. The spore DNA is bound by a group of DNA binding proteins named small acid-soluble proteins (SASPs) that block the access of toxic chemicals to the genomic DNA.4 Furthermore, this SASP-DNA interaction changes the DNA photoreaction pattern. Instead of generating pyrimidine cyclobutane dimers, the spore DNA photoreaction produces a different dimer, 5-thyminyl-5,6-dihydrothymine (commonly called spore photoproduct or SP) as the exclusive UV damage product. Spores express a specific enzyme, spore photoproduct lyase (SPL), to effectively reverse the SP dimer at the early germination phase, allowing resumption of their normal life cycle (Scheme 1). This unique DNA photochemistry and the associated efficient damage repair in spores are suggested to be the keys for their extreme UV resistance; however the reaction mechanism of both processes is not well-understood.5-13
Scheme 1.
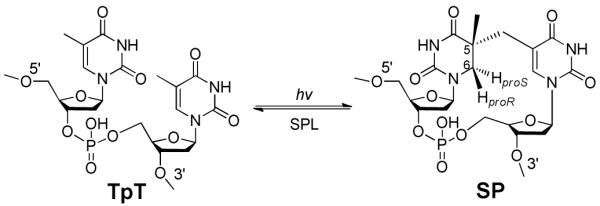
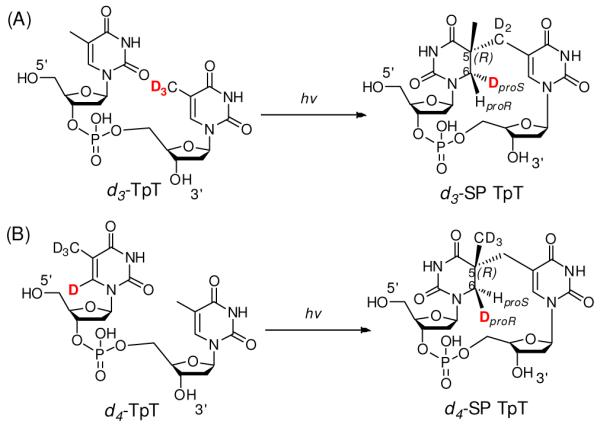
SP was first discovered about half a century ago;14 the mechanism regarding its photo-formation was largely unclear.5,15 As shown in Scheme 1, as a result of SP formation, the SP C5 carbon becomes chiral, and the C6 carbon becomes pro-chiral, possessing two hydrogen atoms with unknown origins. We hypothesized that one of the C6 hydrogen atoms is transferred from the methyl group of the 3′-T, while the other one is retained from the original thymine at that position. To test this assumption, we synthesized two TpT isotopologs: a d3-TpT dinucleotide containing a CD3 moiety in the 3′-T and a d4-TpT dinucleotide with the four carbon-bound protiums in the 5′-T replaced by deuteriums (Scheme 2). After UV radiation, a deuterium was transferred exclusively to the 6-HproS position when d3-TpT was used as the starting material (Scheme 2A). The migrated atom was a protium when d4-TpT was employed (Scheme 2B). These observations, together with the previous NMR study which determined the R configuration of the C5 carbon at the 5′-T,16 enabled us to propose a new mechanism for the SP photoreaction.17
Scheme 2.
SP is repaired by the spore photoproduct lyase (SPL) enzyme at the early bacterial germination phase.5,18,19 SPL is a member of the so-called radical SAM superfamily, which was defined by the characteristic CXXXCXXC motif,20 although recent evidence suggests that other three-cysteine motifs still facilitate the same radical chemistry.18,21,22 The three cysteine residues serve as ligands for each of three irons in the [4Fe-4S] cluster, with the fourth iron being coordinated by the S-adenosylmethionine (SAM) in a bi-dentate manner, with its amino and carboxylate moieties serving as the fourth and fifth ligands to the cluster.23 The cluster at its +1 oxidation state donates an electron to SAM to cleave its C-S bond, generating a 5′-deoxyadenosyl radical (5′-dA•) This 5′-dA radical catalyzes highly diverse biochemical reactions in animals, plants and microorganisms, including steps in metabolism, DNA/RNA modification, and the biosynthesis of vitamins, coenzymes and many antibiotics.18-22,24-36
SPL catalyzes SP repair using a direct reversal strategy via a radical mechanism. The currently hypothesized SPL mechanism is shown in Scheme 3: the 5′-dA radical generated via SAM reductive cleavage first abstracts one of the two hydrogen atoms on the C6 carbon of SP to form a C6 radical and 5′-dA. This C6 radical fragments into one thymine and a thymine radical, which takes the hydrogen back from 5′-dA to yield the repaired T (The thymine methyl radical in Scheme 3 is likely to delocalize to the aromatic ring; the current drawing is a simplified model to facilitate discussion). Such a hypothesis was first suggested by Begley and Mehl based on a modeling study.12 By attaching a phenylthiol moiety at the C6 position of a bipyrimidine analog, clean formation of dimethylthymine was observed under radical generating conditions, indicating that SPL initiates the SP repair by abstracting one of the 6-H atoms. Broderick et.al. later tritiated thymine at either the CH3 (at C5) or the C6 position and introduced the labeled thymine into a pUC18 vector.11 After UV radiation to generate SP in the plasmid DNA, the SP containing plasmid was treated with SPL. Tritium was found to be incorporated into SAM when the C6 position of thymine was tritiated. In contrast, no tritium was found in SAM when the CH3 group was labeled. Such observations supported a proposal that the SPL abstracts the H-atom from the C6 carbon to initiate the SP repair. The abstracted H-atom ended up in SAM, leading to a hypothesis that SAM is regenerated after each reaction cycle, as shown in Scheme 3.
Scheme 3.

A careful analysis of this experimental approach reveals that it was based on the assumption that the 6-H atom from thymine remains at that position after SP formation, which is proved correct from our recent photochemical studies.17 However, we were intrigued by the fact that no tritium was detected in SAM when the thymine methyl group was tritiated as an H atom from the CH3 group of 3′-T migrates to the 6-HproS position of SP after the photoreaction, as illustrated by Scheme 2. Additionally, the C6 carbon in SP has two H atoms and is pro-chiral. Given that enzymatic reactions are usually highly stereoselective, then if the reaction is initiated by H-abstraction at C6, which H-atom is taken?
We therefore prepared several dinucleotide SP TpT substrates with the two 6-H atoms of SP selectively labeled via DNA photochemistry.17 Using the resulting TpT isotopologs to probe the SPL reaction, we are able not only to answer the questions raised above, but also demonstrate that the currently hypothesized SPL mechanism needs significant revision.
Experimental Section
Materials and Methods
Unless otherwise stated all solvents and chemicals used were of commercially available analytical grade. They were purchased from Sigma, Fisher, or VWR and used without further purification except the 3,5-bis(4-chlorobenzoyl)-2-deoxy-α-D-ribofuranosyl chloride (9), which was purchased from ShangHai Hanhong Chemical Co., Ltd at Shanghai, P. R. China. All reactions were carried out using oven or flame-dried glassware under a nitrogen atmosphere in distilled solvents. Dichloromethane and pyridine were distilled over calcium hydride. Purification of reaction products was carried out by flash chromatography using silica gel (Dynamic Adsorbents Inc, 32–63 μm). For TLC analysis, precoated plates (w/h F254, Dynamic Adsorbents Inc, 0.25 mm thick) were used. The 1H and 13C NMR spectra were obtained on a Bruker 500 MHz NMR Fourier transform spectrometer. NMR spectra were recorded in the following solvents: deuterated chloroform (CDCl3), with residual chloroform (δ 77.2 ppm for 13C NMR) and TMS (δ 0 ppm for 1H NMR), deuterated methanol (δ 3.31 ppm for 1H NMR and δ 49.1 ppm for 13C NMR) or deuterated methyl sulfoxide (DMSO-d6), with residual methyl sulfoxide (δ 2.50 ppm for 1H NMR and δ 39.5 ppm for 13C NMR) taken as the standard. The chemical shifts in NMR spectra were reported in parts per million (ppm). Mass spectrometry (MS) analysis was obtained using Agilent 1100 series LC/MSD system with Electrospray Ionization (ESI). The TpT photoreaction was carried out using a Spectroline germicidal UV sterilizing lamp (Dual-tube, 15 w, intensity: 1550 uw/cm2) with samples ~5 cm from the lamp.
All DNA-modifying enzymes and reagents were purchased from Fermentas Life Sciences (Glen Burnie, MD). B. subtilis strain 168 chromosomal DNA was purchased from the ATCC (ATCC 23857D). Oligonucleotide primers were obtained from Integrated DNA Technologies (Coralville, IA). E. Coli BL21(DE3) and expression vector pET-28a were purchased from Novagen (Madison, WI). The construct containing the SPL gene was co-expressed with plasmid pDB1282, which was a generous gift from Prof. Squire Booker at the Pennsylvania State University. 5′-deoxyadenosine (5′-dA) and S-adenosylmethionine (SAM) was purchased from Aldrich and used directly without further purification. 5 Prime Perfectpro* Nickel nitrilotriacetic acid (Ni-NTA) resin was purchased from Fisher Scientific. All other buffers and chemicals were of the highest grade available.
The protein purification as well as the enzyme reactions were carried out under an inert atmosphere using a Coylab anaerobic chamber (Grass Lake, MI) with the H2 concentration around 3%. Product analysis was conducted using a Waters (Milford, MA) breeze HPLC system with a 2489 UV/Visible detector and an Agilent 1100 LC-MS system under both a positive and negative ionization mode. Sonic disruption of E. coli cells was conducted by a Fisher Scientific* Model 500 Digital Sonic Dismembrator. DNA sequencing was performed by the Indiana University Nucleic Acid Core Facility at IU School of Medicine.
Construction of the of SPL expression vector
The splB gene was cloned from the B. subtilis chromosomal DNA (strain 168) using the synthetic oligonucleotide primers 5′-GCAAGTGCGAATTCCAGAACCCATTTGTTCC-3′ (containing an EcoRI site) and 5′-GCAAGCTCAAGCTTTTAAGTGAAATATTCAAT-5′ (containing a HindIII site) and amplified by standard PCR techniques. The resulting PCR product was digested by EcoRI/HindIII and ligated into the same sites in pET-28a, which harbors an in-frame N-terminal hexahistidine tag to facilitate protein purification. The construct was transformed into E. coli 10 G chemically competent cells purchased from Lucigen Corporation (Middleton, WI) for isolation and amplification of the splB gene-containing plasmid DNA. The resulting vector was named SPL-pET28 and co-transformed with a pDB 1282 vector into E. coli BL21(DE3) obtained from Stratagene (La Jolla, CA) for protein overexpression. The pDB 1282 vector confers ampicillin resistance and harbors an E. coli operon that is involved in the biosynthesis of FeS clusters and is thus believed to facilitate incorporation of the FeS clusters into the apoprotein of SPL.21,37
Expression of SPL
A single colony of transformed cells was used to inoculate 5 mL of LB medium containing the appropriate antibiotics to maintain selection for the plasmid. The cultures were grown to saturation at 37 °C with vigorous shaking and then used to inoculate 1 L of antibiotics containing LB medium. Once the cells reached early log phase (OD600 ≈ 0.8), the temperature was decreased to 16 °C and the expression of the genes induced by addition of isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. The expression was allowed to proceed for 12 h and the cells harvested by centrifugation (5000 rpm for 15 min at 4 °C). The medium was decanted and the cell pellet stored at −20 °C.
Purification of SPL protein
To purify SPL, the cell pellet from 1 L of culture was resuspended in 30 mL of lysis buffer containing 25 mM Tris, 300 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol, 1 mM PMSF and 10% glycerol (pH 7.0). The cells were lysed by sonication on ice and centrifuged at 17000 rpm for 20 min at 4 °C. The supernatant was loaded onto a 1.5 mL Ni-NTA-agarose column and moved into a Coy anaerobic chamber. The rest of the protein purification steps were carried out under an inert atmosphere. The column was washed with 10 volumes of the buffer containing 50 mM Tris, 300 mM NaCl, 20 mM imidazole and 10% glycerol (pH 7.0). The tagged SPL protein was eluted with the same buffer but containing 250 mM imidazole. The eluted protein was yellow-brown colored and was determined to be essentially homogeneous by SDS-PAGE gel stained with Coomassie blue. The proteins were dialyzed three times against a buffer containing 25 mM Tris, 300 mM NaCl and 10% glycerol (pH 7.0). The protein was then aliquoted into 50 μl PCR tubes, flash-frozen in liquid N2 and stored at −80 °C.
Protein, iron, and sulfide assays
Routine determinations of protein concentrations were done by the method of Bradford,38 using bovine serum albumin and bovine gamma globulin as protein standards. Protein concentrations were calibrated on the basis of the absorption of aromatic residues at 280 nm in the presence of 6 M guanidine hydrochloride using the method of Gill and von Hippel.39 Iron content was determined using o-bathophenanthroline (OBP) under reductive conditions after digestion of the protein in 0.8% KMnO4 and 1.2 M HCl as described by Fish.40 Iron standards were prepared from commercially available ferric chloride and ferrous ammonium sulfate. Sulfide assays were carried out using the method described by Beinert.41
[4Fe-4S] cluster reconstitution
The as-isolated SPL (300 μL) was taken out of the −80 °C freezer and allowed to thaw inside the Coy chamber at ambient temperature. To this protein solution, 5 μL of 100 mM DTT was added followed by the addition of FeCl3 and Na2S to a final concentration of 100 M each. The SPL was then incubated anaerobically for 1 h at ambient temperature. In the presence of 10% glycerol, the protein was fairly stable and little precipitate was observed after these treatments. Under rare circumstances, centrifugation of the resulting protein solution was needed to remove the precipitated protein; otherwise, the protein can be used directly for the activity assays. For the samples prepared for iron and sulfur content analyses, desalting columns (Thermo Scientific Zeba Spin Desalting Column from Fisher Scientific) were applied to remove the un-reacted small inorganic ions before measurements were conducted.
Preparation of deuterated SP TpT
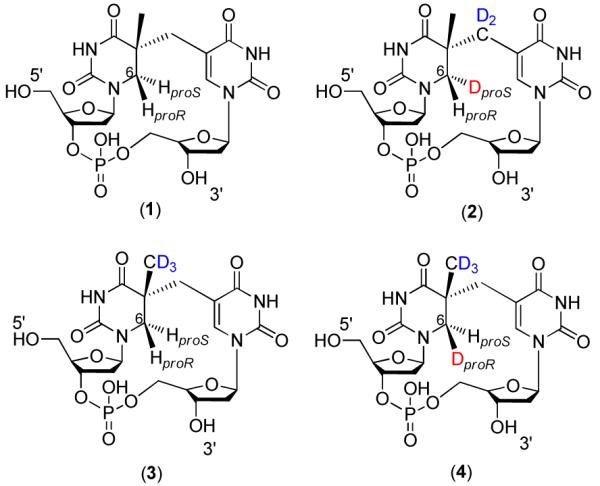
As shown in Chart 1, four different SP TpTs were prepared in this report with deuterium being selectively incorporated into different positions. SPs were prepared in two different ways: organic synthesis or DNA photochemistry.17,42 Among the four SPs shown in Chart 1, compound (1) was prepared via organic synthesis and compounds 2, 3 and 4 were prepared via photochemistry using correspondent deuterated TpT dinucleotides as the starting material. The experimental details in SP syntheses can be found in either the supporting information or our previous publication.17,43 The formed SP TpTs were isolated by a Waters semi-prep HPLC column, characterized via NMR and ESI-MS spectroscopes and kept for enzyme activity studies.
Chart 1.
SPL activity determination
The SPL protein after cluster re-constitution was used immediately to assay the SPL activity. Typically, a reaction mixture contained 30 M SPL, 3 mM correspondent SP TpT substrate, 300 μM SAM, and 2 mM DTT in a final volume of 400 μL of buffer containing 25 mM Tris-HCl, 300 mM NaCl and 10% glycerol at pH 7.0. Sodium dithionite (final concentration 1 mM) was added as a reductant to initiate the enzyme reaction. The reactions were carried out under anaerobic conditions at ambient temperature for various periods of time. Under the conditions of the assay, the formation of TpT was linear with time for up to 15 min. At each time point, 90 μL of the solution was taken out to an Eppendorf tube and quenched by 10 μL of 3 M HCl. Other quenching methods such as heating at 100 °C for 5 minutes or flash-freezing by liquid N2 were also tested and no difference on SPL activity was observed by these quenching means. After removing the protein residues via centrifugation at 15,000 rpm for 20 min, the resulting supernatants were loaded onto HPLC. The yielded TpT from each sample was integrated from HPLC chromatographs, calibrated and plotted against reaction time to generate the reaction rate.
SPL activity examined in the absence of external reductant
The SPL protein after cluster re-constitution as described above was reduced by 1 mM sodium dithionite for 30 min. Excess inorganic ions were removed via a Thermo Scientific Zeba Spin Desalting Column. SP TpT and SAM were then added to a final concentration of 3 mM and 100 M (~ 3-fold of SPL) respectively to initiate the SP repair reaction. At different time points, 50 μL of the solution was taken out and quenched by 5 μL of 3 M HCl and the resulting solution analyzed by HPLC.
SPL activity examined in D2O
To investigate the involvement of the water solvent in SPL reaction, the reconstituted SPL protein was exchanged into a D2O buffer containing 25 mM Tris-HCl, 300 mM NaCl and 5% d8-glycerol at pH 7.0 via an Amicon Ultra-0.5 mL Centrifugal Filter column (Millipore, MA). SP TpT (1), SAM and DTT were subsequently added from the stock solution prepared using the same D2O buffer and the reaction were quenched by HCl at various reaction times. The resulting solution was analyzed by LC-MS.
Solvent isotope effect determination
To minimize the impact of the handling process on the enzyme activity, the SPL enzyme was exchanged into a H2O buffer under identical conditions employed in D2O experiments. SP TpT (1), SAM and dithionite were subsequently added to initiate the SP repair reaction and the reaction quenched by HCl at various reaction times and analyzed via HPLC. The solvent isotope effect was determined by comparing the rate obtained in H2O with that determined in D2O.
Reaction isotope effect determination in D2O
The SPL enzyme was exchanged into D2O buffer as described above followed by addition of SAM and sodium dithionite. SP compound 1 or 4 was subsequently added and the reaction allowed proceeding for varying times, then quenched by HCl and analyzed by HPLC. The isotope effect was calculated via comparing the reaction rates determined with compound 1 and 4 as substrate respectively.
Examination of deuterium incorporation into SAM
The incorporation of deuterium from d4-SP TpT (4) into SAM was examined via a literature procedure with some modifications.26 Briefly, the assay employed 1 mM 4, 200 μM SAM, and 150 μM SPL in a 300 μL buffer which contained 25 mM Tris-HCl, 300 mM NaCl, 1 mM DTT and 10% glycerol at pH 7.0. The reaction was initiated by addition of dithionite to a final concentration of 1 mM and allowed to proceed at ambient temperature for 3 h. All substrate added was consumed at this point as proved by HPLC analysis, suggesting that the SPL enzyme catalyzed at least 6 turnovers. 100 μL of the reaction solution was taken out and quenched by one of several methods: addition of 10 μL 3 M HCl; addition of 10 μL 10% SDS; heating at 100 °C for 30 minutes. The SAM released by the former two methods was analyzed by LC-MS. Thermal decomposition of SAM by the last method leads to the formation of 5′-methylthioadenosine (MTA),44 which was confirmed by co-injection of an authentic sample bought from the Sigma-Aldrich Corporation with the SAM decomposition product. Any deuterium incorporated into SAM is expected to be retained in the resulting MTA, which was analyzed by LC-MS using the protocol developed previously in mechanistic studies of lysine 2,3-aminomutase (LAM).45,46
HPLC assay for product analysis
Chromatography was performed at room temperature with detection at 268 nm using an Agilent Zorbax reverse-phase C-18 column (3.5 μM, 4.6×50 mm). The column was equilibrated in 50 mM triethylammonium acetate (TEAA), pH 6.5, and compounds were eluted with an ascending gradient (0 – 25%) of buffer B which is composed of 50% buffer A and 50% acetonitrile at a flow rate of 1 mL/min. Under this gradient, SP TpT was eluted at 6.0 min, 5′-dA at 7.8 min and TpT at 10.2 min. The identity of the products was confirmed by co-injection of respective authentic samples as well as by LC-MS spectrometry. The area of the product peak was determined after subtraction of the baseline from the t = 0 chromatograph and the amounts of 5′-dA and TpT formed were determined by reference to standard curves constructed with authentic samples.
LC-MS assay for product analysis
Low resolution LC-MS analyses were conducted via an Agilent 6130 Quadrupole LC/MS spectrometer coupled to an Agilent 1100 series chromatography system. The high resolution LC-MS analyses were conducted via an Agilent 6520 Accurate Mass Q-TOF LC/MS coupled to an Agilent 1200 series capillary chromatography system. The high resolution mass data analysis and formula assignments were conducted using Agilent MassHunter software. Due to potential contamination to the LC/MS system, the TEAA buffer system used above was aborted and instead a 0.1% formic acid in H2O was utilized as solvent A and 0.1% formic acid in acetonitrile as solvent B. The Waters X-bridge™ OST C18 column (2.5 μM, 4.6×50 mm) was used for the low resolution LC-MS analysis. The column was equilibrated in solvent A, and compounds were eluted with an ascending gradient (0 – 17%) of solvent B at a flow rate of 1 mL/min in 17 min. Under this gradient, SAM was eluted at 2.1 min, SP TpT at 5.5 min, 5′-dA at 7.1 min and TpT at 8.2 min. The mass signals in positive and negative ion mode were monitored simultaneously in the experiments. For the high resolution mass analysis, an Agilent Poroshell 300SB C18 (5 μM, 1.0×75 mm) column with a flow rate of 0.25mL/min using a gradient of 0 to 30% solvent B over 20 minutes was used and only mass signals under the negative ion mode were monitored.
UV-visible spectroscopy
UV–visible spectra were recorded using UV-Mini 1240 Spectrophotometer in combination with the associated data manager software package. UV-visible spectroscopy was determined using the same buffer described above, which contained 25 mM Tris-HCl, 300 mM NaCl and 10% glycerol at pH 7.0. 300 μL SPL solution was transferred to a UV cuvette with a 1-cm length path (purchased from Fisher Scientific and modified by the glass shop at IU Bloomington) plugged with a rubber septum in the Coy chamber and the UV spectra taken outside the glovebox using a Shimadzu UV-MINI-1240 spectrometer. The cluster reduction was achieved by addition of sodium dithionite to a final concentration of 2 mM in the anaerobic chamber.
EPR experiments
Continuous wave (CW) EPR spectra were recorded on a modified Varian spectrometer at 35 GHz (“Q”-band) and 2 K.47 The as-isolated SPL (400 μM, 3.1 iron/protein) was reduced with 2 mM dithionite inside the anaerobic chamber for 60 min and placed into the EPR tube and immediately frozen in liquid N2. The sample remained frozen for EPR measurements. Under the experimental conditions employed here, which lead to “rapid-passage” effects,48 35 GHz EPR spectra are observed in the dispersion mode and appear as absorption lineshapes, rather than the standard absorption mode detection and first derivative presentation. Digital derivatives were taken to allow conventional presentation. EPR simulations were performed using the program QPOW,49 as modified by J. Telser.
Results
SPL protein expression and purification
The splB gene was cloned into a pET 28a vector which introduces of a hexahistidine tag to facilitate protein purification. Ni-NTA chromatography afforded SPL as a dark-brown solution when purified under a strictly inert atmosphere. The purity of the protein was checked by SDS-PAGE gel to be >95% and the purified SPL exhibited a single band at ~ 40 kDa (Figure 1). A typical yield for such a SPL purification process is ~40 mg protein per liter of LB media.
Figure 1.
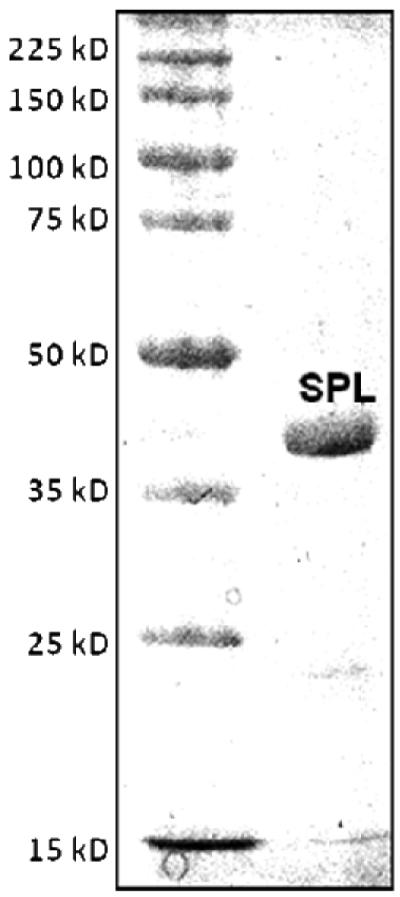
The SDS-PAGE gel of the SPL protein overexpressed from E. coli and purified via Ni-NTA chromatography.
Iron-sulfur content in SPL
The presence of the [4Fe-4S] cluster in SPL was confirmed by the iron-sulfur content analysis. The as-isolated SPL was found to contain 3.1 iron and 2.9 sulfur atoms per protein; these numbers increase to 3.8 iron and 3.7 sulfur atoms after cluster re-constitution. These numbers further support the conclusion that SPL harbors a [4Fe-4S] cluster in its holoenzyme form.
Spectroscopic characterization of purified SPL
The as-isolated SPL protein exhibited a typical UV absorption of a [4Fe-4S]2+ species with a shoulder at 420 nm, suggesting that the majority of the protein-harbored clusters survived during the protein purification process (Figure 2). The UV absorption at 420 nm increased slightly after cluster reconstitution (~10%), suggesting that more [4Fe-4S]2+ cluster was restored back into the protein. The absorption was bleached quickly upon addition of sodium dithionite due to the formation of the [4Fe-4S]1+ cluster (Figure 2). As expected, the reduced cluster destabilizes the protein; a prolonged (1 h) incubation at ambient temperature led to a partial precipitation of the SPL protein.50
Figure 2.

UV-visible absorption spectra of the [4Fe-4S] cluster in SPL at the as-isolated form (solid line) and the reduced form (dashed line). For both spectra, 50 M SPL protein was utilized in a 25 mM Tris buffer containing 300 mM NaCl, 10% glycerol, at pH 7.0. For the reduced protein, the reduction was conducted by 2 mM dithionite for 20 min before the spectrum was taken. The spectra were recorded in a UV cuvette with a 1 cm path length under anaerobic conditions at room temperature.
EPR characterization of the reduced cluster in SPL
After dithionite reduction, the resulting [4Fe-4S]1+ cluster in SPL is EPR active and exhibits an S = 1/2 signal that is similar to that of other members of the radical SAM superfamily, such as pyruvate formate lyase activating enzyme51 (Figure 3). Simulation yields g = [2.030(5), 1.930(5), 1.895(5)], which is equivalent to the g values reported for this center by other workers.7-9 Note that the greater field dispersion of 35 GHz EPR allows resolution of slight rhombic splitting that may not always be resolved in X-band (~9 GHz) studies.9
Figure 3.
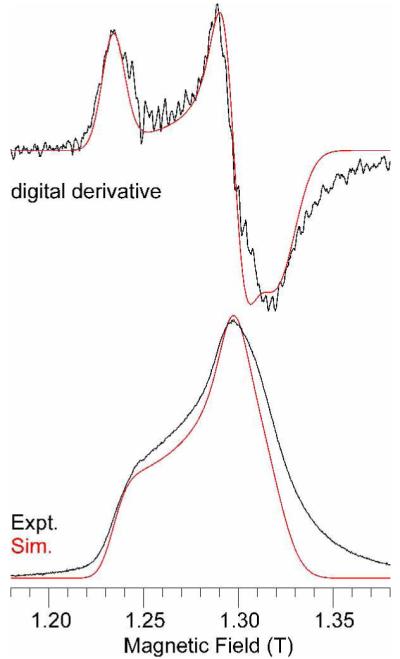
35 GHz EPR spectrum of dithionite-reduced SPL (~ 0.4 mM). Experimental spectrum (black trace): temperature, 2 K; microwave frequency, 35.063 GHz; microwave power, 100 μW (30 dBm); 100 kHz field modulation amplitude, 1.0 G; time constant, 8 ms; scan time, 30 s; average of 4 scans. Simulated spectrum (red trace): S = 1/2, g = [2.030, 1.930, 1.895]; single crystal Gaussian linewidths, W = [200, 180, 300] MHz (half-width at half-maximum). The lower pair of traces are present the absorption lineshape, as experimentally observed under “rapid passage” conditions; the upper pair of traces are digital first derivatives to provide a more familiar EPR presentation. The “tailing” seen at high field may be due to relaxation effects in passage and not to actual spectral intensity.
Quantitation of the EPR signal of reduced SPL is complicated by the nature of the rapid passage EPR phenomenon and the lack of a suitable standard. A true S = 1/2 system such as Cu(II), commonly used as a spin standard, has totally different relaxation behavior than an FeS cluster. The S = 1/2 ground states of [Fe2S2]+ and [Fe4S4]+ are the result of intricate spin coupling mechanisms that also result in many low lying spin excited states.52-55 We have therefore employed another FeS protein, which was available to us, for EPR intensity comparison with reduced SPL. The protein was Fd1 isolated from Aquifex aeolicus by Meyer and co-workers.56 The relaxation behavior of this protein, under identical experimental conditions, is similar to that of reduced SPL, as evidence by the microwave power dependence of their EPR spectra at 2 K.43 At the lowest microwave power for which signals could readily be observed (~10 W), where saturation effects are minimized, the integrated intensity of a 2 mM solution of A. aeolicus Fd1 was ten times that of a solution of reduced SPL. This would indicate that the concentration of SPL containing [4Fe-4S]+ cluster was ~200 M. Considering the facts that the protein concentration was 400 μM and the as-isolated SPL contained 3.1 Fe atoms/protein molecule, this data indicates that the 1 h reduction by dithionite resulted a reduced [4Fe-4S]+ cluster in ~ 70% of our SPL samples, contrasting to the 20% [4Fe-4S]+ cluster observed in PFL-AE.57 Taking into account the partially precipitated protein during the prolonged dithionite incubation, a close to quantitative reduction of the SPL cluster could be obtained if proper conditions are employed.
d3-SP TpT (2) repair by SPL
Under the currently hypothesized mechanism, H atom abstraction from either 6-HproR or 6-HproS position of SP by the 5′-dA radical (Figure 4, pathway A or B) leads to different reaction isotopolog products owing to the fact that only 6-HproS is labeled by a deuterium in 2. Therefore, analyzing the deuterium distribution in the resulting products should subsequently shed light on the SPL mechanism. 5′-dA was isolated and found to exhibit a [M + H]+ signal of 252.2 (calculated 252.15, Figure 5a), implying no deuterium incorporation. The isolated TpT was found to possess a [M − H]− signal of 548.2 (Figure 6a), corresponding to the formation of d3-TpT. These data suggest that reaction possibly occurred under the pathway A shown in Figure 4; no deuterium was abstracted by 5′-dA radical and all deuterium atoms in SP TpT were retained in the repaired TpT.
Figure 4.

Using d3-SP TpT (2) as the enzyme substrate to illustrate the expected reaction products according to the current SPL mechanism shown in Scheme 3. (A): The 5′-dA radical abstracts the protium at the 6-HproR position to initiate the reaction. All three deuterium atoms in compound 2 should be retained in the repair product TpT; thus d3-TpT as well as regular 5′-dA and SAM are expected after the reaction. (B): The 5′-dA radical abstracts the deuterium at the 6-HproS position to initiate the reaction. Correspondingly, a deuterium is incorporated into the resulting 5′-dA and d2/d3-TpT and d1/d0-SAM mixtures are expected by the end of the reaction due to the H or D abstraction from the methyl group of 5′-dA by the TpT radical in the H atom back donation step.
Figure 5.

(a) [M + H]+ signal of the 5′-dA isolated out of the SPL reaction with the d3-SP TpT (2) which contains a deuterium at the 6-HproS position as the enzyme substrate, the 252.1 signal suggests that no deuterium was incorporated into the 5′-dA. (b) [M + H]+ signal of the 5′-dA isolated with d4-SP TpT (4) which contains a deuterium at the 6-HproR position as the enzyme substrate. The observed 253.1 signal suggests that a deuterium atom was incorporated into the 5′-dA.
Figure 6.

(a) The [M − H]− signal of the TpT isolated in the d3-SP TpT (2) reaction, the detected 548.2 signal suggests that all three deuterium atoms in SP were transferred into the formed TpT. (b) The [M − H]− signal of the TpT isolated in the d4-SP TpT (4) reaction. The detected 548.2 signal suggests either a large isotope effect in the H atom back-donation step or a loss of one deuterium during the catalysis. (c) The high-resolution ESI-MS of the TpT generated from the enzyme repair of 4. The formation of a sole d3-TpT species was suggested by the detected isotopic peak distributions. (d) The [M − H]− signal of the TpT isolated with regular SP (1) as the enzyme substrate in H2O buffer. (e) The [M − H]− signal of the TpT isolated with 1 as substrate in 98% D2O buffer. The +1 MS signal observed in (e) relative to that in (d) clearly suggests the incorporation of a deuterium from the D2O buffer into the formed TpT.
d4-SP TpT (4) repair by SPL
Enzyme repair of d4-SP (4) resulted in a 5′-dA species which exhibited a major [M + H]+ signal at m/e = 253.2 (Figure 5b), implying that a deuterium was taken from 4 during the enzyme reaction. The isolated 5′-dA also exhibited a minor peak of 252.2 at roughly one fourth the intensity of the 253.2 signal in the mass spectroscopy, indicating presence of some non-deuterated 5′-dA. However, surprisingly the isolated TpT was found to possess a [M − H]− signal at m/e = 548.2 (Figure 6b), corresponding to the formation of d3-TpT. This result could be reached via two possibilities: (1) A similar reaction as shown in Figure 4 occurred here as well. However, a large isotope effect was involved in the H atom back-donation step between the 5′-dA and the TpT radical, leading to the formation of a small amount of d4-TpT and a majority of d3-TpT. The ESI-MS signal of d4-TpT was obscured by the second isotopic peak of the d3-TpT and thus escaped detection. (2) A different reaction occurred; the deuterium abstracted from 4 was not returned to TpT after enzyme catalysis.
TpT analysis by high resolution ESI-MS
To test if a large kinetic isotope effect (KIE) occurs in the H back-donation step, we re-determined the mass of the resulting TpT out of the 4 repair by high resolution mass spectroscopy. As shown in Figure 6c, the first two isotope peaks exhibit mass of 548.1482 and 549.1511 respectively, corresponding to the first two peaks of d3-TpT species (Calc: 548.1479 and 549.1509 respectively). According to the Agilent MassHunter software, such an isotopic distribution suggests the presence of a single d3-TpT species with a 99% certainty, and furthermore, the software was able to automatically calculate the correct formula of the d3-TpT species with an error of 0.6 ppm. No obvious mass peak was detected around m/e = 549.1536, the calculated mass for the first isotope peak of d4-TpT compound. Moreover, the observed abundance of the second isotope peak was 21.91% of the first one, which is only slightly lower than the theoretical value (23.81%) for a d3-TpT compound. If d4-TpT was produced and co-eluted with d3-TpT on HPLC, then the ratio between the second/first isotope peaks should be increased. We therefore conclude that it is very unlikely that there was any detectable d4-species present in the isolated d3-TpT.
SP TpT (1) repair by SPL in D2O buffer
If a large KIE is not involved, then the abstracted deuterium must be exchanged by a protium during the catalysis. As the most likely protium source in enzyme reaction is the aqueous buffer, we therefore tested if an H atom from the water was incorporated into the repaired TpT. The SP TpT (1) repair was conducted in a buffer containing ~ 98% of D2O. As expected, the isolated TpT was found to possess a [M − H]− signal at m/e = 546.2 (Figure 6e), corresponding to the formation of d1-TpT. In contrast, the reaction in H2O resulted regular TpT, with a [M − H]− signal at m/e = 545.2 (Figure 6d). This observation suggests that a proton from the original SP TpT was exchanged with solvent; thus the reaction pathway represented by Figure 4 was not correct and a solvent exchangeable proton must be involved in enzyme catalysis.
d3-SP TpT (3) repair by SPL
As shown in Chart 1, the differences between compounds 2 and 4 are: (1) A deuterium occupies the 6-HproS position in 2 and the 6-HproR position in compound 4. (2) Compound 2 possesses a CD2 linker and a CH3 moiety while 4 has a CH2 linker and a CD3 moiety. The repair of d4-SP TpT (4) suggests that a deuterium was abstracted by the 5′-dA radical during the SPL mediated repair. To exclude the possibility that the abstracted deuterium was from the CD3 moiety but not from the 6-HproR in 4, a d3-SP TpT (3) which possesses a CD3 moiety but two protium atoms on the C6 carbon (Chart 1) was utilized for the SPL reaction. After a 20-minute enzyme reaction, the LC-MS analyses of the products found that the isolated 5′-dA showed m/e = 252.2 (from [M + H]+) and the repaired TpT m/e = 548.2 (from [M − H]−), with the isotopic patterns identical to what were shown in Figure 5a and 6a respectively. This finding implies that, as expected, the methyl group did not participate in the enzyme reaction; all three deuterium atoms were therefore retained in the resulting TpT. Thus, the deuterium abstracted from 4 must be from the 6-HproR position. The fact that no deuterium abstraction from the 6-HproS position of 2 was observed also suggests that the SPL enzyme abstracts the H atom with an exclusive stereoselectivity.
Reaction rate and isotope effect determination
The SP repair rates with substrates 1, 2 and 4 were determined based on the appearance of TpT in the HPLC chromatographs as illustrated by Figure 7. The formation rate of 5′-dA was also calculated. The SPL reactions were quenched by HCl within the first 3 minutes for compound 1 and 2 and 10 minutes for compound 4 to ensure that both 5′-dA and TpT formation were within the linear region. The resulting 5′-dA and TpT peaks in the HPLC chromatographs were integrated and the integrations plotted against reaction time. Using the HPLC calibration curve generated with authentic 5′-dA and TpT, the formation rates of these two species were calculated and are shown in Table 1.
Figure 7.
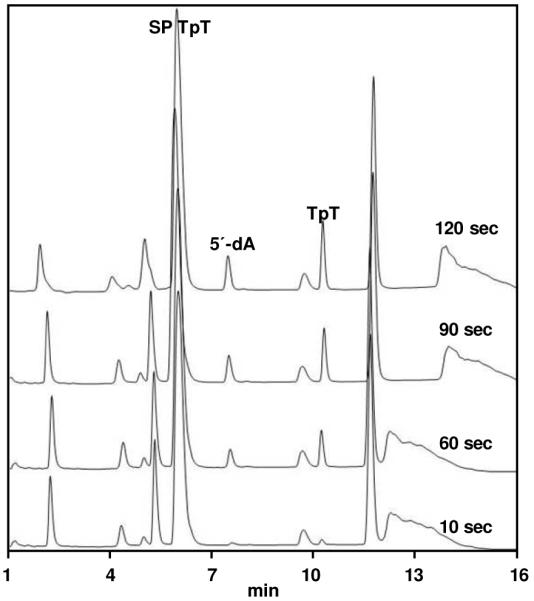
HPLC chromatograph of the SP TpT (1) repair mediated by SPL in the first two minutes of reaction. SP TpT was eluted at 6.0 min, 5′-dA at 7.8 min, and TpT at 10.2 min.
Table 1. The formation rates of 5′-dA and TpT in SPL catalyzed reaction.
| Substrate used | SP TpT (1) | d3-SP TpT (2) | d4-SP TpT (4) |
|---|---|---|---|
| TpT formation rate in H2O buffer (min−1) | 0.35 ± 0.03 | 0.35 ± 0.03 | 0.12 ± 0.01 |
| 5′-dA formation rate (min−1) in H2O buffer | 0.20 ± 0.02 | 0.20 ± 0.02 | 0.09 ± 0.01 |
| TpT formation rate in D2O buffer (min−1) | 0.34 ± 0.03 | N.D. | 0.12 ± 0.01 |
N.D.: not determined
The Km for SP TpT is found to be below 30 μM although the value was not accurately determined due to the slow reaction rate and the instability of the enzyme during the elongated reactions. The Km for SP TpT was reported to be 6 μM by Fontecave and coworkers,8 which is in reasonable agreement with our finding. To ensure that the rates reported in Table 1 reflect the Vmax, the substrate concentration was maintained at ~100-fold of the enzyme concentration and at least 100-fold of Km. Doubling substrate concentration or decreasing its concentration by half did not change the reaction rates, suggesting that the enzyme was saturated and the rates determined truly represented the Vmax.
Examination of Table 1 indicates that both TpT and 5′-dA were formed at almost identical rates regardless of whether 1 or 2 (d3-SP TpT) was used as the enzyme substrate, suggesting that protium abstraction occurred in both cases. Such an observation again suggests that it is the protium at the 6-HproR position of 2 that was abstracted by SPL to initiate the repair reaction. The repair rate of 2 was thus not examined in D2O reaction as a nearly identical rate to that of 1 is expected. Both TpT and 5′-dA were found to form at decreased reaction rates when d4-SP TpT (4) was utilized as the enzyme substrate. Comparing the rates of TpT formation yields a primary KIE of 2.9 ± 0.3 (Figure 8). The formation of 5′-dA was also slowed when 4 was used; however care has to be taken before any conclusion can be drawn due to two reasons: (1) The 5′-dA is a d1/d0 mixture as indicated by the presence of 252.1 peak in Figure 5b; (2) The 5′-dA observed is likely to be a reaction intermediate; its formation rate does not reflect the property of the overall enzyme reaction (see discussion below).
Figure 8.

Determination of the Vmax based on the formation of TpT (A) and 5′-dA (B) in SPL catalyzed dinucleotide SP TpT repair. Enzyme reaction rates were calculated via plotting the amount of TpT isolated versus reaction time. The primary kinetic isotope effect (KIE) for SPL reaction was determined to be 2.9 ± 0.3 by comparing the rates of TpT formation shown in plot A.
Changing the buffer from H2O to D2O has little effect on the reaction rates, as shown in Table 1, suggesting no significant solvent isotope effect is associated with the SPL reaction. Furthermore, the primary KIE was unchanged in D2O as well. Contrasting to the d3-TpT species ([M − H]− signal at 548.2) obtained from the d4-SP TpT (4) repair in H2O buffer, LC-MS analysis of the yielded TpT in D2O reaction revealed a dominant [M − H]− signal at 549.2, suggesting a solvent deuterium was incorporated into the TpT, making it a d4 species.43 This observation complements the previous results in the repair of 1 in D2O buffer, confirming the involvement of an exchangeable proton in SPL catalysis.
SPL reaction in the absence of excess reductant
The observed 1:1.7 ratio between 5′-dA and TpT formed during the first two minutes of 1 repair seems to suggest that SAM plays a catalytic role, with SAM regenerated after each catalytic cycle.58 As suggested by Scheme 3, regeneration of SAM returns an electron to the iron-sulfur cluster, reducing it to the +1 oxidation state and making it ready for the next catalytic cycle. This rationale suggests that a SPL enzyme with an [Fe4S4]+ cluster should be able to catalyze multiple turnovers, similar to what was observed for lysine 2,3-aminomutase (LAM).59 We therefore reduced the iron-sulfur cluster in SPL by dithionite for 0.5 hr and removed the excess reductant via a desalting column. Upon addition of 2-3 times of SAM and substrate 1, the pre-reduced SPL remained active for 3 hrs, catalyzing more than 10 turnovers (Figure 9A). This observation is in line with that found in the previous SPL studies,11 suggesting that SAM is truly catalytic. The pre-reduced enzyme is only slightly less active than that in the presence of excess dithionite especially in the first 15 minutes of the reaction; a similar observation was also made in LAM studies.59
Figure 9.

(A) Formation of TpT during a 3-hr SP repair reaction.  Reaction was conducted under normal conditions: After cluster reconstitution, the reaction was initiated by 1 mM dithionite.
Reaction was conducted under normal conditions: After cluster reconstitution, the reaction was initiated by 1 mM dithionite.  After cluster reconstitution, the enzyme was pre-reduced by 1 mM dithionite for 0.5 hr. The excess inorganic ions were then removed by a desalting treatment. Dithionite was re-supplemented to the resulting protein solution and the reaction initiated by addition of SAM and SP.
After cluster reconstitution, the enzyme was pre-reduced by 1 mM dithionite for 0.5 hr. The excess inorganic ions were then removed by a desalting treatment. Dithionite was re-supplemented to the resulting protein solution and the reaction initiated by addition of SAM and SP.  After cluster reconstitution, cluster pre-reduction by dithionite and desalting treatment, SP and SAM were added to start the reaction. (B) 5′-dA formation observed in the acid-quenched SPL reaction at various time. The reaction was carried out under normal conditions as described above.
After cluster reconstitution, cluster pre-reduction by dithionite and desalting treatment, SP and SAM were added to start the reaction. (B) 5′-dA formation observed in the acid-quenched SPL reaction at various time. The reaction was carried out under normal conditions as described above.
5′-dA generation
As shown in Figures 7 and 8, a linear formation of 5′-dA was observed in the first two minutes of the reaction. A prolonged reaction however revealed that the 5′-dA formation gradually slowed down and reached its maximum at about 15 minutes (Figure 9B). Surprisingly, after 15 minutes, the amount of 5′-dA observed in the acid-quenched reaction gradually decreased and fell into the basal level in ~ 1 hr. In radical SAM enzymes, 5′-dA is often observed as a side product from the abortive SAM cleavage reaction.10,13,60,61 Such a product should accumulate in the solution, resulting in an increase in the amount of 5′-dA observed during the course of the reaction. The observed increase-first-followed-by-decrease pattern in Figure 9B is highly unusual in radical SAM reactions, suggesting that the 5′-dA observed is not the abortive product, but a true reaction intermediate. The fact that TpT formation remains linear in the first 15 minutes of the reaction followed by a gradual slow-down, with only a very small amount of TpT being formed after 1 hr, suggests that the formation of 5′-dA and TpT are concerted processes, further supporting this conclusion.
Furthermore, a lack of 5′-dA formation was often observed when DTT was omitted in SPL reaction; formation of TpT was however unaffected. This observation further supports the conclusion that SAM is possibly regenerated in SPL reaction. It also appears to suggest that the presence of DTT may slow down this regeneration process, enabling trapping of 5′-dA as a reaction intermediate. However, the SAM regeneration and/or 5′-dA formation seem to be controlled by other factors as well, which we do not fully understand at this point. For instance, the protein preparation seems also to affect the amount of 5′-dA generated in the reaction; protein purified in air generated much more 5′-dA than protein purified anaerobically. Further work is needed to decipher this intriguing SAM regeneration process.
No deuterium incorporation into SAM
Although SAM seems regenerated after each SPL catalytic cycle, LC-MS analysis of the isolated SAM exhibits a MS signal at m/e = 399.2 (calculated 399.15) regardless of the SP TpT substrates employed.43 Although 5′-dA radical is proved to abstract the deuterium from the 6-HproR position of 4, after 6 enzyme turnovers in the presence of 1.5-fold of SAM, there was still no obvious deuterium incorporation into SAM. This result contradicts an observation in the previous tritium labeling experiment.11 Interestingly in LAM where SAM also appears regenerated after each catalytic cycle, no deuterium transfer from lysine to SAM was observed either,45 although a more sensitive tritium transfer experiment revealed ~ 1 - 6% of tritium incorporation from the lysine substrate into SAM when large excess of SAM was adopted.46
Discussion
Protein expression and purification
It is known that co-expression of iron-sulfur proteins with a pDB1282 plasmid which encodes a set of E. coli proteins involved in iron-sulfur cluster biogenesis increases the yield of the soluble protein by facilitating cluster incorporation into the newly synthesized apoprotein.21,37,62 The incorporated cluster serves as a structural scaffold to help the protein fold correctly, therefore increasing the yield of the soluble portion during expression. The pDB1282 was therefore introduced into SPL expression and the yield of soluble SPL at least doubled.43 The resulting SPL exhibits the highest activity among the SPL enzymes purified to date when dinucleotide SP is used as substrate,6,8 highlighting the importance of cluster incorporation to the enzyme activity.
Substrate preparation and enzyme activity assays
The dinucleotide SP TpT substrate can be prepared via organic synthesis as shown by Begley and co-workers.42 Among the four SP TpTs shown in Chart 1, two of them, compounds 1 and 3 can be readily synthesized via this route. However, such a method is not suitable to synthesize the selectively labeled compounds 2 and 4, as the synthesis includes a hydrogenation step which is catalyzed by Rh/Al2O3, resulting in the addition of H2 to the C=C bond from either side of the thymine plane. Thus, if D2 is used in hydrogenation, the resulting SP will be a mixture of two configurational isomers with deuterium occupying either the 6-HproR or 6-HproS position. Such a mixture cannot be separated via chromatographic means, and thus cannot provide any useful mechanistic information.
In contrast, photoreaction has proved a reliable means to prepare SP substrate with exclusive stereoselectivity.17 Due to the nature of this process, SP can be generated only via a solid phase reaction which restricts the thermal motion and conformational change of TpT so that only thymine residues adopting the “right” conformation dimerize, while the vast majority of excited molecules are thermally quenched.63 Thus deuterated SP is obtained at only 0.1 – 0.4 % yields depending on whether a primary deuterium isotope effect is encountered in photoreaction. However, despite the low yields, the stereoselectively labeled SP compounds 2 and 4 can be readily produced by this photochemical means.
Although our purified SPL enzyme exhibits the highest activity (0.35 ± 0.03 min−1) with 1 as the enzyme substrate,6,8 the reaction is still slow probably due to the weak binding affinity of this minimally sized dinucleotide substrate to the enzyme. In contrast, with a SP-containing GGTTGG hexamer or an SP-containing plasmid DNA, the SP repair rate increased 50 to 500 times due to the improved substrate binding.7,11 Our SPL reaction rate is also comparable with other known radical SAM enzymes characterized to date.26,37,64,65
SPL mechanism - H atom abstraction by 5′-dA radical
The reductive cleavage of SAM generates a 5′-dA radical, which abstracts an 6-H atom from SP to form 5′-dA. Analysis of the 5′-dA isotopomers/isotopologs that result from use of selectively labeled substrates 2 and 4 provides the most direct evidence to reveal which of the C6 H-atoms is abstracted during the reaction.
As shown in Figure 5, when 2, which contains a deuterium at the 6-HproS position, was used for the SPL reaction, a protium was abstracted by the 5′-dA radical. In contrast, the abstracted atom was a deuterium when 4, which contains a deuterium at the 6-HproR position, was used. The fact that a protium was again abstracted when 3, which contains a CD3 group but two C6 protiums, was employed firmly supports the proposal that the abstracted H atom is from the C6 carbon. Taken together, these results clearly demonstrate that SPL abstracts the 6-HproR atom, leaving a C6 radical behind to initiate the SP repair process.
The equation shown in Scheme 1 is often used to illustrate the reactions of SP formation and repair in literature.8,9 Such an equation seems to imply that the SP repair catalyzed by SPL is a simple reverse process of SP photo-formation. Our observations prove that this assumption is overly simplistic. As described in Scheme 2, an H atom from the CH3 moiety of the 3′-T migrates to the 6-HproS position in the SP formation and the original H atom at the C6 of 5′-T remains on that carbon, becoming the 6-HproR atom of SP.17 In contrast, the migrated atom becomes the 6-HproR during the enzyme repair; while 6-HproS subsequently becomes the C6 proton in the repaired thymine.
Such a conclusion also rationalizes part of the observations previously made by Broderick and co-workers.11 In their experiments, tritium was incorporated into either the methyl or the C6 carbon of thymine. Our photochemical studies show that labeling the –CH3 moiety of thymine would result in tritium incorporation into the 6-HproS position of SP; while labeling the thymine C6 carbon would incorporate tritium into the 6-HproR position. The SPL enzyme abstracts the 6-HproR atom, leaving behind the 6-HproS atom at the C6 carbon of repaired 5′-thymine. Therefore, in the previous study, although the SP 6-HproS could bear a tritium originating from the –CH3 moiety of thymine before the photoreaction, that atom was not abstracted and had no chance to enter the catalytic cycle of SP repair. In contrast, the tritium at the thymine C6 carbon migrated to the 6-HproR position of SP, and was then abstracted by the 5′-dA radical to initiate the SP repair process. Thus, the stereoselectivity in H atom abstraction was reflected in Broderick’s earlier work as well; however it was not recognized due to a lack of full understanding of SP photochemistry at that time.
The primary KIE of 2.9 ± 0.3 exhibited by the SPL reaction suggests that the H atom abstraction mediated by the 5′-dA radical is involved in the enzyme rate-determining steps. The presence of this relatively large KIE further confirms the stereo configuration assignment that the abstracted H atom is from the 6-HproR position. As expected, the formation of 5′-dA was slowed down as well owing to this KIE when 4 was employed for SPL reaction.
SPL mechanism - H atom back donation
Comparing with the radical initiation step, our results on the H atom back-donation step are quite surprising. The current reaction mechanism predicts the H atom abstracted by the 5′-dA radical would be donated back to the repaired TpT, however the abstracted deuterium from 4 was not returned. Carefully analyzing the MS isotopic pattern of the resulting TpT excludes the possible involvement of large KIE; while the deuterium incorporation from the D2O buffer into TpT strongly suggests that the TpT radical takes an H atom from an unknown source which is able to exchange proton with the buffer.
Although these results are inconsistent with an earlier work,11 they do agree with an observation by Fontecave and co-workers.10 By employing a SAM molecule with a deuterated methylene moiety, they found no evidence for those deuterium atoms to be transferred into the resulting TpT. This result, together with ours, suggests that no direct H atom exchange occurs between the 5′-dA and TpT radical. Recently the cysteine 141 residue of the SPL from Bacillus strains has drawn much attention due to its potential involvement in SPL catalysis.10,66 Distinct from the cysteines in the CXXXCXXC SAM motif, the C141 residue does not participate in the [4Fe-4S] cluster formation and subsequent SAM redox chemistry. However, mutation of this residue to alanine in B. subtilis abolished the SPL activity, leading to cell death under UV radiation with a similar efficiency as mutations of cysteine residues involved in the SAM motif.66 Fontecave and co-workers revisited this mutation in an in vitro enzymology study and found that instead of forming TpT, the mutant yielded a TpT-SO2 molecule as the major repair product, which was generated through a reaction between the TpT radical and dithionite, the reductant added to reduce the [4Fe-4S]2+ cluster to initiate the SPL reaction.10 Although the role of C141 is not clearly defined to date, the formation of TpT-SO2 strongly indicates that this residue is associated with the H atom back-donation step of the enzymatic reaction.
Using the cysteine residue as the H atom donor (instead of the 5′-dA) is more reasonable considering the different bond strengths involved. A calculation predicts the C-H bond dissociation energy (BDE) of 5′-dA to be 12.2 kcal/mol higher than that for the methyl moiety of thymine (98.2 vs 86.0 kcal/mol); in contrast, the BDE of the S-H bond in cysteine is suggested to be around 82 kcal/mol;67,68 which is line with other computational results that indicate the allylic C-H bond to be ~3-4 kcal/mol stronger than the S-H bond in a thiol compound like methylthiol.69,70 It is therefore logical for SPL to use the cysteine S-H moiety instead of the C-H bond of 5′-dA as the direct H-atom donor, greatly lowering the energy barrier for this H-atom back-transfer step.
The involvement of the cysteine residue also solves the space problem raised by the current mechanism.67 The current mechanism requires 5′-dA to be associated with both H-atom transfer steps. The first step involves the 5′-dA radical and the 6-HproR on the 5′-thymine residue of SP. Should the resulting 5′-dA donate the H atom back to the thymine allyl radical at the 3′ end of the SP, it has to move down one nucleoside, roughly 3-4 Ǻ vertical distance. This movement requires a major protein conformational change, which is unfavorable in energy.67 In contrast, such a conformational change is no longer necessary should the cysteine residue serve as the H-atom donor; all the H atom transfer steps involved in catalysis can be readily accommodated within the protein framework.
The H atom back-transfer step from the cysteine to the thymine allyl radical is unlikely to be involved in the enzyme rate-limiting steps, as indicated by the absence of a solvent isotope effect. The unchanged KIE exhibited by SPL reaction in D2O buffer also supports this conclusion.
SPL mechanism – SAM regeneration
The results from the SAM regeneration step are also surprising. In the previous studies, the conclusion of SAM regeneration was drawn after the tritrium label was found to be transferred from the SP into SAM.11 However, examining the isolated SAM from SPL reaction with ~ 1.5-fold SAM and 6-fold deuterated substrate 4 found no apparent deuterium incorporation even after all the SP substrate was consumed. This result supports the hypothesis of SAM regeneration but SAM may not be regenerated via a simple one-step reaction as shown in Scheme 3.
In radical SAM enzymes, the ratio of 5′-dA to product is of significance. A 1 : 1 ratio implies SAM serves as a co-substrate as one molecule of substrate modified consumes one molecule of SAM, while a 1 : X ( X >> 1) ratio suggests SAM serves as a co-factor as only a catalytic amount is needed.58 As revealed in Figure 9B, the ratio between 5′-dA and TpT produced after a 2-hr reaction is greater than 1 : 500. This observation further supports a catalytic role for SAM, suggesting SAM to be regenerated at the end of each catalytic cycle.
Carefully examining the SPL reaction reveals that although 5′-dA was observed in the early stage of the reaction; these 5′-dA molecules were not accumulating in the solution. This observation suggests that the 5′-dA observed in the quenched reaction is actually a true reaction intermediate on the way to SAM regeneration, not the abortive product of the uncoupled SAM cleavage reaction as commonly observed in other radical SAM enzymes.10,13,60,61 Furthermore, the amount of 5′-dA observed appears to be coupled to the rate of TpT formation, further supporting our conclusion. The observed 5′-dA formation rate is always smaller than that of TpT formation, suggesting that the assumption of 5′-dA is the faster step in enzyme catalytic cycle. Therefore, these 5′-dA molecules are kinetically competent to serve as reaction intermediate as well.
When a thiol containing compound such as DTT is removed from the reaction system, the TpT formation seems to be relatively undisturbed, however the amount of 5′-dA observed often decreases into the basal level. The fact that 5′-dA formation can be disturbed by other processes such as protein purification clearly suggests that the 5′-dA formation/SAM regeneration is likely to be more complex than currently thought and more work is needed before this process is understood.
The putative H-atom back transfer from the C141 residue to the thymine methyl radical yields a thiyl radical, which thus must be involved in the SAM regeneration process. The simplest model to accommodate it is to insert this cysteine residue between the thymine allyl radical and 5′-dA in the reaction pathway. However, as rationalized above, the thiyl radical is more stable than the allylic radical in thymine, which makes the direct H-atom transfer from 5′-dA to the cysteine thiyl radical even more unfavorable thermodynamically. Even so, a similar reaction was proposed in class II ribonucleotide reductase (RNR),71 where the catalytic thiyl radical does abstract an H atom back from 5′-dA to regenerate a 5′-dA radical. This 5′-dA• subsequently recombines with the Cob(II)alamin species to regenerate the adenosylcobalamin. Such a reaction is suggested to proceed via a concerted mechanism: the un-favored H atom abstraction by the thiyl radical is coupled with the formation of a stable C-Co bond in adenosylcobalamin, providing an energy offset for the former process.71 As the SAM formation from 5′-dA and methionine is also heavily favored thermodynamically,72,73 coupling the H atom abstraction by the thiyl radical with the SAM regeneration process may provide a explanation for the SPL reaction as well.
Although it is possible for the single cysteine residue to regenerate SAM, if cysteine is the only protein residue involved, then one should expect production of a significant amount of abortive product 5′-dA under competition of DTT, should the reaction site be ready accessible to this small molecule. The fact that little abortive 5′-dA was generated in the presence of DTT suggests that the enzyme possesses some kind of “error-prevention” function. Such a function is difficult to achieve by a single cysteine and is more likely to be realized via the involvement of other amino acid residues. Taken together, our data suggests that there may be a cysteine-containing electron-transfer pathway involved in SAM regeneration, which possibility will be tested in future studies.
Newly Hypothesized SPL mechanism
All of these findings imply that the original mechanistic hypothesis for SPL11 needs to be refined by including protein residues in the enzyme catalysis. We therefore tentatively propose a modified mechanism as shown in Figure 10. After the reductive SAM cleavage reaction with an electron provided by the [4Fe-4S]1+ cluster, the resulting 5′-dA radical abstracts the 6-HproR atom to yield the SP radical on the C6 carbon. Subsequent fragmentation of SP leads to the formation of a delocalized methyl radical on the 3′-T. Instead of abstracting an H atom back from the 5′-dA, the TpT radical draws an H atom from an unidentified protein residue, with the C141 the most likely candidate, to yield the repaired TpT. The resulting thiyl radical on this cysteine could abstract an H atom back from 5′-dA; or it may oxidize its neighboring residue(s) to generate another protein radical species before it reacts with 5′-dA. Recombination of the 5′-dA radical and the methionine regenerates SAM at the end of each catalytic cycle.
Figure 10.

Newly proposed reaction mechanism for SPL catalyzed SP dimer repair. The 5′-dA radical generated from SAM reductive cleavage reaction takes the 6-HproR atom to yield a C6 radical on SP, the SP methylene bridge subsequently undergoes a hemolytic cleavage to give a thymine methyl radical. (Note: This allyl radical is likely to delocalize to the thymine aromatic ring; the current drawing as a methyl radical is a simplified model to facilitate discussion) This radical abstracts an H atom back from an unknown protein residue, presumably C141, to generate a thiyl radical, releasing the repaired TpT. This thiyl radical either takes an H-atom back from the 5′-dA directly, or it reacts with other protein residues before the 5′-dA is re-oxidized to the radical form again. The resulting 5′-dA radical recombines with methionine to regenerate SAM and finish one catalytic cycle.
One problem posed by this mechanism is to explain the tritium interchange between 5′-dA radical (SAM) and thymine substrate observed by Broderick and co-workers.9,11 A recent observation made by Eguchi and co-workers in their studies of BtrN suggested that the hydrogen atom abstraction step between the 5′-dA radical and the enzyme substrate is reversible.65 Such a reversible H atom interchange step was proposed by Liu and co-workers in their studies of DesII as well.26 If a similarly reversible step is involved between 5′-dA radical and the SP substrate as shown in Figure 9, the tritium exchange observed between 5′-dA radical (SAM) and thymine can also be rationalized. Additionally, no data was shown on how much tritium was exchanged between the reaction partners in the radio-labeling experiments.9,11 As scintillation counting is a relatively sensitive method, a rather low percentage of tritium incorporation can be detected.
The immediate task to firmly establish the hypothesized reaction mechanism is to confirm that the cysteine 141 indeed functions as the H atom donor. The radical intermediates involved in the reaction pathway may also be trapped and characterized via similar approaches conducted by Frey and co-workers in their mechanistic studies of LAM.74 These experiments as well as a search for possible protein residues that may participate in the SAM regeneration process are currently in progress.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Professor Eric Long at IUPUI for helpful discussions, the National Institute of Environmental Health Sciences (R00ES017177) as well as IUPUI startup funds for financial support. The NMR and MS facilities at IUPUI are supported by National Science Foundation MRI grants CHE-0619254 and DBI-0821661, respectively. We thank Professor Brian M. Hoffman, Northwestern University, for use of the 35 GHz EPR spectrometer, which is funded by NSF grant MCB-0316038. We thank Prof. Jacques Meyer, CEA-Grenoble, France, for providing samples of reduced Aquifex aeolicus Fd1. We thank Prof. Squire J. Booker, the Pennsylvania State University, for the generous gift of plasmid pDB1282.
Footnotes
Supporting Information Available: Syntheses and characterizations of deuterated SP substrates, TpT photochemistry, ESI-MS spectra of the isolated SAM, comparison EPR spectra of Aquifex aeolicus Fd1 and reduced SPL. This information is available free of charge via the internet at http://pubs.acs.org.
References
- (1).Prescott LM, Harley JP, Klein DA. Microbiology. 6th ed. McGraw-Hill Higher Education; Dubuque, IA: 2005. [Google Scholar]
- (2).Nicholson W, Munakata N, Horneck G, Melosh H, Setlow P. Microbiol. Mol. Biol. Rev. 2000;64:548. doi: 10.1128/mmbr.64.3.548-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Setlow P. Environ. Mol. Mutagen. 2001;38:97. doi: 10.1002/em.1058. [DOI] [PubMed] [Google Scholar]
- (4).Setlow P. Annu. Rev. Microbiol. 1988;42:319. doi: 10.1146/annurev.mi.42.100188.001535. [DOI] [PubMed] [Google Scholar]
- (5).Desnous CL, Guillaume D, Clivio P. Chem. Rev. 2010;110:1213. doi: 10.1021/cr0781972. [DOI] [PubMed] [Google Scholar]
- (6).Silver S, Chandra T, Zilinskas E, Ghose S, Broderick W, Broderick J. J. Biol. Inorg. Chem. 2010;15:943. doi: 10.1007/s00775-010-0656-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Pieck J, Hennecke U, Pierik A, Friedel M, Carell T. J. Biol. Chem. 2006;281:36317. doi: 10.1074/jbc.M607053200. [DOI] [PubMed] [Google Scholar]
- (8).Chandor A, Berteau O, Douki T, Gasparutto D, Sanakis Y, Ollagnier-De-Choudens S, Atta M, Fontecave M. J. Biol. Chem. 2006;281:26922. doi: 10.1074/jbc.M602297200. [DOI] [PubMed] [Google Scholar]
- (9).Buis J, Cheek J, Kalliri E, Broderick J. J. Biol. Chem. 2006;281:25994. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- (10).Chandor-Proust A, Berteau O, Douki T, Gasparutto D, Ollagnier-De-Choudens S, Fontecave M, Atta M. J. Biol. Chem. 2008;283:36361. doi: 10.1074/jbc.M806503200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cheek J, Broderick J. J. Am. Chem. Soc. 2002;124:2860. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- (12).Mehl RA, Begley TP. Org. Lett. 1999;1:1065. doi: 10.1021/ol9908676. [DOI] [PubMed] [Google Scholar]
- (13).Rebeil R, Nicholson WL. Proc. Natl. Acad. Sci. 2001;98:9038. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Donnellan JE, Jr., Setlow RB. Science. 1965;149:308. doi: 10.1126/science.149.3681.308. [DOI] [PubMed] [Google Scholar]
- (15).Cadet J, Vigny P. Bioorganic Photochemistry: Photochemistry and the Nucleic Acids. Vol. 1. Wiley; New York: 1990. The photochemistry of nucleic acids; p. 1. [Google Scholar]
- (16).Mantel C, Chandor A, Gasparutto D, Douki T, Atta M, Fontecave M, Bayle PA, Mouesca JM, Bardet M. J. Am. Chem. Soc. 2008;130:16978. doi: 10.1021/ja805032r. [DOI] [PubMed] [Google Scholar]
- (17).Lin G, Li L. Angew. Chem. Int. Ed. 2010;49:9926. doi: 10.1002/anie.201005228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Marsh ENG, Patterson DP, Li L. ChemBioChem. 2010;11:604. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Frey PA, Hegeman AD, Ruzicka FJ. Crit. Rev. Biochem. Mol. Biol. 2008;43:63. doi: 10.1080/10409230701829169. [DOI] [PubMed] [Google Scholar]
- (20).Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucl. Acids Res. 2001;29:1097. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chatterjee A, Li Y, Zhang Y, Grove TL, Lee M, Krebs C, Booker SJ, Begley TP, Ealick SE. Nat. Chem. Biol. 2008;4:758. doi: 10.1038/nchembio.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Paraskevopoulou C, Fairhurst SA, Lowe DJ, Brick P, Onesti S. Mol. Microbio. 2006;59:795. doi: 10.1111/j.1365-2958.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- (23).Walsby CJ, Ortillo D, Yang J, Nnyepi MR, Broderick WE, Hoffman BM, Broderick JB. Inorg. Chem. 2005;44:727. doi: 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- (24).Vey JL, Drennan CL. Chem. Rev. 2011;111:2487. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ruszczycky MW, Choi S-H, Liu H-W. J. Am. Chem. Soc. 2010;132:2359. doi: 10.1021/ja909451a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Szu P-H, Ruszczycky MW, Choi S-H, Yan F, Liu H-W. J. Am. Chem. Soc. 2009;131:14030. doi: 10.1021/ja903354k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mulder DW, Boyd ES, Sarma R, Lange RK, Endrizzi JA, Broderick JB, Peters JW. Nature. 2010;465:248. doi: 10.1038/nature08993. [DOI] [PubMed] [Google Scholar]
- (28).Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y. Nature. 2010;463:554. doi: 10.1038/nature08732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Driesener R, Challand M, Mcglynn S, Shepard E, Boyd E, Broderick J, Peters J, Roach P. Angew. Chem. Int. Ed. 2010;49:1687. doi: 10.1002/anie.200907047. [DOI] [PubMed] [Google Scholar]
- (30).Grove TL, Ahlum JH, Sharma P, Krebs C, Booker SJ. Biochem. 2010;49:3783. doi: 10.1021/bi9022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yan F, Lamarre JM, Roì˄Hrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. J. Am. Chem. Soc. 2010;132:3953. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wecksler SR, Stoll S, Tran H, Magnusson OT, Wu S-P, King D, Britt RD, Klinman JP. Biochem. 2009;48:10151. doi: 10.1021/bi900918b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Grove TL, Lee K-H, Clair J, Krebs C, Booker SJ. Biochem. 2008;47:7523–7538. doi: 10.1021/bi8004297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Frey PA, Magnusson OT. Chem. Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- (35).Farrar CE, Siu KKW, Howell PL, Jarrett JT. Biochem. 2010;49:9985. doi: 10.1021/bi101023c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. Science, ASAP. doi: 10.1126/science.1200877. [DOI] [PubMed] [Google Scholar]
- (37).Cicchillo RM, Iwig DF, Jones AD, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, Booker SJ. Biochem. 2004;43:6378. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- (38).Bradford MM. Anal. Biochem. 1976;72:248. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- (39).Gill SC, Von Hippel PH. Anal. Biochem. 1989;182:319. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- (40).Fish WW. Methods Enzymol. 1988;158:357. doi: 10.1016/0076-6879(88)58067-9. [DOI] [PubMed] [Google Scholar]
- (41).Beinert H. Anal. Biochem. 1983;131:373. doi: 10.1016/0003-2697(83)90186-0. [DOI] [PubMed] [Google Scholar]
- (42).Kim SJ, Lester C, Begley TP. J. Org. Chem. 1995;60:6256. [Google Scholar]
- (43).see supporting information.
- (44).Iwig DF, Booker SJ. Biochem. 2004;43:13496. doi: 10.1021/bi048693+. [DOI] [PubMed] [Google Scholar]
- (45).Aberhart DJ. J. Chem. Soc. Perkin Trans. 1. 1988:343. [Google Scholar]
- (46).Kilgore JL, Aberhart DJ. J. Chem. Soc. Perkin Trans. 1. 1991:79. [Google Scholar]
- (47).Werst MM, Davoust CE, Hoffman BM. J. A. Chem. Soc. 1991;113:1533. [Google Scholar]
- (48).Mailer C, Taylor CPS. Biochim. Biophys. Acta. 1973;322:195. doi: 10.1016/0005-2795(73)90293-6. [DOI] [PubMed] [Google Scholar]
- (49).Belford RL, Nilges MJ. EPR Symposium, 21st Rocky Mountain Conference Denver; Colorado. 1979.Aug, [Google Scholar]
- (50).Ugulava NB, Gibney BR, Jarrett JT. Biochem. 2001;40:8343. doi: 10.1021/bi0104625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cheek J, Krebs C, Huynh B, Broderick J. J. Inorg. Biochem. 2001;86:176. [Google Scholar]
- (52).Bertrand P. Inorg. Chem. 1993;32:741. [Google Scholar]
- (53).Bertrand P, Gayda J-P, Fee JA, Kuila D, Cammack R. Biochim. Biophys. Acta. 1987;916:24. doi: 10.1016/0167-4838(87)90206-8. [DOI] [PubMed] [Google Scholar]
- (54).Bertrand P, Guigliarelli B, More C. New. J. Chem. 1991;15:445. [Google Scholar]
- (55).Bertrand P, More C, Guigliarelli B, Fournel A, Bennett B, Howes B. J. Am. Chem. Soc. 1994;116:3078. [Google Scholar]
- (56).Meyer J, Clay MD, Johnson MK, Stubna A, Munck E, Higgins C, Wittung-Stafshede P. Biochem. 2002;41:3096. doi: 10.1021/bi015981m. [DOI] [PubMed] [Google Scholar]
- (57).Henshaw TF, Cheek J, Broderick JB. J Am. Chem. Soc. 2000;122:8331. [Google Scholar]
- (58).Marsh E, Patwardhan A, Huhta M. Bioorg. Chem. 2004;32:326. doi: 10.1016/j.bioorg.2004.06.001. [DOI] [PubMed] [Google Scholar]
- (59).Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. Biochem. 1998;37:2578. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- (60).Duschene KS, Broderick JB. FEBS Lett. 2010;584:1263. doi: 10.1016/j.febslet.2010.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Padovani D, Thomas F, Trautwein AX, Mulliez E, Fontecave M. Biochem. 2001;40:6713. doi: 10.1021/bi002936q. [DOI] [PubMed] [Google Scholar]
- (62).Kriek M, Peters L, Takahashi Y, Roach PL. Protein Express. Purif. 2003;28:241. doi: 10.1016/s1046-5928(02)00680-0. [DOI] [PubMed] [Google Scholar]
- (63).Schreier WJ, Schrader TE, Koller FO, Gilch P, Crespo-Hernandez CE, Swaminathan VN, Carell T, Zinth W, Kohler B. Science. 2007;315:625. doi: 10.1126/science.1135428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Ballinger MD, Reed GH, Frey PA. Biochem. 1992;31:949. doi: 10.1021/bi00119a001. [DOI] [PubMed] [Google Scholar]
- (65).Yokoyama K, Numakura M, Kudo F, Ohmori D, Eguchi T. J Am. Chem. Soc. 2007;129:15147. doi: 10.1021/ja072481t. [DOI] [PubMed] [Google Scholar]
- (66).Fajardo-Cavazos P, Rebeil R, Nicholson W. Curr. Microbiol. 2005;51:331. doi: 10.1007/s00284-005-0052-8. [DOI] [PubMed] [Google Scholar]
- (67).Guo J, Luo Y, Himo F. J. Phys. Chem. B. 2003;107:11188. [Google Scholar]
- (68).Himo F. BBA-Bioenerg. 2005;1707:24. doi: 10.1016/j.bbabio.2004.04.006. [DOI] [PubMed] [Google Scholar]
- (69).S., Jursic B. J. Chem. Soc., Perkin Trans. 2. 1999:369. [Google Scholar]
- (70).Blanksby SJ, Ellison GB. Acc. Chem. Res. 2003;36:255. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- (71).Lawrence CC, Stubbe J. Curr. Opin. Chem. Biol. 1998;2:650. doi: 10.1016/s1367-5931(98)80097-5. [DOI] [PubMed] [Google Scholar]
- (72).Wang SC, Frey PA. Trends Biochem. Sci. 2007;32:101. doi: 10.1016/j.tibs.2007.01.002. [DOI] [PubMed] [Google Scholar]
- (73).Wang SC, Frey PA. Biochem. 2007;46:12889. doi: 10.1021/bi701745h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Frey PA. Annu. Rev. Biochem. 2001;70:121. doi: 10.1146/annurev.biochem.70.1.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.