

Abstract
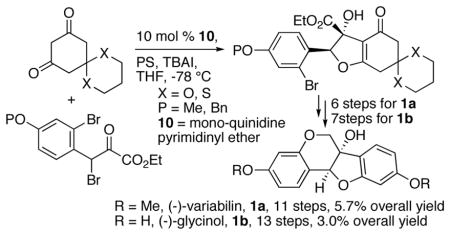
Total syntheses of (−)-variabilin and (−)-glycinol have been accomplished, using the catalytic, asymmetric “interrupted” Feist-Bénary reaction (IFB) as the key transformation to introduce both stereogenic centers. A monoquinidine pyrimidinyl ether catalyst affords the IFB products in over 90% ee in both cases. Other key steps include an intramolecular Buchwald-Hartwig coupling and a nickel-catalyzed aryl tosylate reduction.
Variabilin (1a) and glycinol (1b) are members of the phytoalexins family produced by certain plants in response to fungal infection.1,2 Naturally occurring variabilin occurs as the (+)- and (−)-enantiomers, both of which show antifungal activity towards Monilinia fructicola (ED50 < 2×10−5 M).3 (−)-Glycinol was first isolated in 1978 by Lyne and Mulheirn from coppertreated soybean cotyledons.4 It is believed that glycinol is the key precursor in the biosynthesis of glyceollins.5 Burow et al. have recently shown that glycinol exhibits a strong in vitro estrogenic effect by binding to both estrogen receptor ERα (IC50 = 13.75 nM) and ERβ (IC50 = 9.05 nM).6
To date, only one asymmetric total synthesis of each target has been accomplished, both of which set the stereogenic centers by asymmetric dihydroxylation in the presence of stoichiometric amounts of OsO4 and quinine or quinidine derivatives.7 Here, we report total syntheses of both 1a and 1b, using the catalytic, asymmetric “interrupted” Feist-Benary (IFB) reaction developed by Calter et al. to install the chirality into the target molecules (Scheme 1).8 Completion of the synthesis would then involve aromatization, intramolecular Buchwald-Hartwig reaction to form the benzopyranoid ring, tosylate reduction and finally deprotection in the case of glycinol. The IFB reactions would require dione nucleophiles 2a or 2b and the β-bromo-α-ketoester electrophiles 3a or 3b, which were all either known compounds or close analogues.
Scheme 1.

Retrosynthetic Plan for 1a and 1b
The synthesis of dithiane 2a began with the addition of lithium propiolate with propylene oxide in the presence of BF3 · Et2O to afford alkynyl alcohol 4 (Scheme 2). Oxidation of 4 with the Swern reagent or the Dess-Martin periodinane provided multiple products. A successful oxidant was silica gel-supported Jones reagent (SJR), which gave a mixture of alkyne 5 and allene 6 in quantitative yield after filtration. Compound 5 was the major initial product of the reaction, but converted into 6 at −25 °C in under 12 hours. Addition of 1,3- propanedithiol to the mixture of 5 and 6 in the presence of base produced keto ester 7, which was subjected to Dieckmann condensation to give 2a in 47% overall yield.
Scheme 2.

Synthesis of Dithiane 2a
Pyruvate 3a was synthesized according to a well documented procedure beginning with the lithium trimethylethylenediamine-directed ortho-bromination of p-anisaldehyde to produce bromoaldehyde 8 (Scheme 3).9 Submission of 8 to a Darzens reaction led to epoxide 9, which was converted into β-bromo-α-ketoester 3a by treatment with MgBr2. 10
Scheme 3.

Synthesis of Pyruvate 3a
With 2a and 3a in hand, the IFB reaction proceeded smoothly with mono-quinidine pyrimidinyl ether 10 as a catalyst and provided 11 as an inseparable 8.5:1 E:Z mixture of diastereomers (Scheme 4). Exposure of 11 to iodobenzene bis(trifluoromethylacetate) (PIFA) produced the corresponding, unstable dimethoxy acetal,11 which was subjected to LiHMDS to give unstable phenol 13. Both 12 and 13 were prone to elimination to form the corresponding furans. Tosylation of 13 afforded tosylate 14, still as a mixture of diastereomers. However, we were able to use analytical HPLC to determine the enantio- and diastereopurity of 14.
Scheme 4.

Synthesis of Tosylate 14
We were initially surprised with the (E)-selectivity of the IFB reaction of 2a and 3a, as previous examples had provided (Z)-selectivity, but further analysis of factors governing the diastereoselectivity of the IFB reaction provided a rationale (Figure 2). In general, the IFB reaction of substituted bromoketoesters proceeds via a dynamic, kinetic resolution, with tetrabutylammonium iodide (TBAI) serving to convert the bromide into the corresponding iodide and then constantly racemize this compound. The previous examples using unsubstituted aryl substituents yielded the (Z)-R,R-product, with both the catalyst and the substrate favoring attack, via a transition state favored by the Felkin-Nguyen model with a strongly electron-withdrawing substituent, on the Re-face of the (S)-enantiomer of the haloketeoester.
Figure 2.

Transition-state models for the IFB reaction. Catalyst omitted for clarity.
However, the ortho-substituent in the current substrate sterically disfavors this mode of attack, so the substrate-catalyst combination now favors Re-face attack on the R-enantiomer, leading to the (E)-R,S-stereoisomer. The ortho-bromine cannot rotate away from this interaction, as to do so would place the bromine in an electronically unfavorable interaction with the carbonyl oxygen or the iodine.
Continuing with the synthesis, the reduction of 14 first provided the aldehyde, which was then immediately resubmitted to a second reduction to afford 1,2-diol 15 (Scheme 5). Exposure of 15 to modified Buchwald-Hartwig coupling conditions gave dihydrobenzopyran 18, which we were able to isolate in high diastereo-purity.12 Initial attempts to reduce 18 using the conditions of Lipshutz and Kogan afforded 1a, but with irreproducible conversions.13 After experimenting with different hydride and nickel sources, we found that use of the THF-soluble LiBH4 and the formation of nickel(0) from in situ reduction of NiCl2 reliably afforded variabilin 1a. The analytical data for the product matched those reported previously for (−)-variabilin.7a In total, this synthesis required 11 steps with 5.7% overall yield. The bis-demethylation of variabilin 1a into glycinol 1b afforded the furan under Lewis acidic conditions, and gave low and irreproducible conversion under nucleophilic conditions. Therefore, we developed an alternative synthesis of glycinol that employed more easily removable protecting groups from the beginning.
Scheme 5.

Synthesis of Variabilin 1a
This synthesis of glycinol began with the construction of 1,3-dioxane 2b. Treatment of acetonedicarboxylic acid 19 with TMSO(CH2)3OTMS in the presence of a catalytic amount of TMSOTf yielded 20.14 Exposure of 20 to MeMgBr in CH2Cl2 at −78 °C for 12 h afforded keto ester 21 in moderate conversion, but we were able to recover the unreacted starting material. Dieckmann condensation of 21 promoted by LDA provided 2b in a similar fashion to the cyclization of 7 to 2a.
We prepared β-bromo-α-ketoester 3b through known bromoaldehyde 22 (Scheme 7). 15 Addition of N,N′-dimethylethylenediamine to 4-hydroxylbenzaldehyde in refluxing toluene afforded the aminal. ortho-Metallation, bromination and hydrolysis provided aldehyde 22, which was benzylated to give 23. Submission of 23 to Darzens reaction and subsequent ring-opening afforded 3b.
Scheme 7.

Synthesis of Bromoketone 3b
The IFB reaction of 2b and 3b proceeded smoothly with 10 as the catalyst and provided 25 in a 10:1 E:Z ratio (Scheme 8). In this series chromatography allowed us to remove most of the (Z)-diastereomer. Aromatization of 25 with tBuOK followed by tosylation afforded tosylate 27. We were able to use analytical HPLC to determine the enantio- and diasteropurity of this compound.
Scheme 8.

Synthesis of Tosylate 27
Two-stage reduction of 27 with LiBH4 gave diol 28, which was subjected to our modified Buchwald-Hartwig conditions to give dihydropyran 29 (Scheme 9). Reduction of the aryl tosylate afforded alcohol 30. We accomplished removal of the propylene glycol group from 30 by oxidation with DMP followed by elimination with NEt3. Finally, deprotection of the aryl benzyl group by transfer hydrogenation provided (−)-glycinol 1b in 90% yield. The analytical data for this product matched those reported previously for (−)-glycinol.4,7b This synthesis required 13 steps with 3.0% overall yield.
Scheme 9.

Synthesis of Glycinol 1b
In conclusion, we have accomplished the catalytic, asymmetric total syntheses of (−)-variabilin and (−)- glycinol by employing the “interrupted” Feist-Benary reaction as the key step to introduce both stereogenic centers simultaneously. Both syntheses proceed in high enantio- and diastereoselectivity. The methodology developed should be applicable to the syntheses of other natural products with dihydrobenzofuran moieties.
Supplementary Material
Figure 1.
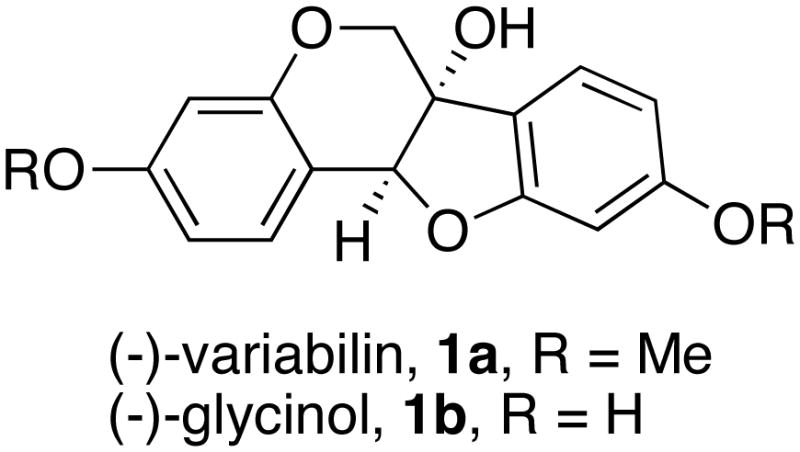
Structures of Variabilin (1a) and Glycinol (1b)
Scheme 6.

Synthesis of Dioxane 2b
Acknowledgments
The authors thank the NIH for financial support of this project. We acknowledge Dr. Ryan Phillips (IRIX Pharmaceuticals) for preliminary experiments.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Bilton JN, Debnam JR, Smith IM. Phytochemistry. 1976;15:1411–1412. [Google Scholar]; (b) Kurosawa K, Ollist WD, Sutherland IO, Gottlieb OR. Phytochemistry. 1978;17:1417–1418. [Google Scholar]; (c) Magalhães AF, Tozzi AMGA, Magalhães EV, Sannomiya M, Soriano MDPC, Perez MAF. An Acad Bras Cienc. 2007;79:351–367. doi: 10.1590/s0001-37652007000300001. [DOI] [PubMed] [Google Scholar]
- 2.Banks SW, Dewick PM. Phytochemistry. 1983;22:2729–2733. [Google Scholar]
- 3.Perrin DR, Cruickshank IAM. Phytochemistry. 1969;8:971–978. [Google Scholar]
- 4.Lyne RL, Mulheirn LJ. Tetrahedron Lett. 1978;34:3127–3128. [Google Scholar]
- 5.(a) Keen NT, Zaki AI, Sims JJ. Phytochemistry. 1972;11:1031–1039. [Google Scholar]; (b) Zähringer U, Ebel J, Mulheirn LJ, Lyne RL, Grisebach H. FEBS Lett. 1979;101:90–92. doi: 10.1016/0014-5793(79)81301-0. [DOI] [PubMed] [Google Scholar]; (c) Hagmann M-L, Heller W, Grisebach H. Eur J Biochem. 1984;142:127–131. doi: 10.1111/j.1432-1033.1984.tb08259.x. [DOI] [PubMed] [Google Scholar]; (d) Ebel J. Ann Rev Phytopathol. 1986;24:235–264. [Google Scholar]; (e) Matthews DE, Plattner RD, VanEtten HD. Phytochemistry. 1989;28:113–115. [Google Scholar]
- 6.Boue SM, Tilghman SL, Elliott S, Zimmerman MC, Williams KY, Payton-Stewart F, Miraflor AP, Howell MH, Shih BY, Carter-Wientjes CH, Segar C, Beckman BS, Wiese TE, Cleveland TE, McLachlan JA, Burow ME. Endocrinology. 2009;150:2446–2453. doi: 10.1210/en.2008-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) van Aardt TG, van Rensburg H, Ferreira D. Tetrahedron. 2001;57:7113–7126. [Google Scholar]; (b) Luniwal A, Khupse RS, Reese M, Fang L, Erhartdt PW. J Nat Prod. 2009;72:2072–2075. doi: 10.1021/np900509f. [DOI] [PubMed] [Google Scholar]
- 8.Calter MA, Phillips RM, Flaschenriem C. J Am Chem Soc. 2005;127:14566–14567. doi: 10.1021/ja055752d. [DOI] [PubMed] [Google Scholar]
- 10.(a) Merz KH, Muller T, Vanderheiden S, Eisenbrand G, Marko D, Brase S. Synlett. 2006;20:3461–3463. [Google Scholar]; (b) Lear Y, Durst T. Can J Chem. 1997;75:817–824. [Google Scholar]; (c) Comins DL, Brown JD. J Org Chem. 1984;49:1078–1083. [Google Scholar]
- 9.(a) Tsuboi S, Furutani H, Takeda A, Kawazoci K, Sato S. Bull Chem Soc Jpn. 1987:2475–2480. [Google Scholar]; (b) Coutrot P, Legris C. Synthesis. 1975;2:118–120. [Google Scholar]
- 11.Stork G, Zhao K. Tetrahedron Lett. 1989;30:287–290. [Google Scholar]
- 12.(a) Shelby Q, Kataoka N, Mann G, Hartwig J. J Am Chem Soc. 2000;122:10718–10719. [Google Scholar]; (b) Torraca KE, Kuwabe SI, Buchwald SL. J Am Chem Soc. 2000;122:12907–12908. doi: 10.1021/ja012046d. [DOI] [PubMed] [Google Scholar]
- 13.(a) Lipshutz BH, Frieman BA, Butler T, Kogan V. Angew Chem Int Ed. 2006;45:800–803. doi: 10.1002/anie.200502887. [DOI] [PubMed] [Google Scholar]; (b) Kogan V. Tetrahedron Lett. 2006;47:7515–7518. [Google Scholar]
- 14.Arnott G, Heaney H, Hunter R, Page PCB. Eur J Org Chem. 2004:5126–5234. [Google Scholar]
- 15.(a) Bevan MF, Euerby RM, Qureshi JS. J Chem Res (M) 1989:901–920. [Google Scholar]; (b) Couture A, Deniau E, Lebrum S, Hoarau C, Grandclaudon P. Nat Prod Lett. 1999;13(1):33–40. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.