

Abstract
Raloxifene hydrochloride (R-HCl), a BCS class II drug, remains a mainstay in the prevention and pharmacologic therapy of osteoporosis. Its absolute bioavailability, however, is 2% due to poor solubility and extensive first pass metabolism. The present study describes two simultaneous approaches to improve its bioavailability, complexation of R-HCl with cyclodextrin(s), and formulation of mucoadhesive microspheres of the complex using different proportions of carbopol and HPMC. Microspheres were pale yellow in color, free-flowing, spherical, and porous in outline. The particle size ranged between 3 and 15 μm, and entrapment efficiency was found to be within 81.63% to 87.73%. A significant improvement in the solubility of R-HCl was observed, and it differed with the combination of excipients used. X-ray diffraction and differential scanning calorimetry studies revealed that enhancement in drug solubility was resulted due to a change in its crystallinity within the formulation. Microspheres possessed remarkable mucoadhesion and offered controlled drug release, lasting up to 24 h. They produced a sharp plasma concentration–time profile of R-HCl within 30 min post-administration to Wistar rats. [AUC]0–24 h was found to be 1,722.34 ng h/ml, and it differed significantly to that of pure drug powder (318.28 ng h/ml). More than fivefold increase in AUC and more than twofold increase in MRT were observed. FT-IR studies evidenced no interaction among drug and excipients. The results of this study showed that mucoadhesive microspheres could be a viable approach to improve the pharmacokinetic profile of R-HCl.
KEY WORDS: BCS class II, mucoadhesive microsphere, osteoporosis, raloxifene hydrochloride, spray drying
INTRODUCTION
Osteoporosis poses a significant health problem worldwide; a 50-year-old postmenopausal woman has a 40–50% risk of having osteoporotic fracture in her lifetime (1). Lifestyle changes, weight-bearing exercise, calcium and vitamin D supplementation, and pharmacologic therapy are the general measures for the prevention of postmenopausal osteoporosis (2). Anti-resorptives are the major pharmacologic agents for osteoporosis which inhibit the development and/or action of osteoclasts. They include bisphosphonates [selective estrogen receptor modulator (SERM)], hormone replacement therapy, and calcitonin. Raloxifene hydrochloride (R-HCl) is an FDA-approved SERM for the prevention and treatment of osteoporosis. It is a BCS class II drug, with slight aqueous solubility (627.4 ± 132.0 μg ml−1; 3). Absolute bioavailability of R-HCl is 2% due to its poor water solubility and extensive first-pass metabolism. Also, it exhibits a high inter-subject variability (4,5). Various strategies have been reported to improve the solubility and bioavailability of R-HCl, such as lipid-based delivery systems (6), inclusion complexes (7), and co-grinding (6,8). Co-grinding suffers from the demerit of higher polymer consumption, whereas lipid-based carriers are often limited in terms of drug-to-lipid mass ratio, which interferes with their stability aspect (9). Besides, they provide a mere two- to threefold enhancement in bioavailability (7).
The present study describes two simultaneous approaches to improve the bioavailability of R-HCl. First, inclusion complex of R-HCl was prepared with cyclodextrin (CD) and then the complex was formulated as bioadhesive microspheres, using different proportions of carbopol (CP) and HPMC. Inclusion in CDs is a convenient alternative to improve the stability and solubility of poorly water soluble drugs (10). Osmotic effect, induced by them, helps in pore formation and contributes to effective drug mobility within the hydrated matrix (11). Moreover, microsphere formulation of the complexed drug would further enhance the solubility of drug, reduce the likelihood of dose dumping, minimize dose-related variations, and improve patient compliance (12–14).
Carbopol has been widely used for the preparation of mucoadhesive drug delivery systems. It, however, swells excessively upon being exposed to pH above 6.0 (15). Moreover, it causes irritation at the mucosal surface (16). It was, therefore, combined with HPMC to optimize the mucoadhesion and swelling characteristics of microspheres and simultaneously, reduce its irritancy (17,18). As of the date, no such combination has been reported for mucoadhesive microspheres of R-HCl.
MATERIALS AND METHODS
Materials
R-HCl was received as a gift sample from Dr. Reddys Lab, Hyderabad, India. CD derivatives (β, γ, and hydroxypropyl-β) and HPMC K15M were kindly gifted by Lupin Research Park, Pune, India. Carbopol 974, heparin, acetonitrile, and fluorescein sodium (FS) were purchased from Lobachemie Pvt. Ltd., Biologicals E Ltd., Merck Ltd., and Hi Media Ltd., Mumbai, respectively. All other chemicals and solvents were of analytical grade and used as received.
Methods
Phase Solubility and Equilibrium Solubility Studies
Complexation of R-HCl with various CD derivatives (α, β, hydroxypropyl-β, and γ) was studied by phase solubility study (19). An excess amount of drug along with the appropriate amount of CD was added to 10 ml of triple-distilled water, and the suspension was subjected to magnetic stirring for 72 h at 37.0 ± 1.0°C. Preliminary studies showed that equilibrium was reached after this stirring period. The suspension was then centrifuged (15,000 rpm/5 min), and the supernatant was analyzed spectrophotometrically for drug content at 285 nm (Shimandzu 7800, Tokyo, Japan).
Apparent solubility of R-HCl was determined as a function of added ligand concentration, i.e., CD (20). Apparent solubility (or formation) constant, Ka, was calculated using the following equation:
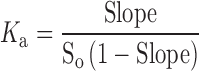 |
1 |
Inherent solubility (So) of R-HCl was determined in pure water under identical experimental conditions. Equilibrium solubility in different solvents was also determined by the same method.
Drug–Polymer Compatibility Studies
FT-IR spectra of the samples were recorded by FT-IR spectrophotometer (Schimadzu, Model 8400, Japan). Samples were prepared by grinding with anhydrous KBr powder and compressing them to form pellets. They were subsequently observed over the range of 4,000–400 cm−1 with a resolution of 4 cm−1 up to 50 scans.
Differential Scanning Calorimetry and X-ray Diffraction Studies
Solid state interaction of physical mixture of drug and excipients was studied using differential scanning calorimetry (DSC) and X-ray diffraction (X-RD). DSC thermogram was recorded by scanning the sample in a hermetic pan made of aluminum, at a heating rate of 10°C/min over the range of 25–300°C. An empty aluminum pan was used as reference (DU-PONT, Model 9900, USA). Heating was performed under nitrogen atmosphere at a flow rate of 50 ml/min.
X-RD pattern was traced using Ni-filtered Cu–K radiation at a voltage of 40 kV and a current of 20 mA (Rigaku Powder X-ray Diffractometer, Japan). The sample was gently compressed into sample holder, and the powder surface was smoothed with a flat perspex block. Radiations scattered by sample(s) in the crystalline region were recorded. Diffraction patterns were obtained using a step width of 0.04° with a detector resolution in 2θ (diffraction angle) of 10–70° at a scan rate of 1.0°/s.
Formulation of Mucoadhesive Microspheres of R-HCl
Microspheres, composed of different combination of excipients and drug (Table I), were prepared by spray drying of polymeric dispersion with a standard 0.5-mm nozzle. Liquid dispersions, containing a solid feed concentration of 2.5% w/v, were fed to the nozzle with a peristaltic pump, atomized and blown together with hot air to the chamber wherein solvent was evaporated and dried product was collected. For all the batches, experimental conditions were selected as follows: ethanol–water (4: 1) as solvent system, feed concentration of 2.5% w/v, inlet air temperature of 110°C, outlet air temperature of 51°C, pump setting of 12 ml/min, and 2 atm pressure.
Table I.
Formulation Composition of Microspheres
| Batch code | R-HCl (mg) | γ-CD (mg) | CP971 (mg) | HPMC-K4M (mg) |
|---|---|---|---|---|
| F1 | 40 | 40 | 15 | 105 |
| F2 | 40 | 40 | 30 | 90 |
| F3 | 40 | 40 | 45 | 75 |
| F4 | 40 | 40 | 60 | 60 |
| F5 | 40 | 40 | 75 | 45 |
Characterization of the Prepared Microspheres
Particle size and size distribution of the microspheres were estimated by laser particle size analyzer, containing helium–neon laser of wavelength 632.8 nm and radiant power 5 mW (Akesmind 4800S, Japan). Relative frequency of diameter of particles was obtained by calculations based on volume distribution. Particle size at 10% (D10), 50% (D50), and 97% (D97) of total fraction was obtained. Particle size at 97% of total fraction was reported. Shape and surface morphology of the prepared microspheres were observed by field emission scanning electron microscope (S-4700, Hitachi, Japan). The sample was mounted onto an aluminum stub and sputter coated for 120 s with platinum particles in an argon atmosphere.
Percentage Yield and Entrapment Efficiency
For determination of entrapment efficiency (EE), a weighed amount of formulation was added to a 100-ml phosphate buffer (pH 6.8) containing 0.5% v/v polysorbate 80. The sample was solubilized under magnetic stirring for 24 h at room temperature (RT), following which it was centrifuged (15,000 rpm/5 min) and filtered through a 0.45-μm pore size filter. The filtrate was then diluted with the same solvent, and the drug was quantified. The following equations were used:
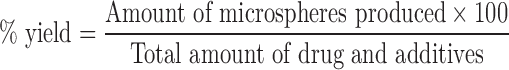 |
2 |
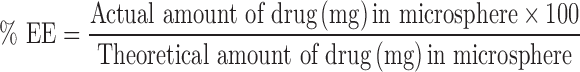 |
3 |
In Vitro and In Vivo Mucoadhesion Studies
Excised mucosa of stomach and duodenal region of Wistar rats were used for in vitro mucoadhesion studies. Microspheres equivalent to 5 mg drug were placed on mucosal surface, the latter being fixed over a glass plate. The assembly was maintained at RT and 70% relative humidity (RH). The plate was inclined at 45°, and tissues were thoroughly washed with buffer solution using a peristaltic pump operating at the rate of 10 ml/min for 2 min (21). Drug concentration in the perfusate was determined using UV spectrophotometry. Microsphere amount corresponding to the drug amount in perfusate was calculated. Adhered microsphere amount was estimated from the difference between the initial amount and the amount in perfusate. Percent mucoadhesion was calculated using the formula:
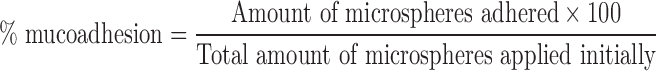 |
In vivo mucoadhesion was evaluated in Wistar rats using a modified fluorescence-labeled method (22). Fluorescence labeling was performed by incorporating FS into the formulation. Animals were fasted overnight with water ad libitum and then randomly divided into two groups (n = 6). Group 1 was orally administered with blank microspheres, whereas group 2 was given the equivalent amount of formulation incorporated with FS. At specified time intervals (2, 3, and 4 h), animals were sacrificed; stomach and small intestine were removed, cut into segments, and homogenized separately. After being subjected to centrifugation (15,000 rpm/15 min), fluorescence of each sample was measured in a spectrofluorimeter at excitation and emission wavelengths of 485 and 520 nm, respectively (Shimadzu RF-1501, Tokyo, Japan). The amount of microspheres adhering to the mucosal surface was calculated by extrapolating these values to the calibration curve of FS. Percentage mucoadhesion onto different segments was calculated.
In Vitro Drug Release Study
The formulation equivalent to 5 mg drug was placed in dialysis bag (Sigma Aldrich, molecular weight 12,000 Da), and it was secured along the paddle of dissolution apparatus with the help of a rubber band. The study was performed in 900 ml release media, maintained at 37 ± 0.5°C and stirred at 75 rpm (USP-XXIV, Campbell Electronics, Mumbai, India). With a viewpoint to mimic the fate of drug in the body, three dissolution media were used: acidic buffer (pH 1.2) for the first 2 h, acetate buffer (pH 4.5) for the next 2 h, and phosphate buffer (pH 6.8) for rest of the study period. Polysorbate 80 (0.5% v/v) was used in each case to maintain sink condition. Five-milliliter aliquots were withdrawn at prespecified time intervals and were immediately replenished with the same volume of fresh media. The drug was quantified spectrophotometrically at 285 nm.
Pharmacokinetic Studies
A series of standard solutions of R-HCl were prepared in methanol in the range of 0.050–200 μg/ml. Standard calibration samples were prepared by adding 100 μl standard solution of drug and 300 μl acetonitrile to 100 μl of blank plasma. Final standard R-HCl concentrations in plasma were 0.010–40.0 μg/ml. Sample extraction was performed by adding 50 μl of methanol and 200 μl of acetonitrile to the mixture (23). The resulting solution was vortex mixed for 10 s and then centrifuged (15,000 rpm/10 min). Twenty microliters of the filtered (0.22 μm pore size) supernatant was injected to HPLC system (Cecil CE4201, Cambridge, UK), equipped with C-18 column (Phonomenex, 250 × 4.60 mm, particles size 5 μm) and UV visible detector.
The separation of R-HCl from endogenous substances was optimized by using 50% v/v acetonitrile with 0.05 M ammonium acetate as mobile phase, adjusted to pH 4.0 using acetic acid (23). Precision and accuracy of the method were evaluated by adding quality control samples (0.2, 0.8, and 5.0 μg/ml) to the blank plasma. Extraction efficiency was calculated by comparing the mean peak height of three extracted plasma samples with those of the three non-extracted standard solutions of the same concentration. Linearity, limit of quantification, and inter-day and intra-day variability were also calculated.
In vivo pharmacokinetic study was conducted in Wistar rats (of either sex) as per the protocol approved by the Institutional Animal Ethical Committee, Banaras Hindu University (Approval Number Dean/10-11/162). Animals were provided free access to tap water and standard pelleted diet. They were housed in cage and maintained on a 12-h light/dark cycle at RT and RH of 45–55%. They were acclimatized to laboratory conditions for 1 week prior to dosing (6). Animals were divided into two groups (n = 6), fasted 10 h prior to dose administration and for 4 h post-dose with free access to water. Equivalent oral dose of the test (optimized batch F4) and reference samples (5 mg/kg free base) were dispersed in 0.3% carboxymethyl cellulose and administered to the animal using an oral feeding canula. Serial blood samples (0.3 ml) were withdrawn from the retro-orbital plexus under mild ether anesthesia and collected in labeled tubes containing 50 microunits of heparin. The following time intervals were used: pre-dose, 0.25, 0.5, 1, 1.5, 2, 4, 8, 12, and 24 h post-dose. Blood samples were centrifuged immediately; plasma was transferred to the individual Eppendorf tubes and stored below −70°C until extraction. After extraction, the samples were analyzed by the above stated standardized HPLC method.
The estimation of pharmacokinetic parameters was performed using Kinetica 5.0 software. Maximum plasma concentration (Cmax), time to achieve maximum plasma concentration (Tmax), and mean residence time (MRT) were calculated. Area under plasma concentration–time curve [AUC]0–24 was determined by trapezoidal method till last measurement point and was extrapolated to infinity [AUC]0–∞.
RESULTS AND DISCUSSION
Solubility Studies
R-HCl showed a linear increase in solubility as the concentration of CD was increased. The linearity corresponded to AL-type of curve with slope value <1, signifying that complexation took place in stoichiometric ratio of 1:1 (20,24). Apparent complexation constant (Ka) was found to be 1,438.3 ± 141.2 M−1 for β-CD, 3,311.2 ± 126.5 M−1 for γ-CD, 3,430.6 ± 101.8 M−1 for HP-β-CD, and 3,556.6 M−1 for γ-CD with 0.1% v/v HPMC. The maximum solubility and highest Ka value were observed in HP-β-CD, whereas minimum corresponding values were observed for β-CD. Solubility and Ka of γ-CD drug complex were comparable to those of HP-β-CD (Fig. 1a). Incorporation of HPMC with γ-CD resulted into further enhancement in drug solubility (Fig. 1b), whereas other derivatives did not provide significant enhancement of drug solubility in combination with HPMC (data not shown). γ-CD being less toxic than other derivatives (25) was, therefore, used as a complexing agent in the proposed formulation.
Fig. 1.

Phase solubility studies of R-HCl with different cyclodextrin derivatives
R-HCl showed pH-dependent solubility, an increase in solubility was observed when pH was increased from 1.2 to 4.5. However, drug solubility decreased with increase in pH beyond 4.5. Lowest solubility at pH 1.2 could be attributed to the common ion effect. Addition of polysorbate resulted into increased solubility at all pH values. Equilibrium solubility of all the formulations was found to be higher than the physical mixture of similar composition.
FT-IR Study
Interaction among drug and excipients results into shifting of functional group peaks in IR spectra. FT-IR spectra of R-HCl exhibited characteristic peaks at 1,643.41 (C=O stretching), 1,597.33 (–C–O–C– stretching), 1,464.02 (–S–benzothiofuron), and 908.57 cm−1 (benzene ring). They were well preserved in its physical mixture with excipients as well as in microsphere formulation (Fig. 2). These results are indicative that no interaction occurred between drug and excipients.
Fig. 2.

FT-IR spectra of a) pure R-HCl powder, b) physical mixture of R-HCl with excipients, and c) R-HCl-loaded microspheres (batch F4)
DSC and X-RD Studies
R-HCl exhibited a single sharp melting endothermic peak at 269°C (Fig. 3(A)). Drug-free microsphere showed a broad endothermic peak in the range from 25°C to 87°C, which may be because of endothermic relaxation of polymeric chain (8). Thermograms indicated that polymers are amorphous and hydrated compounds (Fig. 3(B)). Decrease in the intensity of melting endothermic peak (as evidenced by change in their enthalpies (ΔHm)) was observed for drug-loaded microspheres than pure drug (Fig. 3(C)). This is indicative of change in the crystallinity of drug upon incorporating it into the formulation.
Fig. 3.

DSC thermograms of a) R-HCl, b) drug-free microspheres (batch F4), and c) drug-loaded microspheres (batch F4)
X-RD studies were performed in conjunction with DSC to verify the reduction of crystallinity of R-HCl within the formulation. Diffraction spectrum of drug sample showed distinct peaks at 2θ of 12.85°, 14.43°, 15.65°, 19.11°, 22.70°, and 25.7° (Fig. 4(a)). All these peaks, though of relatively lesser intensity, were observed to be in the same position in its inclusion complex with γ-CD. However, a relative reduction of diffraction intensity was observed (Fig. 4(b)). However, in case of drug-loaded microspheres, a drastic reduction in the peak intensity and number of peaks was observed, which is indicative of change in the crystallinity of drug (Fig. 4(c)). It was thus concluded that the drug was converted from crystalline to amorphous state upon inclusion complex formation and incorporation of the complex into microsphere formulation (7,26).
Fig. 4.

X-RD spectra of a) pure R-HCl, b) R-HCl complexed with γ-CD, and c) microspheres composed of complexed drug along with excipients (batch F4)
Characterization of the Prepared Microspheres
Spray-dried microspheres were pale yellow in color and free-flowing. The yield value of different batches varied from 28% to 30%, which is fairly less. Poor yield could be attributed to the loss of smaller particles which could not be collected. Particle size (D97) of all batches ranged between 3 and 15 μm (Table II). Microspheres with greater diameters resulted upon increasing the proportion of carbopol which could be attributed to the increment in viscosity of feed dispersion imparted by it. Higher viscosity results into the formation of larger droplets (14). Particle size of F4 and F5 was significantly smaller than the other batches (one-way ANOVA, p < 0.05). This effect could be due to decrease in the viscosity of dispersion in presence of γ-CD. Hydrogen bonding of carbopol gets weakened in presence of CD molecules (27). EE of all the batches ranged between 82% and 88%. The addition of γ-CD to carbopol–HPMC combination resulted up to 5% increment in EE (data not shown). However, no correlation between the ratio of carbopol to HPMC and EE was observed.
Table II.
Evaluation of the Prepared Microspheres
| Batch code | Particles size D 97 (μm) | Entrapment efficiency (%) | Yield (%) |
|---|---|---|---|
| F1 | 14.69 | 87.73 ± 0.12 | 28.78 ± 0.24 |
| F2 | 10.03 | 84.67 ± 0.26 | 29.65 ± 0.82 |
| F3 | 8.23 | 85.46 ± 0.86 | 30.21 ± 0.67 |
| F4 | 4.31 | 82.12 ± 0.98 | 29.82 ± 0.27 |
| F5 | 3.07 | 81.63 ± 0.31 | 27.81 ± 0.54 |
Microspheres possessed almost spherical shape (Fig. 5a). Their surface was rough and some pores could be observed on it (Fig. 5b). Pores would have formed during quick evaporation of solvent, and this might be advantageous as it would offer greater surface area for dissolution (28) and help in the penetration of solvent across the matrix. Keeping in view the amorphous nature of drug within the microsphere matrix, it could be anticipated to be in an easy dissolvable form throughout its transit in the gastrointestinal tract.
Fig. 5.

Scanning electron microscopic view of a) prepared microspheres, b) surface of the microspheres of batch F4
In Vitro and In Vivo Mucoadhesion Studies
Mucoadhesive behavior of CP is affected by factors such as pH and ionic strength (29). At pH ≤ 5.0, the lower degree of ionization of carboxyl groups results in less swelling of CP and consequently, its stronger interaction with polysaccharides present at the mucosal surface (30). These effects were reflected in this study. A proportional increase in mucoadhesive strength of the formulation was observed with increase in the ratio CP to HPMC (CP/HPMC) up to 1. Mucoadhesion differed significantly among F1 to F4 batches for any possible combination. It, however, decreased to a significant level when the proportion of CP was increased beyond 1 (p < 0.05, one-way ANOVA; Fig. 6a). This could be due to the fact that hydration of CP would have outweighed its mucoadhesive force (16). Further, except for batch F2, the percentage of microspheres retained in stomach was higher than those in the intestine.
Fig. 6.

a) In vitro and b) in vivo mucoadhesion tests of the prepared mucoadhesive microspheres upon stomach and intestinal mucosa of Wistar rats
In vivo mucoadhesion test, performed on batches F2, F3, and F4, correlated well with in vitro results (Fig. 6b). After 4 h, about 60% of microspheres from batch F4 were found to be retained within the stomach. Batches F3 and F5 showed significantly lower mucoadhesion in both stomach and intestinal mucosa (p < 0.05, one-way ANOVA). These results indicated that the microspheres constituted of CP and HPMC in the ratio of 1:1 underwent optimum hydration in vivo and therefore, were adhered to the stomach mucosa for extended period of time. This indeed is an advantageous effect since pH of stomach favors the solubility of R-HCl. Thus, the retention of formulation within stomach could possibly help in reducing the lag time of drug release.
In Vitro Drug Release
R-HCl has the pKa values of 8.95, 9.83, and 10.91 (3), and it showed pH-dependent solubility. Formulations, however, did not exhibit pH-dependent in vitro dissolution behavior (data not shown). In vitro release from pure R-HCl powder was found to be only 5.14% during the first 2 h, and a total of 25.81% was released at the end of 24 h, suggesting a strong need of improving its dissolution rate. Complexation with γ-CD and formulating it as microspheres resulted into significant improvement in its dissolution rate. This agreed well to the previously reported findings (31,32).
CP and HPMC, both have tendency of quick hydration at the entire physiological pH range (18,33). Therefore, microspheres from different batches showed difference in their release rates (Fig. 7). Batches F1 and F2 composed of CP/HPMC of 1:7 and 1:3, respectively, provided significantly lower drug release (p < 0.05) in comparison to the other batches. HPMC in higher proportion results into swelling of microspheres which in turn increases the diffusional path length of drug molecules, thus lowering their release rate (16). Likewise, batch F5 provided significantly lower drug release during the first 2 h, which demonstrated that CP/HPMC > 1 would result into profuse swelling of microspheres, thus reducing their release rate. Batch F3 provided a release pattern similar to that of F4, except for the first 3 h during which drug release from the former was significantly lower (p < 0.05). Batch F4, containing CP and HPMC in equivalent proportion, afforded a fairly controlled release pattern for the entire study period and its release rate conformed best to the mucoadhesive performance. Its composition was, therefore, considered to be optimized, and it was selected for performing pharmacokinetic studies.
Fig. 7.

In vitro drug release profile of different batches of the prepared microspheres
Pharmacokinetic Studies
R-HCl was found to be well separated under HPLC conditions used. Retention time was 3.32 ± 0.013 min (n = 6). No endogenous plasma components eluted at the retention time of drug. The limits of quantification and detection were 0.050 and 0.014 μg/ml, respectively. The calibration curve of drug in rat plasma was found to be linear (R2 = 0.9924) at concentrations ranging from 0.050 to 5.0 μg/ml. The extraction efficiencies were 85.2 ± 16.1, 79.7 ± 5.7, and 90.1 ± 6.4 (n = 3) at concentrations of 0.2, 0.8, and 5.0 μg/ml, respectively, which were indicative of little drug loss during extraction.
Microspheres produced a sharp plasma concentration–time profile of R-HCl within 30 min post-administration. Cmax of 358.44 ng/ml was observed at 2 h as against to 8 h provided by drug powder. More than fivefold increase in AUC and more than twofold increase in MRT were observed (Table III, Fig. 8). Another study, conducted in Wistar–Hannover rats with raloxifene-hydroxybutenyl-β-CD complex, reported twofold increase in Cmax and threefold increase in AUC as compared to pure drug (7). Thus, we concluded from our study that microsphere formulations could result into significant improvement in the bioavailability of R-HCl (one-way ANOVA, p < 0.05), which may subsequently help in its dose reduction to a remarkable level.
Table III.
Comparative Study of the Pharmacokinetic Parameters of Optimized Formulation (Batch F4) and Pure Drug Powder
| Sample | Pharmacokinetic parameters | ||||
|---|---|---|---|---|---|
| T max | C max | [AUC]0–24h | [AUC]0–∞ | MRT0–t | |
| Batch F4 | 2.0 h | 358.44 ng/ml | 1,722.34 ng h/ml | 1,773.53 ng h/ml | 12.51 h |
| Pure R-HCl | 8.0 h | 36.36 ng/ml | 318.68 ng h/ml | 361.38 ng h/ml | 6.43 h |
Fig. 8.

Plasma concentration vs. time (h) profile of the optimized formulation (batch F4) and pure drug powder following oral administration to Wistar rats
CONCLUSION
Results of this study showed that mucoadhesive microspheres composed of inclusion complex of R-HCl with γ-CD could be a radical approach to improve the bioavailability of R-HCl. DSC and X-RD studies provided the evidence that enhancement in solubility of drug resulted due to the change in its crystallinity within the formulation. More than 60% of oral dose was retained up to 4 h in stomach which provided much favorable pH conditions for dissolution of drug. The release rate of R-HCl from the prepared microspheres was controlled and extended up to 24 h. A significant improvement in the value of Tmax, Cmax, AUC, and MRT was obtained, which is indicative of improved absorption, higher bioavailability, and sustained effect of drug. The formulation could help in dose reduction of R-HCl to a remarkable level. However, it can be ascertained only after the clinical evaluation of the developed formulation.
Acknowledgments
The first author acknowledges the financial assistance of the University Grants Commission, New Delhi, in carrying out this research work. The support of Prof. O.N. Srivastava, Department of Physics, Banaras Hindu University, is thankfully acknowledged for providing the facility of SEM.
References
- 1.Epstein S. Update of current therapeutic options for the treatment of postmenopausal osteoporosis. Clin Ther. 2006;28:151–73. doi: 10.1016/j.clinthera.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 2.Vik SA, Maxwell CJ, Hanley-David A. Treatment of osteoporosis in an older home care population. BMC Musculoskel Disord. 2005;6:7. doi: 10.1186/1471-2474-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teeter JS, Meyerhoff RD. Environmental fate and chemistry of raloxifene hydrochloride. Environ Toxicol Chem. 2002;21:729–36. doi: 10.1002/etc.5620210407. [DOI] [PubMed] [Google Scholar]
- 4.Celnikier DH. Pharmacokinetics of raloxifene and its clinical application. Eur J Obs Gyn Reprod Biol. 1999;85:23–9. doi: 10.1016/S0301-2115(98)00278-4. [DOI] [PubMed] [Google Scholar]
- 5.Mizuma T. Intestinal glucuronidation metabolism may have a greater impact on oral bioavailability than hepatic glucuronidation metabolism in humans: a study with raloxifene, substrate for UGT1A1, 1A8, 1A9, and 1A10. Int J Pharm. 2009;378:140–1. doi: 10.1016/j.ijpharm.2009.05.044. [DOI] [PubMed] [Google Scholar]
- 6.Jagadish B, Yelchuri R, Bindu K, Tangi H, Maroju S, Rao VU. Enhanced dissolution and bioavailability of raloxifene hydrochloride by co-grinding with different superdisintegrants. Chem Pharm Bull. 2010;58:293–300. doi: 10.1248/cpb.58.293. [DOI] [PubMed] [Google Scholar]
- 7.Michael FW, Wacher VJ, Ruble KM, Ramsey MG, Edgar KJ, Buchanan NL, et al. Pharmacokinetics of raloxifene in male Wistar–Hannover rats: influence of complexation with hydroxybutenyl-beta-cyclodextrin. Int J Pharm. 2008;346:25–37. doi: 10.1016/j.ijpharm.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Garg A, Singh S, Rao VU, Bindu K, Balasubramaniam J. Solid state interaction of raloxifene HCl with different hydrophilic carriers during co-grinding and its effect on dissolution rate. Drug Dev Ind Pharm. 2009;35:455–70. doi: 10.1080/03639040802438365. [DOI] [PubMed] [Google Scholar]
- 9.Skalko N, Brandl M, Bedirevic-Lacan M, Filipovic-Grcic J, Jalsenjak I. Liposomes with nifedipine and nifedipine-cyclodextrin complex: calorimetrical and plasma stability comparison. Eur J Pharm Sci. 1996;4:359–66. doi: 10.1016/S0928-0987(96)00180-7. [DOI] [Google Scholar]
- 10.Dhanaraju D, Kumaran KS, Baskaran T, Moorthy MSR. Enhancement of bioavailability of griseofulvin by its complexation with beta-cyclodextrin. Drug Dev Ind Pharm. 1998;24:583–7. doi: 10.3109/03639049809085663. [DOI] [PubMed] [Google Scholar]
- 11.Cappello B, Rosa GD, Giannini L, Rotonda MIL, Mensitieri G, Miro A, et al. Cyclodextrin-containing poly(ethyleneoxide) tablets for the delivery of poorly soluble drugs: potential as buccal delivery system. Int J Pharm. 2006;319:63–70. doi: 10.1016/j.ijpharm.2006.03.031. [DOI] [PubMed] [Google Scholar]
- 12.Melia CD, Washington N, Wilson CG. Advantages and disadvantages of multiparticulate delivery systems. In: Melia CD, Washington N, Wilson CG, editors. Multiparticulate oral dosage forms: technology and biopharmaceutics. Edinburgh: Scottish Academic Press; 1994. pp. 135–40. [Google Scholar]
- 13.Tao Y, Lu Y, Sun Y, Gu B, Lu W, Pan J. Development of mucoadhesive microspheres of acyclovir with enhanced bioavailability. Int J Pharm. 2009;378:30–6. doi: 10.1016/j.ijpharm.2009.05.025. [DOI] [PubMed] [Google Scholar]
- 14.Wong SM, Kellaway IW, Murdan S. Enhancement of the dissolution rate and oral absorption of a poorly water soluble drug by formation of surfactant-containing microparticles. Int J Pharm. 2006;317:61–8. doi: 10.1016/j.ijpharm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 15.Myung KC, Hongkee S, Hoo-Kyung C. Preparation of mucoadhesive microspheres containing antimicrobial agents for eradication of H. pylori. Int J Pharm. 2005;297:172–9. doi: 10.1016/j.ijpharm.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Tiwari S, Singh S, Rawat M, Tilak R, Mishra B. L9 orthogonal design assisted formulation and evaluation of chitosan-based buccoadhesive films of miconazole nitrate. Curr Drug Deliv. 2009;6:305–16. doi: 10.2174/156720109788680921. [DOI] [PubMed] [Google Scholar]
- 17.Khanna R, Agarwal SP, Ahuja A. Mucoadhesive buccal tablets of clotrimazole for oral candidiasis. Drug Dev Ind Pharm. 1997;23:831–7. doi: 10.3109/03639049709150554. [DOI] [Google Scholar]
- 18.Li S, Lin S, Daggy BP, Mirchandani HL, Chien YW. Effect of HPMC and carbopol on the release and floating properties of gastric floating drug delivery system using factorial design. Int J Pharm. 2003;253:13–22. doi: 10.1016/S0378-5173(02)00642-7. [DOI] [PubMed] [Google Scholar]
- 19.Jug M, Beirevi-Laan M, Bengez S. Novel cyclodextrin-based film formulation intended for buccal delivery of atenolol. Drug Dev Ind Pharm. 2009;35:796–807. doi: 10.1080/03639040802596212. [DOI] [PubMed] [Google Scholar]
- 20.Gibaud S, Zirar SB, Mutzenhardt P, Isabelle F, Astier A. Melarsoprol–cyclodextrin inclusion complexes. Int J Pharm. 2005;306:107–21. doi: 10.1016/j.ijpharm.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Patil SB, Murthy RSR. Preparation and in vitro evaluation of mucoadhesive chitosan microspheres of amlodipine besylate for nasal administration. Indian J Pharm Sci. 2006;68:64–7. doi: 10.4103/0250-474X.22966. [DOI] [Google Scholar]
- 22.Albrecht K, Greindl M, Kremser C, Wolf C, Debbage P, Bernkop-Schnürch A. Comparative in vivo mucoadhesion studies of thiomer formulations using magnetic resonance imaging and fluorescence detection. J Control Release. 2006;115:78–84. doi: 10.1016/j.jconrel.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 23.Yang ZY, Zhang ZF, He XB, Zhao GY, Zhang YQ. Validation of a novel HPLC method for the determination of raloxifene and its pharmacokinetics in rat plasma. Chromatographia. 2007;65:197–201. doi: 10.1365/s10337-006-0123-4. [DOI] [Google Scholar]
- 24.Koester LS, Bertuol JB, Groch KR, Xavier CR, Moellerke R, Mayorga P, et al. Bioavailability of carbamazepine: β-cyclodextrin complex in beagle dogs from hydroxypropylmethylcellulose matrix tablets. Eur J Pharm Sci. 2004;22:201–7. doi: 10.1016/j.ejps.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Marcon F, Mathiron D, Pilard S, Lemaire-Hurtel AS, Dubaele JM, Djedaini-Pilard F. Development and formulation of a 0.2% oral solution of midazolam containing γ-cyclodextrin. Int J Pharm. 2009;379:244–50. doi: 10.1016/j.ijpharm.2009.05.029. [DOI] [PubMed] [Google Scholar]
- 26.Vippangunta SR, Maul KA, Tallavajhala S, Grant DJW. Solid state characterization of nifedipine solid dispersion. Int J Pharm. 2002;236:111–26. doi: 10.1016/S0378-5173(02)00019-4. [DOI] [PubMed] [Google Scholar]
- 27.Bilensoy E, Rouf MA, Vural I, Şen M, Hıncal AA. Mucoadhesive, thermosensitive, prolonged-release vaginal gel for clotrimazole: β-cyclodextrin complex. AAPS PharmSciTech. 2006;7:E3–7. doi: 10.1208/pt070238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaefer MJ, Singh J. Effect of isopropyl myristic acid ester on the physical characteristics and in vitro release of etoposide from PLGA microspheres. AAPS PharmSciTech. 2000;1:article 32. doi: 10.1208/pt010432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singla AK, Chawla M, Singh A. Potential applications of carbomer in oral mucoadhesive controlled drug delivery system: a review. Drug Dev Ind Pharm. 2000;26:913–24. doi: 10.1081/DDC-100101318. [DOI] [PubMed] [Google Scholar]
- 30.Chun M, Kwak B, Choi CH. Preparation of buccal patch composed of carbopol, poloxamer and hydroxypropyl methylcellulose. Arch Pharm Res. 2003;26:973–8. doi: 10.1007/BF02980208. [DOI] [PubMed] [Google Scholar]
- 31.Han RY, Fang JY, Sung KC, Hu OYP. Mucoadhesive buccal disks for novel nalbuphine prodrug controlled delivery: effect of formulation variables on drug release and mucoadhesive performance. Int J Pharm. 1999;177:201–9. doi: 10.1016/S0378-5173(98)00343-3. [DOI] [PubMed] [Google Scholar]
- 32.Gavini E, Rassu G, Haukvik T, Lanni C, Racchi M, Giunchedi P. Mucoadhesive microspheres for nasal administration of cyclodextrins. J Drug Target. 2009;17:168–79. doi: 10.1080/10611860802556842. [DOI] [PubMed] [Google Scholar]
- 33.Park SH, Chun MK, Choi HK. Preparation of an extended-release matrix tablet using chitosan/carbopol interpolymer complex. Int J Pharm. 2008;347:39–44. doi: 10.1016/j.ijpharm.2007.06.024. [DOI] [PubMed] [Google Scholar]