

Abstract
As the ultimate electron acceptor in oxidative phosphorylation, oxygen plays a critical role in metabolism. When oxygen levels drop, heterodimeric hypoxia-inducible factor (Hif) transcription factors become active and facilitate adaptation to hypoxia. Hif regulation by oxygen requires the protein von Hippel-Lindau (pVhl) and pVhl disruption results in constitutive Hif activation. The liver is a critical organ for metabolic homeostasis, and Vhl inactivation in hepatocytes results in a Hif-dependent shortening in life span. While albumin-Cre;VhlF/F mice develop hepatic steatosis and impaired fatty acid oxidation, the variable penetrance and unpredictable life expectancy has made the cause of death elusive. Using a system in which Vhl is acutely disrupted and a combination of ex vivo liver perfusion studies and in vivo oxygen measurements, we demonstrate that Vhl is essential for mitochondrial respiration in vivo. Adenovirus-Cre mediated acute Vhl disruption in the liver caused death within days. Deprived of pVhl, livers accumulated tryglicerides and circulating ketone and glucose levels dropped. The phenotype was reminiscent of inborn defects in fatty acid oxidation and of fasted PPARα-deficient mice and while death was unaffected by pharmacologic PPARα activation, it was delayed by glucose administration. Ex vivo liver perfusion analyses and acylcarnitine profiles showed mitochondrial impairment and a profound inhibition of liver ketone and glucose production. By contrast, other mitochondrial functions, such as ureagenesis, were unaffected. Oxygen consumption studies revealed a marked suppression of mitochondrial respiration, which, as determined by magnetic resonance oximetry in live mice, was accompanied by a corresponding increase in liver pO2. Importantly, simultaneous inactivation of Hif-1β suppressed liver steatosis and rescued the mice from death. These data demonstrate that constitutive Hif activation in mice is sufficient to suppress mitochondrial respiration in vivo and that no other pathway exists in the liver that can allow oxygen utilization when Hif is active precluding thereby metabolic collapse.
Keywords: Vhl, Hif, liver steatosis, mitochondrial respiration
Introduction
The von Hippel-Lindau (VHL) gene functions as a tumor suppressor. Inactivating germline VHL mutations predispose to renal cell carcinoma (RCC) of clear-cell type (ccRCC) (Latif et al 1993), and somatic VHL mutations frequently occur in sporadic ccRCC (Gnarra et al 1994, Nickerson et al 2008), where they are accompanied by loss of heterozygosity. The VHL protein (pVHL) functions as the substrate recognition subunit of an E3 ubiquitin ligase complex that targets, among others, hypoxia-inducible factor (HIF) α subunits for degradation (Cockman et al 2000, Kamura et al 2000, Maxwell et al 1999, Ohh et al 2000, Tanimoto et al 2000). The levels of HIF-α subunits are regulated by pVHL in an oxygen-dependent manner (Maxwell et al 1999). When oxygen abounds, HIF-α subunits are hydroxylated at specific prolyl residues creating high affinity binding sites for pVHL with resultant polyubiquitylation and degradation (Ivan et al 2001, Jaakkola et al 2001, Masson et al 2001, Yu et al 2001). In contrast, when oxygen levels are low, HIF-α subunits remain unmodified, evade pVHL recognition, interact with HIF-β leading to the formation of heterodimeric transcription factors that translocate to the nucleus where they regulate transcription (Ozer and Bruick 2007). Both HIF-1α and HIF-2α subunits interact with Hif-1β and are regulated by pVHL (Kaelin and Ratcliffe 2008). When pVHL is disrupted, HIF-1α and HIF-2α regulation becomes uncoupled from oxygen levels, leading to constitutive expression of their target genes (Ohh 2006).
Vhl function has been extensively studied in both epithelial and mesenchymal tissues (Kapitsinou and Haase 2008). Homozygous Vhl mutant embryos die in midgestation seemingly from abnormal placental vascularization (Gnarra et al 1997). Vhl inactivation at embryonic day (E)10.5 using a tamoxifen inducible Cre results in liver necrosis and death around E15 (Hong et al 2006). Vhl is essential for liver function, and Vhl disruption in adult hepatocytes results in premature death, typically within weeks (Haase et al 2001, Kim et al 2006b, Park et al 2007, Peyssonnaux et al 2007, Rankin et al 2009). However, despite extensive studies (Chen et al 2010, Haase et al 2001, Hong et al 2006, Kim et al 2006b, Park et al 2007, Peyssonnaux et al 2007, Rankin et al 2007, Rankin et al 2009) how Vhl is important for liver function and survival is not known. In adult hepatocytes, Vhl loss results in increased erythropoietin production with consequent polycythemia (Peyssonnaux et al 2007, Rankin et al 2007), glycogen accumulation (Park et al 2007) and the accumulation of neutral lipids (Haase et al 2001, Kim et al 2006b, Peyssonnaux et al 2007, Rankin et al 2009). However, none of these functions have been linked to the death of mice. In addition, liver biosynthetic function appears to be preserved (Peyssonnaux et al 2007). Recently, pVhl was shown to be required for β-oxidation of fatty acids, and this may explain the steatotic phenotype (Rankin et al 2009). However, liver steatosis per se is unlikely to cause death. In all adult models, variable penetrance and unpredictable survival have made unraveling the cause of death impossible.
Here, we acutely inactivated Vhl in hepatocytes with an adenovirus coding for Cre recombinase (Ad-Cre) and using a variety of ex vivo and in vivo approaches, we demonstrate that Vhl is required for mitochondrial respiration in hepatocytes in vivo and that Vhl loss blocks ketone and glucose production leading to hypoketonemia, hypoglycemia and death within days. As evidenced by simultaneous Hif-1β inactivation, the deleterious effects induced by Vhl loss are Hif-dependent. Constitutive Hif activation in hepatocytes blocked mitochondria from utilizing available oxygen indicating that no other pathway in the liver exists that can overcome a perceived state of hypoxia imposed by Hif. To our knowledge, this is the first report to show that Hif activation is sufficient to block mitochondrial respiration in vivo.
Results
Acute Vhl inactivation in the liver leads to hepatic steatosis, hyperlipidemia, and death
Vhl is essential for normal liver function and survival. Consistent with previous studies (Haase et al 2001, Kim et al 2006b, Peyssonnaux et al 2007, Rankin et al 2009), albumin-Cre; VhlF/Fmice develop liver steatosis and exhibit a shortened life span (Figure 1). We were interested in understanding what function Vhl played in hepatocytes that was essential for survival, but the variability in life expectancy across individual mice made it impossible to establish with certainty the cause of death (Figure 1B). Whereas some mice would succumb before 4 weeks of age, others survived past 10 weeks. Furthermore, when moribund mice were evaluated it was impossible to determine whether the abnormalities observed were the cause of illness or simply indicators of their moribund state.
Figure 1.
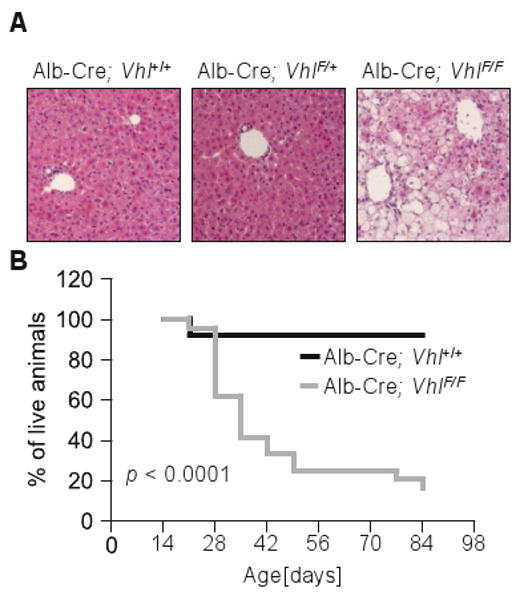
Albumin-Cre; VhlF/F mice develop liver steatosis and exhibit a shortened but unpredictable survival. (A) Microscopic sections of representative liver areas from 3-week old animals (original magnification × 200). (B) Survival curves of Albumin-Cre; Vhl+/+ and Albumin-Cre; VhlF/F animals (n = 50).
We reasoned that abrupt Vhl disruption using Ad-Cre may result in an accelerated and more synchronous phenotype amenable to longitudinal studies and experimental dissection. Ad-Cre would not only result in acute loss of pVhl, but also induce a state of stress providing thereby a sensitized background for analysis. Tail vein injection of Ad-Cre resulted in efficient VhlF allele recombination in the liver (Figure 2A). By contrast, Ad-Cre had little effect on VhlF in other organs (Figure 2A, Figure S1A). Three days after Ad-Cre, approximately 70% of the VhlF allele in liver extracts was recombined and considering that hepatocytes account for 70-80% of the liver, it was estimated that Vhl was inactivated in almost all targeted cells. VhlF recombination was accompanied by a decrease in pVhl, which was undetectable 3 days after Ad-Cre (Figure 2B). As expected, pVhl depletion activated Hif leading to the upregulation of both Hif-1α liver specific targets such as Pdk1 (Rankin et al 2007) as well as Hif-2α targets such as Vegf (Rankin et al 2008) (Table 1 and 2).
Figure 2.
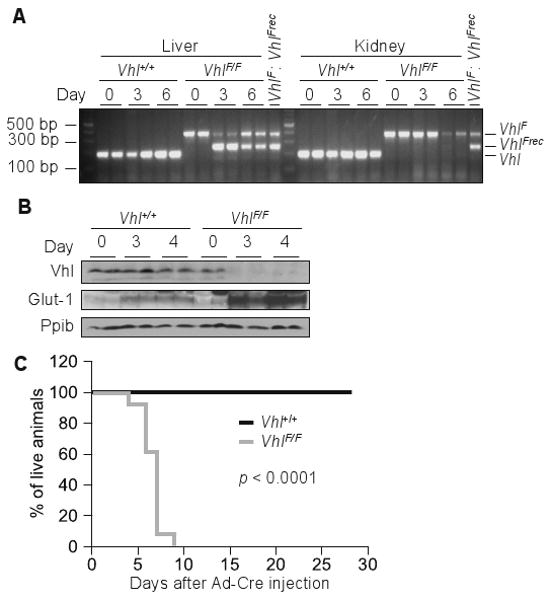
Acute Vhl inactivation in hepatocytes is lethal. (A) PCR analysis of genomic DNA isolated from tissues of Vhl+/+ and VhlF/F animals at the indicated number of days post Ad-Cre injection; 1:1 mixture of genomic DNA containing unrecombined (VhlF) and completely recombined (VhlFrec) alleles was used as amplification control. (B) Western blot analysis of liver lysates from 24 hours fasted animals of the indicated genotypes following the stated number of days after Ad-Cre. (C) Survival curve of Ad-Cre injected animals of the indicated genotypes (n = 21).
Table 1.
qRT-PCR analysis of gene expression in mice of the indicated genotypes at the indicated number of days following Ad-Cre injection (n = 3 for genotype and timepoint). * p < 0.05, ** p < 0.01.
| Fold change VhlF/F / Vhl+/+ | |||
|---|---|---|---|
| Day 0 | Day 3 | Day 4 | |
| Hif targets | |||
| Vegf | 1.23 | 7.95** | 13.73** |
| Glut-1 | 1.09 | 2.43 | 6.45 |
| SREBP pathway | |||
| SCAP | 1.14 | 1.14 | 1.37 |
| Insig-1 | 1.12 | 1.3 | 0.76 |
| Insig-2a | 0.81 | 4.93** | 11.56** |
| Insig-2b | 1.22 | 1.07 | 1.86 |
| SREBP-1a | 1.28 | 1.44* | 1.53 |
| SREBP-1c | 1.5 | 0.83 | 0.04* |
| SREBP-2 | 1.2 | 1.22 | 1.28 |
| Cholesterol synthesis | |||
| FPP Synthase | 0.89 | 0.67 | 0.43 |
| Squalene Syntase | 1.04 | 0.68 | 0.32 |
| HMG-CoA Synthase 1 | 1.1 | 0.83 | 0.53 |
| HMG-Coa Reductase | 1.59 | 0.72 | 0.69 |
| Cholesterol degradation and transport | |||
| ABCA1 | 1.13 | 2.66* | 3.83** |
| ABCG5 | 1.11 | 0.92 | 0.22* |
| CYP7A1 | 1.08 | 0.51 | 0.02* |
| LDL Receptor | 1.01 | 0.76 | 0.44 |
| VLDL Receptor | 1.96 | 3.26* | 15.07* |
| Triglyceride synthesis | |||
| GPAT | 0.93 | 1.55* | 1.38 |
| DGAT1 | 1.04 | 0.8 | 1.1 |
| DGAT2 | 1.14 | 0.9 | 0.28* |
| AGPAT1 | 1.13 | 1.52 | 1.75 |
| AGPAT2 | 1.0 | 1.42 | 0.94 |
| AGPAT3 | 0.94 | 1.4 | 2.16* |
| AGPAT4 | 0.94 | 1.71 | 2.06** |
| Nuclear Receptors | |||
| PPARα | 1.06 | 1.68 | 2.44 |
| PPARδ | 1.32 | 3.15 | 6.23* |
| PPARγ | 1.24 | 1.07 | 1.43 |
| LXRα | 1.22 | 1.31 | 1.04 |
| Fatty acid synthesis | |||
| Malic enzyme | 1.04 | 0.59 | 0.73 |
| Acyl-CoA Synthase | 0.83 | 1.19 | 0.64 |
| Acetyl-CoA Carboxylase 1 | 1.06 | 0.58 | 0.33* |
| Fatty Acid Synthase | 1.06 | 0.61 | 0.24* |
| Fatty acid oxidation | |||
| Acyl-CoA Oxidase 1 | 0.87 | 1.19 | 0.79 |
| Acetyl-CoA Carboxylase 2 | 0.85 | 1.35 | 1.02 |
| Carnitine Palmitoyl Transferase 2 | 0.98 | 1.16 | 1.5 |
Table 2.
qRT-PCR analysis of gene expression in Ad-Cre injected animals, fasted for 24 hours (n = 4 for genotype and timepoint).* p < 0.05, ** p < 0.01, *** p < 0.001.
| Fold change VhlF/F / Vhl+/+, fasted | |||
|---|---|---|---|
| Day 0 | Day 3 | Day 4 | |
| Hif targets | |||
| Glut-1 | 1.18 | 2.21** | 4.68* |
| Pdk1 | 1.06 | 4.18*** | 6.66*** |
| Nuclear Receptors | |||
| PPARα | 1.16 | 1.63*** | 1.41 |
| RXRα | 0.98 | 0.67** | 0.83 |
| Fatty acid oxidation | |||
| Acyl-CoA Oxidase 1 | 1.36 | 0.68** | 1.09 |
| Carnitine palmitoyl transferase 1a | 1.13 | 0.45** | 0.37*** |
| Carnitine palmitoyl transferase 2 | 1.03 | 1 | 1.33 |
| Mcad | 1.34 | 1.18 | 1.17 |
| Lcad | 1.09 | 1.04 | 1.51 |
| Ketogenesis | |||
| HMG-CoA synthase 2 | 1.33 | 0.99 | 1.18 |
| Gluconeogenesis | |||
| Pepck | 1.14 | 0.44** | 0.49 |
| G6Pase | 0.96 | 0.72 | 1.2 |
| PGC-1α | 0.91 | 0.45* | 0.5 |
| Lipid droplet proteins | |||
| Adfp | 1.05 | 1.22 | 1.71 |
Ad-Cre mediated Vhl inactivation was lethal within days. Median survival of Ad-Cre VhlF/F mice was 6 days and all mice were dead by day 10 (Figure 2C). By contrast, survival was unaffected in similarly Ad-Cre injected Vhl+/+ animals. The differences between VhlF/F and Vhl+/+ mice could not be attributed to differences in Ad-Cre as both strains showed similar changes in liver function tests and weight (data not shown). In addition, both VhlF/F and Vhl+/+ livers exhibited a similar infiltration of immune cells (Figure S1B). The immune cell infiltration was manifested in VhlF/F livers also by an increase in unrecombined Vhl allele starting at day 4 after Ad-Cre injection (Figure 2A, Figure S1A). While the rapidity of the death may be exacerbated by the adenoviral infection, the death phenotype was clearly Vhl-dependent and none of the wild-type mice died (Figure 2C). The brisk and nearly synchronous death of VhlF/F mice provided a suitable experimental system to dissect the reason why Vhl function was essential for liver function and survival. In addition, by comparing to similarly Ad-Cre injected mice, we were able to exclude confounding effects from Ad-Cre.
This experimental paradigm allowed us to study the changes induced by Vhl loss longitudinally over time and obtain measurements before the mice became moribund. At day 3 post-Ad-Cre injection, both VhlF/F and wild-type livers exhibited mild microsteatosis (Figure S1B). However, whereas the steatosis improved in wild-type animals, it worsened in VhlF/F (Figure 3A and B, Figure S1B). Oil red O staining indicated the accumulation of neutral lipids in Vhl-deficient livers (Figure 3B, Figure S1B). Triglycerides were increased in Ad-Cre VhlF/F livers (Figure 3C) and the mice developed hypertriglyceridemia (Figure 3D). By contrast, liver cholesterol content was comparable in all animals (Figure 3E), and circulating cholesterol was only mildly elevated in Ad-Cre VhlF/F mice (Figure 3F).
Figure 3.
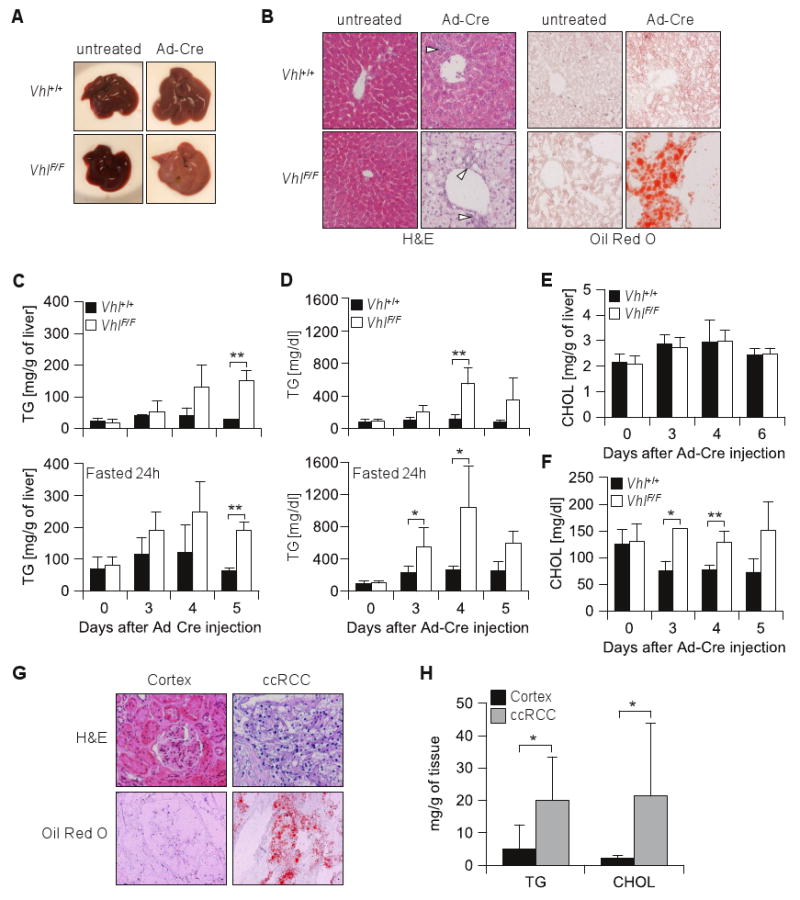
Acute Vhl inactivation in the liver leads to hepatic steatosis reminiscent of the lipid accumulation in ccRCC. (A) Macroscopic images and (B) microscopic sections of representative liver areas (original magnification × 200) of mice fasted for 24 hours of the indicated genotypes, either untreated or 5 days after Ad-Cre injection; open arrow heads, inflammatory cells; H&E, hematoxylin eosin. (C) Triglyceride composition of livers from animals of the indicated genotypes, either nonfasted or fasted for 24 hours (n = 2-4 for each genotype and timepoint). (D) Circulating triglycerides from nonfasted or 24 hours fasted animals (n = 3-6 for each genotype and timepoint). Cholesterol levels in nonfasted animals (as in C) in liver (E) or serum (F). (G) Representative images of clear-cell renal cell carcinoma (ccRCC) and normal renal cortex sections stained by H&E and Oil red O with a hematoxylin counterstain (original magnification × 200). (H) Lipid composition analysis of ccRCC and normal renal cortex (n = 9-10 per group). *, p < 0.05; **, p < 0.01.
As fasting may cause mild liver steatosis and to adjust for potential differences in food intake between Ad-Cre injected VhlF/F and Vhl+/+ mice, similar studies were performed after a 24 hour fast. While fasting resulted in an increase in baseline TG in the liver and to some extent in the plasma, higher TG levels were again observed in VhlF/F mice (Figure 3C and D).
Interestingly, the lipid accumulation in Vhl-deficient livers was reminiscent of ccRCC which accumulate neutral lipids giving raise to the clear-cell histological appearance (Figure 3G). However, while cholesterol esters are known to be increased in ccRCC (Gebhard et al 1987, Tosi et al 2004), whether TG accumulate is less clear. We analyzed the lipid composition of a set of ccRCCs and compared the results to samples of normal renal cortex, from where ccRCCs are thought to arise (Zambrano et al 1999). Our data show that ccRCCs accumulate not only cholesterol but, like hepatocytes, also TG (Figure 3H).
The rapid accumulation of TG in the liver of Ad-Cre VhlF/F mice suggested that the role of pVhl in survival may be linked to its role in lipid metabolism. To obtain insight into the metabolic abnormalities underlying the steatosis phenotype, we performed extensive gene expression studies of ∼40 genes implicated in lipid metabolism (Table 1 and 2). In addition, we also directly measured the production by the liver of key metabolites by performing ex vivo liver perfusion experiments. qRT-PCR studies were performed in VhlF/F and Vhl+/+ livers on days 3 and 4 following Ad-Cre injection. The cyclophilin gene (Ppib) was chosen for normalization because, unlike other housekeeping or cytoskeletal genes, its expression was not altered by Ad-Cre (data not shown). In uninjected VhlF/F and Vhl+/+ livers gene expression was, as expected, very similar (Day 0, Table 1 and 2). However, marked differences were observed between VhlF/F and Vhl+/+ livers following Ad-Cre (Table 1 and 2). Hif target genes, including the recently described Insig-2a (Nguyen et al 2007), were induced up to 13 fold in Vhl-deficient livers (Table 1 and 2). Among the genes that were markedly upregulated following Vhl disruption was the VLDL receptor (Table 1), which has been recently proposed to be regulated by Hif-1 (Kasper et al 2005, Manalo et al 2005). In addition, SREBP-1c, an important activator of fatty acid (FA) synthesis genes (Horton et al 2002), was downregulated over 20-fold in Vhl-deficient livers (Table 1). By contrast, the expression of PPARγ was not significantly altered (Table 1). With respect to genes involved in triglyceride synthesis there was no uniform trend; while the mRNAs for acyl glycerol-3-phosphate acyltransferase 3 and 4 were modestly increased, expression of diacylglycerol acyltransferase 2 was decreased, and the expression of several other genes was unchanged (Table 1). Overall the data suggested that the accumulation of TG in the liver was not due to increased lipogenic gene expression.
Inhibition of ketone production in Vhl-deficient livers
To evaluate the cause of death, we measured plasma ketone levels, a key energy source for peripheral tissues during fasting (Wakil and Abu-Elheiga 2009). As expected in uninjected, fasted, VhlF/F and Vhl+/+ mice, plasma ketone levels were indistinguishable (Figure 4A, day 0). Following Ad-Cre injection, circulating ketone levels remained constant in Vhl+/+ mice, but dropped significantly in VhlF/F mice (Figure 4A). As early as day 3 post Ad-Cre, circulating ketones were almost 2-fold lower in VhlF/F compared to Vhl+/+ mice and the levels progressively decreased over time (Figure 4A). Next, we evaluated ketone body production in ex vivo perfusion experiments of intact livers. The levels of ketone bodies produced by Vhl-deficient livers were substantially lower than those produced by wild-type livers (Figure 4B). These data indicated that disruption of pVhl resulted in an inhibition of liver ketogenesis markedly reducing the amounts of circulating ketone bodies.
Figure 4.

Vhl inactivation blocks hepatic ketone production leading to decreased circulating ketone levels and death which is refractory to PPARα activation. (A) Levels of plasma ketones (acetoacetate plus 3-hydroxybutyrate) in animals fasted for 24 hours (for both genotypes, n = 2 for day 0, n = 4-8 for other time points). (B) Ketone body production by perfused livers isolated from 24 hours fasted animals at day 4 post Ad-Cre injection (n = 6 for each genotype); TK, total ketones; BHB, β-hydroxybutyrate; ACAC, acetoacetate. (C) In vitro fatty acid oxidation rates measured as 14CO2 production in liver homogenates from 24 hours fasted animals at day 3 post Ad-Cre injection (n = 2). (D) qRT-PCR analysis of PPARα target genes (or controls) normalized to Ppib in animals of the indicated genotypes 3 days after Ad-Cre injection and following 4 days of WY-14643 (WY), olive oil (vehicle), or no treatment (NT) (for both genotypes, n = 4-5 for each treatment). *, p value comparing VhlF/F vs. Vhl+/+; #, p value comparing WY to vehicle within the same mouse strain. (E) Survival curve for Ad-Cre injected animals treated with olive oil (vehicle) or WY-14643 (n = 5 for each genotype and treatment group). (F) Analysis of plasma triglycerides, same animals as in D (n = 3 per group). * and #, p < 0.05; ** and ##, p < 0.01.
Low ketone production was not caused by decreased expression of the rate limiting enzyme, 3-hydroxy-3-methylglutaryl-CoA synthase 2 (Table 2). However, we observed a 2-fold decrease in FAO rates in liver homogenates from VhlF/F animals as early as day 3 post Ad-Cre injection (Figure 4C). In addition, the expression of several PPARα target genes was downregulated even when PPARα itself was not (Table 2). Notably, PPARα activity has been shown to be downregulated by HIF-1α (Belanger et al 2007, Huss et al 2001) and the phenotype observed was reminiscent of fasted PPARα-deficient mice (Mandard et al 2004). Thus, we sought to activate PPARα in VhlF/F mice and used a pharmacological activator, WY-14643. As determined by the expression of PPARα target genes such as Cyp4a14 and Lcad, WY-14643 led to PPARα activation in the livers of both wild-type and VhlF/F Ad-Cre injected mice (Figure 4D). However, despite PPARα activation and increased expression of lipolytic genes, WY-14643 did not seemingly affect liver steatosis or prolong the survival VhlF/F mice (Figure 4E and data not shown). Incidentally, during the experiments with WY-14643, we noted that in contrast to wild-type mice, the administration of either WY-14643 or olive oil (vehicle) to VhlF/F animals caused the plasma to appear turbid and this was accompanied by a trend towards marked hypertrygliceridemia (Figure 4F).
Decreased liver gluconeogenesis contributes to the death of VhlF/F animals
Next we evaluated glucose levels. At day 5 following Ad-Cre, glucose levels were significantly lower in VhlF/F than in wild-type mice (Figure 5A). However, whereas differences in circulating ketones were observed already at day 3, differences in glucose levels were not observed prior to day 5. Nevertheless, already at day 4, very profound differences were observed in liver glucogeneogenesis. As determined by ex vivo liver perfusion experiments, Vhl-deficient livers produced 70% less glucose than livers from wild-type mice (Figure 5B). These findings were accompanied by a trend towards decreased expression of the major gluconeogenic regulator, peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase (Pepck) (Table 2).
Figure 5.

Decreased hepatic glucose production with consequent hypoglycemia contributes to the death of VhlF/F animals. (A) Plasma glucose levels after a 24 hour fast. (B) Glucose and (C) urea production by perfused livers from animals fasted ∼24 hours on day 4 post Ad-Cre injection (n = 6 for each genotype). (D) Survival curves of VhlF/F animals treated with glucose (or saline) intraperitoneally at 8 hour intervals starting when indicated (n = 7 for each treatment). *, p < 0.05; ***, p < 0.001.
Overall, a profound disruption was observed in both glucose and ketone production by Vhl-deficient livers. To determine whether this represented a general disruption of liver metabolic functions, we measured urea production in perfused livers. Despite impaired gluconeogenesis and ketogenesis, urea production was not significantly different between Vhl-deficient and wild-type livers (Figure 5C). Thus, the defects observed in glucose and ketone production were not the consequence of an overall impairment of liver function. Because ureagenesis requires mitochondria, the data also indicated that the alterations observed were not due to a global impairment of mitochondria.
Next, we sought to determine whether glucose supplementation would prolong the survival of VhlF/F mice. For these experiments, VhlF/F mice were given intraperitoneal glucose every 8 hours starting 70 hours after Ad-Cre injection. By comparison to saline treated VhlF/F mice, the administration of glucose resulted in a modest, but statistically significant improvement in median survival (p = 0.025) (Figure 5D). Taken together these data show that pVhl loss results in an inhibition of liver gluconeogenesis and that low glucose levels contribute to the death of the mice.
Inhibition of mitochondrial respiration by pVhl disruption
To analyze the liver metabolic phenotype further, we measured acylcarnitine levels. Free carnitine levels were lower in Vhl-deficient livers compared to wild-type in both fed and fasted states (Figure 6A, Table S1). Lower free carnitine levels in Vhl-deficient livers correlated with an accumulation of short and medium chain acylcarnitines with an even number of carbon residues. Acetyl (C2) and butyryl (C4) carnitines were 2-3 fold increased in pVhl-depleted livers. In addition, under fasting conditions octanoylcarnitine (C8) was similarly increased in pVhl-depleted livers (Figure 6A). These data indicated that Vhl-deficient livers failed to completely oxidize fatty acids.
Figure 6.
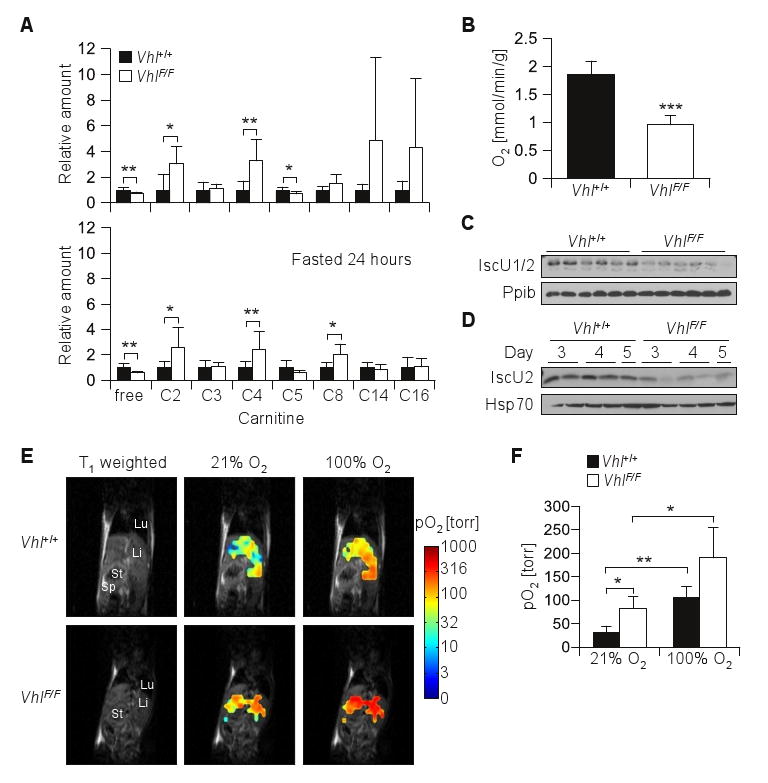
Accumulation of short acylcarnitines and inhibition of oxygen consumption in Vhl-deficient livers. (A) Acylcarnitine levels in extracts from livers of fed or fasted animals sacrificed at day 4 after Ad-Cre injection, (n = 6-7 for each genotype in fed or fasted conditions; for absolute values see Table S1). (B) Oxygen consumption rates of perfused livers from animals fasted ∼24 hours and sacrificed on day 4 post Ad-Cre injection (n = 6 for each genotype). (C) Western blot analysis of perfused livers (as in B) or crude mitochondria (D) isolated from livers of fasted animals at the indicated days after Ad-Cre injection. (E) Liver oximetry using 19F MRI of Oxypherol (perfluorobutylamine emulsion) in Vhl+/+and VhlF/F mice on day 4 post Ad-Cre injection. Representative T1-weighted 1H MRI coronal scans illustrating anatomy of mouse torso showing lung (Lu), liver (Li), stomach (St) and spleen (Sp) from a representative pair of animals. Overlay on anatomical images of pO2 maps (logarithmic color scale) obtained while breathing air or 100% oxygen. (F) Summary of in vivo pO2 measurement (n = 3 for each genotype). *, p < 0.05; **, p < 0.01; ***, p < 0.001, paired t test was used when comparing 21% vs. 100% O2.
To further evaluate the impairment in mitochondrial oxidative capacity, we measured oxygen consumption in intact livers. By comparison to wild-type livers from similarly Ad-Cre injected mice, oxygen consumption was reduced in Vhl-deficient livers. Vhl-deficient livers exhibited an approximately 2-fold decrease in oxygen extraction (Figure 6B). Because mitochondria account for more than 90% of the oxygen consumed by tissues (Taylor 2008), these data indicated that pVhl disruption led to a striking suppression of mitochondrial respiration. Multiple factors are likely to contribute to the decreased mitochondrial respiration in Vhl-deficient livers. Hif-1 regulates pyruvate dehydogenase kinase (Pdk1) (Kim et al 2006a, Papandreou et al 2006) and Pdk1 expression was elevated in Vhl-deficient livers 4-6 fold (Table 2). In addition, the Fe-S cluster assembly proteins IscU1/2 were recently shown to be regulated by pVhl (Chan et al 2009) and IscU1/2 levels were reduced in the livers from Ad-Cre injected VhlF/F mice (Figure 6C and D).
We sought to extend our observations in perfused livers to live mice and measured pO2 in the liver of anesthetized animals using fluorine (19F) magnetic resonance imaging (MRI). For these experiments, Oxypherol, an MRI pO2 reporter probe that is sequestered in the liver, was administered to Ad-Cre injected VhlF/F and Vhl+/+ mice and the mice were evaluated by 19F MRI. pO2 maps were acquired and overlaid onto T1-weighted 1H MRI anatomical images (Figure 6E). Pronounced pO2 differences were observed between wild-type and Vhl-deficient livers. Whereas in wild-type livers hepatic pO2 was 31±14 torr, in Vhl-deficient livers pO2 was 83±24 torr (p < 0.05; Figure 6F). Thus, the 2-fold decrease in oxygen consumption in perfused Vhl-deficient livers previously observed correlated with a reciprocal increase of a similar magnitude in oxygen tension. Further experiments were performed with the administration of 100% O2. Increasing the inspired O2 concentration resulted in a similar fold increase in pO2 in both wild-type and Vhl-deficient livers (Figure 6F). Thus, the delivery of oxygen to Vhl-deficient livers was intact. In summary, acute pVhl disruption resulted in a profound suppression of mitochondrial respiration and this is accompanied by a corresponding increase in pO2.
Rescue of Ad-Cre VhlF/F mice by Hif-1β disruption
Finally, we sought to determine whether the phenotype observed was dependent on Hif. Because both Hif-1α and Hif-2α have been implicated in liver steatosis (Kim et al 2006b, Rankin et al 2009), we chose to inactivate both of them by eliminating the shared Hif-1β subunit (Tomita et al 2000). Mice carrying a VhlF allele were interbred with mice carrying a Hif-1βF allele to generate VhlF/F; Hif-1βF/F mice. VhlF/F; Hif-1βF/F and VhlF/F mice were subsequently injected with Ad-Cre and efficient loxP site recombination was observed by PCR (Figure 7A). Importantly, in the absence of Hif-1β, Vhl inactivation did not lead to hepatic steatosis or hypertriglyceridemia (Figure 7B and C). Moreover, Hif-1β inactivation rescued VhlF/F mice from death (Figure 7D). These data show that Hif-1β is required for the deleterious effects of Vhl loss and implicates Hif in the inhibition of mitochondrial functions observed following pVhl inactivation. Together with our oxygen measurement studies, these data show that Hif activation in the liver uncoupled from oxygen sensing is sufficient to block mitochondrial respiration and cause death.
Figure 7.
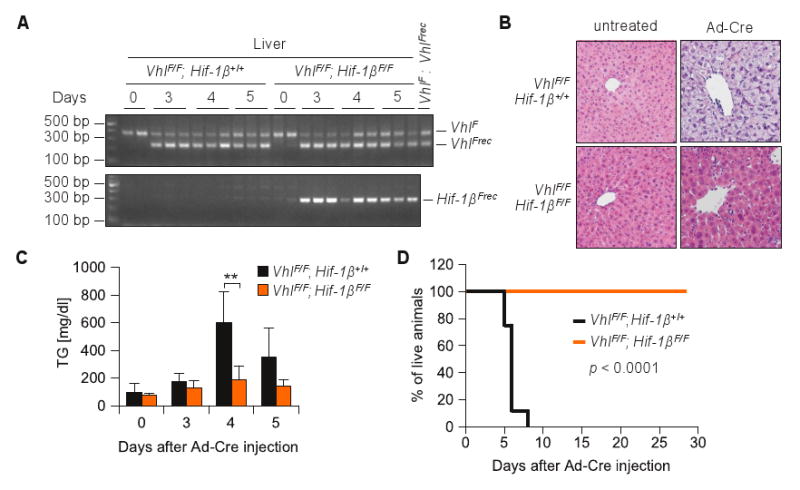
Rescue of Ad-Cre VhlF/F mice by Hif-1β loss. (A) PCR analysis of genomic DNA isolated from livers of VhlF/F animals either Hif-1β+/+ or Hif-1βF/F at the indicated number of days post Ad-Cre injection. (B) H&E staining of representative liver sections from VhlF/F; Hif-1β+/+ livers (and controls) analyzed at day 5 post Ad-Cre injection (original magnification × 200). (C) Plasma triglyceride levels in Ad-Cre treated animals (n = 3-4 for each genotype and timepoint). **, p < 0.01. (D) Survival curve for Ad-Cre treated animals of the indicated genotypes (n = 8-9 per group).
Discussion
Here, we exploited an experimental system in which acute Vhl disruption in mature hepatocytes caused death within days. Vhl inactivation blocked fatty acid oxidation causing liver steatosis and the accumulation of triglycerides in both the liver and plasma. Using liver perfusion experiments we determined that Vhl loss inhibited the production of glucose and ketone bodies likely explaining the reduced circulating ketone and, eventually, glucose levels, leading thereby to the death of mice. Most importantly, Vhl inactivation suppressed mitochondrial respiration and mice were rescued from the lethal effects of Vhl loss by simultaneous inactivation of Hif-1β.
Vhl-deficient livers used approximately 50% less oxygen than their wild-type counterparts indicating a profound inhibition of mitochondrial respiration. Loss of Vhl did not affect all mitochondrial functions however, and urea production was normal. Compellingly, the reduction in O2 extraction in perfused Vhl-deficient livers was associated with a corresponding increase of a similar magnitude in liver pO2 in live mice. These data show that Vhl loss blocks mitochondrial respiration in the liver in vivo. This defect likely underlies the metabolic and death phenotypes. Specifically, gluconeogenesis is strongly dependent on the TCA cycle and mitochondrial respiration (Burgess et al 2006, Burgess et al 2007). Even in mice with otherwise normal gluconeogenic gene expression, impaired hepatic TCA cycle and oxidative phosphorylation disrupts gluconeogenesis (Burgess et al 2006).
To our knowledge, this is the first study to show that Vhl loss and constitutive Hif activation is sufficient to block oxygen utilization and mitochondrial respiration in live animals. Despite oxygen availability, its utilization was blocked by Hif. We conclude that there is no other pathway in the liver that can thwart the effects of Hif on oxygen disposition. Incidentally, the adverse effects of sustained Hif activation in the liver may explain why, even under conditions of prolonged hypoxia, Hif-1α upregulation in the liver is short-lived (Stroka et al 2001).
These results are in contrast to those from Rankin et al. who using isolated mitochondria from Alb-Cre; VhlF/F livers and succinate or glutamate as substrates reported that Vhl loss in hepatocytes did not affect oxygen consumption (Rankin et al 2009). We speculate that the differences observed between our study and that by Rankin et al. arose because in organello measurements do not fully reproduce the fine metabolic autoregulation that occurs in the closed metabolic pathways in intact organs. Our findings highlight the importance of ex vivo perfusion experiments and in vivo oxygen measurements.
The particular roles of Hif-1α and Hif-2α in the metabolic derangement observed are unknown. Rankin and colleagues showed that inactivation of Hif-2α (but not Hif-1α) prevented steatosis in Alb-Cre; VhlF/F livers (Rankin et al 2009). However, Kim et al. showed that overexpression of a constitutively active form of Hif-2α did not result in liver steatosis and that by contrast, liver steatosis was caused by a stable form of Hif-1α (Kim et al 2006b). In the studies by Kim et al., the severe steatosis observed in Alb-Cre; VhlF/F livers was most closely recapitulated by simultaneous activation of both Hif-1α and Hif-2α proteins and both Hif-1α and Hif-2α proteins may be similarly implicated in Ad-Cre VhlF/F liver steatosis.
Extensive changes in gene expression were observed following Vhl inactivation and it seems unlikely that changes in a single gene would account for the profound suppression in mitochondrial respiration observed. Mitochondrial biogenesis (Zhang et al 2007, Zhang et al 2008) and autophagy (Lei et al 2008, Zhang et al 2008) are regulated by Hif-1. However, since urea production was unaffected, it is improbable that the decrease in oxygen consumption observed resulted from decreased mitochondrial mass. One mechanism may involve the Fe-S cluster assembly proteins IscU1/2, which are regulated by pVhl in a miR-210-dependent manner (Chan et al 2009), and which we found to be downregulated in Ad-Cre VhlF/F livers. Low IscU1/2 levels have been implicated in reducing aconitase and complex I activities (Chan et al 2009) and may also directly affect β-oxidation, which requires Fe-S cluster containing electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO). Another factor contributing to decreased mitochondrial respiration may be Pdk1, a Hif-1 target gene (Kim et al 2006a, Papandreou et al 2006) which was induced in Vhl-deficient livers, and which would downregulate the amount of pyruvate-derived acetyl-CoA available for the TCA cycle.
The phenotype of Ad-Cre VhlF/F animals resembled human inborn fatty acid oxidation defects which are associated with liver steatosis and hypoketotic hypoglycemia (Wood 1999). In patients, death may be precipitated by a viral illness, and in mice, death may have been hastened by the adenoviral infection. Humans with inborn errors in FAO are treated with glucose (Saudubray et al 1999), and exogenous glucose administration prolonged the survival of mice. However, whereas in humans supportive treatment during an acute illness may avert death, glucose administration to mice delayed survival only by a short period of time indicating that the underlying deficit may be more severe.
The finding that Vhl disruption is sufficient to suppress mitochondrial respiration may be amenable to therapeutic exploitation as it would suggest that VHL-deficient tumors may be exquisitely dependent on glycolysis for ATP generation. While the effects of Vhl loss on mitochondrial respiration may be tissue-specific, a similar suppression of mitochondrial functions may also occur in ccRCC. Indeed, ccRCCs have low respiratory chain protein content (Simonnet et al 2002) and impaired mitochondrial function may contribute to the accumulation of TG in this tumor type. In fact, miR-210 is one of the most upregulated microRNAs in ccRCC (Chow et al 2010, Juan et al 2010, Jung et al 2009, Liu et al 2010) and this would be expected to downregulate IscU1/2 proteins. Thus inhibitors of glycolysis may be particularly efficacious against this tumor type.
In summary, our data show that proper Hif regulation is essential for oxygen homeostasis in the liver. Unchecked, Hif is sufficient to suppress mitochondrial respiration in vivo leading to impaired fatty acid oxidation and reduced ketone and glucose production ultimately causing death. Despite oxygen availability, Hif blocks oxygen utilization. Thus, no other oxygen sensing pathway exists in hepatocytes that can substantially modulate the effects of Hif.
Materials and methods
Animals
All animal experiments were approved by IACUC. Two to six month old animals of a mixed background were used for Ad-Cre experiments. VhlF mice (Haase et al 2001) or intercrossed with animals bearing Hif-1βF allele (Tomita et al 2000). Adenovirus coding for Cre recombinase (Ad-Cre) was injected by tail vein, 6×109 pfu/animal. Efficiency of recombination of VhlF and Hif-1βF alleles was tested by PCR using genomic DNA and primers listed in Table S2. Albumin-Cre animals were genotyped at 14 days of age and at that point the survival study was started.
Human tissue
Clear-cell renal cell carcinoma and normal renal cortex samples flash frozen in liquid nitrogen and stored at -80°C were obtained from UT Southwestern tissue bank. Only those samples were used that were devoid of surrounding fat tissue and necrosis, and in case of carcinomas, which exhibited tumor content higher than 70% (inferred from an analysis by a pathologist from H&E of immediate flanking sections).
Glucose and WY-14643 treatment
Glucose (25% in water) or equivalent volume of normal saline (0.9%, Baxter) was given by intraperitoneal injection (IP), 2 mg/g of body weight every 8 hours starting 70 hours after Ad-Cre injection and continued until the last VhlF/F animal in the group died. WY-14643 in olive oil (5 mg/ml) or equivalent volume of vehicle was given by oral gavage at 50 mg/kg of body weight starting 24 hours before Ad-Cre injection. In the PPARα activation study, animals were given five doses every 24 hours and sacrificed 6 hours after the last gavage. In the survival study, the first four doses were given every 24 hours and then every 48 hours; last dose was given to WT animals at day 8 after Ad-Cre injection. Because compared to females, PPARα appears to be more active in males (Ciana et al 2007), males were used for the WY-14643 study.
Blood and tissue analysis
Animals were anesthetized by inhalation of isofluorane (Aerrane, Baxter). Blood was collected from the left ventricle after opening the chest. To obtain plasma, blood was transferred immediately into heparin coated tubes. Plasma and serum were analyzed using Vitros 250 Chemistry System (Johnson and Johnson) except for ketone bodies, which were tested by Autokit Total Ketone Bodies (Wako). Tissues were removed immediately after blood collection and were flash-frozen in liquid nitrogen or fixed in formalin for histological analysis. ccRCC samples were treated similarly. Oil red O staining (Poly Scientific) of frozen sections was performed overnight and was followed by hematoxylin counterstaining. Tissue lipid composition analysis was performed by UT Southwestern Mouse Metabolic Phenotyping Core. Lipids were extracted from frozen tissues as previously described (Folch et al 1957). Briefly, a piece of tissue (100–200 mg) was homogenized in 5 ml of chloroform:methanol (2:1, v/v) using a homogenizer, 1 ml of saline was added and the mixture was vortexed vigorously. The phases of the Folch extraction were separated by centrifugation at 3000 × g for 20 min, the organic phase was transferred to a 5 ml volumetric flask, and the final volume was brought up to 5 ml with chloroform. TG and cholesterol in the chloroform phase were measured using enzymatic assays (Thermo Scientific) (Kuriyama et al 2005).
Isolation of liver mitochondria
Crude mitochondria were isolated as previously described (McDonald et al 2009).
Western blot analysis
Tissue lysates were prepared using QIAshredder columns (Qiagen) as described elsewhere (Peña-Llopis et al., in preparation). Antibodies were obtained from the following sources: Novus Biologicals: Glut-1; Santa Cruz: IscU1/2, pVhl, donkey anti-goat HRP-IgG; Abcam: Ppib; Thermo Scientific: Hsp70; Pierce: goat anti rabbit HRP-IgG.
Quantitative reverse transcribed real-time PCR
cDNA was synthesized from 2-4 μg of total RNA using random primers (Invitrogen) and M-MLV-RT (Invitrogen). PCR was run using iTag Fast SYBR Green Supermix with ROX (BioRad) or Express SYBR GreenER qPCR Supermix Universal (Invitrogen) and a 7500 Real Time PCR System (Applied Biosystems). Primers are listed in Table S3.
Liver perfusion
Livers from animals fasted for ∼24 hours on day 4 after Ad-Cre injection were perfused without recirculation for 60 min as previously described (Burgess et al 2004) except that the perfusion media contained FFAs instead of octanoate and was supplemented with 0.1 mM glutamate, 1.0 mM glutamine, and 1.0 mM alanine. Effluent perfusate was collected for assays of glucose, ketones and urea. Glucose was measured using the hexokinase/glucose-6-phosphate dehydrogenase coupled assay (Bergemeyer et al 1974), ketones by Autokit 3-HB (Wako) and Autokit Total Ketone Bodies (Wako), and urea by an analytical kit (BioAssay Systems, Hayward, CA). Oxygen consumption was calculated based on the difference in oxygen tension in afferent and efferent perfusate measured by a blood gas analyzer (Instrumentation Laboratory, Lexington, MA).
In vitro fatty acid oxidation
Livers from Ad-Cre injected mice on day 3 after injection were removed and immediately submerged in an ice cold reaction solution (Dohm et al 1972) buffered with 100 mM HEPES pH 7.3. Liver specimens (∼500 mg) were minced in reaction buffer and then homogenized with 8 strokes of a hand operated Potter-Elvehjem homogenizer. [1-14C]-oleate (Perkin Elmer) was added to 200 μl of homogenate to a final concentration of 50 μM and the tube was immediately inserted into a vial with silicone septa containing filter paper soaked in hyamine hydroxide at 37°C. Reactions were terminated after 5, 10, 15 and 20 minutes by injection of 100 μl of 7% perchloric acid. After 60 minutes, the filter paper with the captured 14CO2 was transferred into a vial with 4 ml of scintillation fluid and counts were measured for 1 minute. Results are expressed as μmoles of 14CO2 per minute for mg of liver.
Hepatic acylcarnitine profile
Twenty to thirty mg liver samples from fed and 24 hours fasted mice were homogenized in acetonitrile and acylcarnitines were isolated and quantified as previously described (Millington et al 1990) with some minor modifications. An API 3200 triple quadrupole LC-MS/MS mass spectrometer (Applied Biosystems/Sciex Instruments) in positive ionization MRM mode was used to detect carnitines after LC separation. Free carnitine was monitored using the 176 to 117 MRM transition. Acylcarnitines were monitored using a precursor of 99 Da. Acylcarnitines were quantified by comparison of the individual ion peak area with that of an internal 13C standard (Cambridge Isotope Laboratories, Inc.).
In vivo liver pO2 analysis
Oxypherol (perfluorotributylamine emulsion, Alpha Therapeutics Corp., FTBA, 1ml/day) was administered by IP injection to Vhl+/+ and VhlF/F mice for 3 consecutive days starting 24 hours post Ad-Cre injection. At day 4 after Ad-Cre injection, 24 hours after the final administration of Oxypherol, NMR/MRI studies were performed on a 4.7 T Varian Unity INOVA scanner. Mice were anesthetized (isoflurane) and placed into a 3 cm diameter home-built volume coil (tunable from 188.2 MHz for 19F to 200.1MHz for 1H) that covered the torso of the animal, and was centered on the liver. Animal temperature was maintained at 36 ± 1°C by a pad with circulating warm water. Shimming and pulse calibration were performed on the tissue water proton signal and T1-weighted anatomical 1H images were acquired (acquisition parameters: FOV 50 mm × 50 mm, matrix 128 × 128, slice thickness 1 mm, TR/TE = 500 ms/12 ms). 19F NMR spectra were obtained after tuning the coil to 19F resonance frequency. Each spectrum was acquired in 8 s (acquisition parameters: pulse width 40 μs, 8 acquisitions, 60 Hz exponential line broadening) and was used to locate the most upfield resonance frequency, which is assigned to CF3 resonance of FTBA (Mason 1994). T1 values of the CF3 resonance in liver were measured using chemical shift selective imaging with varying repetition times (acquisition parameters: FOV 50 mm × 50 mm, matrix 32 × 32, slice thickness 10 mm). Images with different repetition times were fit with the standard 3-parameter saturation recovery model using the Image Browser software on the Varian scanner to generate maps of T1 and ΔT1 (where ΔT1 is the standard error in T1 curve fit for voxel), R1(=1/T1) maps were converted to pO2 maps using published calibration curves R1 = A + (B * pO2), where A = 0.684 s-1 and B = 0.305 × 10-2 (torr × s)-1 (Wilson et al 1992), using MATLAB software routines. Only voxels from the liver region that were consistently present on both air-breathing and oxygen-breathing T1 maps and had a ΔT1/T1 < 25% were used for further analysis. Final pO2 value for each animal was calculated as median of pooled voxels.
Statistical analysis
All data are presented as means with standard deviations unless otherwise specified. p values are calculated by two tailed Student's t test assuming equal variances unless otherwise specified. For the survival curves a Log-rank test was used. SAS 9.1.3 Service Pack 3 XP_PRO platform was used for the analysis except the in vivo pO2 measurements, where Origin 6.1 software was used.
Supplementary Material
Acknowledgments
We thank Drs Frank J. Gonzalez, Volker H Haase, William Y. Kim and William G. Kaelin Jr for mouse strains and reagents, Dr. Michael Brown for discussions, and Dr. Jay D. Horton for critically reading the manuscript, TianTeng He for help with liver perfusion experiments, and members of the Brugarolas lab for discussions. This work was supported by the following grants to J.B: K08NS051843, American Cancer Society Research Scholar Grant 115739, and RO1CA129387 as well as an RO1DK078184 to S.B. MRI was performed in conjunction with R01CA139043 (R.P. M.) R21CA132096 (V.D.K.) and the UT Southwestern small animal imaging research program U24 CA126608 and P41 RR02584. S.P.-L. was supported in part by a fellowship from the Generalitat Valenciana (Spain). J.B. is a Virginia Murchison Linthicum Scholar in Medical Research at UT Southwestern. Authors declare that there are no conflicts of interest. The content is solely the responsibility of the authors and does not represent official views from any of the granting agencies.
References
- Belanger A, Luo Z, Vincent K, Akita G, Cheng S, Gregory R, et al. Hypoxia-inducible factor 1 mediates hypoxia-induced cardiomyocyte lipid accumulation by reducing the DNA binding activity of peroxisome proliferator-activated receptor alpha/retinoid X receptor. Biochem Biophys Res Commun. 2007;364:567–572. doi: 10.1016/j.bbrc.2007.10.062. [DOI] [PubMed] [Google Scholar]
- Bergemeyer H, Bernt H, Schmidt F, Stork H. Methods in Enzymatic Analysis. Academic Press; London: 1974. pp. 1196–1201. [Google Scholar]
- Burgess S, Hausler N, Merritt M, Jeffrey F, Storey C, Milde A, et al. Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J Biol Chem. 2004;279:48941–48949. doi: 10.1074/jbc.M407120200. [DOI] [PubMed] [Google Scholar]
- Burgess S, Leone T, Wende A, Croce M, Chen Z, Sherry A, et al. Diminished hepatic gluconeogenesis via defects in tricarboxylic acid cycle flux in peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha)-deficient mice. J Biol Chem. 2006;281:19000–19008. doi: 10.1074/jbc.M600050200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, He T, Yan Z, Lindner J, Sherry A, Malloy C, et al. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5:313–320. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Zhang Y, Hemann C, Mahoney C, Zweier J, Loscalzo J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009;10:273–284. doi: 10.1016/j.cmet.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sanford C, Sun J, Choi V, Van Dyke T, Samulski R, et al. VHL and PTEN loss coordinate to promote mouse liver vascular lesions. Angiogenesis. 2010;13:59–69. doi: 10.1007/s10456-010-9164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow T, Youssef Y, Lianidou E, Romaschin A, Honey R, Stewart R, et al. Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin Biochem. 2010;43:150–158. doi: 10.1016/j.clinbiochem.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Ciana P, Biserni A, Tatangelo L, Tiveron C, Sciarroni A, Ottobrini L, et al. A novel peroxisome proliferator-activated receptor responsive element-luciferase reporter mouse reveals gender specificity of peroxisome proliferator-activated receptor activity in liver. Mol Endocrinol. 2007;21:388–400. doi: 10.1210/me.2006-0152. [DOI] [PubMed] [Google Scholar]
- Cockman M, Masson N, Mole D, Jaakkola P, Chang G, Clifford S, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- Dohm GL, Huston RL, Askew EW, Weiser PC. Effects of exercise on activity of heart and muscle mitochondria. Am J Physiol. 1972;223:783–787. doi: 10.1152/ajplegacy.1972.223.4.783. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley G. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Gebhard R, Clayman R, Prigge W, Figenshau R, Staley N, Reesey C, et al. Abnormal cholesterol metabolism in renal clear cell carcinoma. J Lipid Res. 1987;28:1177–1184. [PubMed] [Google Scholar]
- Gnarra J, Tory K, Weng Y, Schmidt L, Wei M, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85–90. doi: 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- Gnarra J, Ward J, Porter F, Wagner J, Devor D, Grinberg A, et al. Defective placental vasculogenesis causes embryonic lethality in VHL-deficient mice. Proc Natl Acad Sci U S A. 1997;94:9102–9107. doi: 10.1073/pnas.94.17.9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase V, Glickman J, Socolovsky M, Jaenisch R. Vascular tumors in livers with targeted inactivation of the von Hippel-Lindau tumor suppressor. Proc Natl Acad Sci U S A. 2001;98:1583–1588. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Furihata M, Baba M, Zbar B, Schmidt L. Vascular defects and liver damage by the acute inactivation of the VHL gene during mouse embryogenesis. Lab Invest. 2006;86:664–675. doi: 10.1038/labinvest.3700431. [DOI] [PubMed] [Google Scholar]
- Horton J, Goldstein J, Brown M. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss J, Levy F, Kelly D. Hypoxia inhibits the peroxisome proliferator-activated receptor alpha/retinoid X receptor gene regulatory pathway in cardiac myocytes: a mechanism for O2-dependent modulation of mitochondrial fatty acid oxidation. J Biol Chem. 2001;276:27605–27612. doi: 10.1074/jbc.M100277200. [DOI] [PubMed] [Google Scholar]
- Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- Jaakkola P, Mole D, Tian Y, Wilson M, Gielbert J, Gaskell S, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C, et al. Identification of a microRNA panel for clear-cell kidney cancer. Urology. 2010;75:835–841. doi: 10.1016/j.urology.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Jung M, Mollenkopf H, Grimm C, Wagner I, Albrecht M, Waller T, et al. MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med. 2009;13:3918–3928. doi: 10.1111/j.1582-4934.2009.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin W, Ratcliffe P. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway R, Conaway J. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitsinou P, Haase V. The VHL tumor suppressor and HIF: insights from genetic studies in mice. Cell Death Differ. 2008;15:650–659. doi: 10.1038/sj.cdd.4402313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper L, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005;24:3846–3858. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Tchernyshyov I, Semenza G, Dang C. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006a;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Kim W, Safran M, Buckley M, Ebert B, Glickman J, Bosenberg M, et al. Failure to prolyl hydroxylate hypoxia-inducible factor alpha phenocopies VHL inactivation in vivo. EMBO J. 2006b;25:4650–4662. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama H, Liang G, Engelking L, Horton J, Goldstein J, Brown M. Compensatory increase in fatty acid synthesis in adipose tissue of mice with conditional deficiency of SCAP in liver. Cell Metab. 2005;1:41–51. doi: 10.1016/j.cmet.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Latif F, Tory K, Gnarra J, Yao M, Duh F, Orcutt M, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- Lei L, Mason S, Liu D, Huang Y, Marks C, Hickey R, et al. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol Cell Biol. 2008;28:3790–3803. doi: 10.1128/MCB.01580-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Brannon A, Reddy A, Alexe G, Seiler M, Arreola A, et al. Identifying mRNA targets of microRNA dysregulated in cancer: with application to clear cell Renal Cell Carcinoma. BMC Syst Biol. 2010;4:51. doi: 10.1186/1752-0509-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manalo D, Rowan A, Lavoie T, Natarajan L, Kelly B, Ye S, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- Mandard S, Müller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci. 2004;61:393–416. doi: 10.1007/s00018-003-3216-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason R. Non-invasive physiology: 19F NMR of perfluorocarbons. Artif Cells Blood Substit Immobil Biotechnol. 1994;22:1141–1153. doi: 10.3109/10731199409138809. [DOI] [PubMed] [Google Scholar]
- Masson N, Willam C, Maxwell P, Pugh C, Ratcliffe P. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J. 2001;20:5197–5206. doi: 10.1093/emboj/20.18.5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell P, Wiesener M, Chang G, Clifford S, Vaux E, Cockman M, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- McDonald J, Ramsey J, Miner J, Nielsen M. Differences in mitochondrial efficiency between lines of mice divergently selected for heat loss. J Anim Sci. 2009;87:3105–3113. doi: 10.2527/jas.2009-1935. [DOI] [PubMed] [Google Scholar]
- Millington D, Kodo N, Norwood D, Roe C. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13:321–324. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- Nguyen A, McDonald J, Bruick R, DeBose-Boyd R. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor-mediated induction of insigs. J Biol Chem. 2007;282:27436–27446. doi: 10.1074/jbc.M704976200. [DOI] [PubMed] [Google Scholar]
- Nickerson M, Jaeger E, Shi Y, Durocher J, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–4734. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohh M, Park C, Ivan M, Hoffman M, Kim T, Huang L, et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- Ohh M. Ubiquitin pathway in VHL cancer syndrome. Neoplasia. 2006;8:623–629. doi: 10.1593/neo.06442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer A, Bruick R. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3:144–153. doi: 10.1038/nchembio863. [DOI] [PubMed] [Google Scholar]
- Papandreou I, Cairns R, Fontana L, Lim A, Denko N. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Park S, Haase V, Johnson R. von Hippel Lindau tumor suppressor regulates hepatic glucose metabolism by controlling expression of glucose transporter 2 and glucose 6-phosphatase. Int J Oncol. 2007;30:341–348. [PubMed] [Google Scholar]
- Peyssonnaux C, Zinkernagel A, Schuepbach R, Rankin E, Vaulont S, Haase V, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin E, Biju M, Liu Q, Unger T, Rha J, Johnson R, et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117:1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin E, Rha J, Unger T, Wu C, Shutt H, Johnson R, et al. Hypoxia-inducible factor-2 regulates vascular tumorigenesis in mice. Oncogene. 2008;27:5354–5358. doi: 10.1038/onc.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin E, Rha J, Selak M, Unger T, Keith B, Liu Q, et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29:4527–4538. doi: 10.1128/MCB.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudubray J, Martin D, de Lonlay P, Touati G, Poggi-Travert F, Bonnet D, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999;22:488–502. doi: 10.1023/a:1005556207210. [DOI] [PubMed] [Google Scholar]
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Béroud C, Demont J, et al. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- Stroka D, Burkhardt T, Desbaillets I, Wenger R, Neil D, Bauer C, et al. HIF-1 is expressed in normoxic tissue and displays an organ-specific regulation under systemic hypoxia. FASEB J. 2001;15:2445–2453. doi: 10.1096/fj.01-0125com. [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CT. Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J. 2008;409:19–26. doi: 10.1042/BJ20071249. [DOI] [PubMed] [Google Scholar]
- Tomita S, Sinal C, Yim S, Gonzalez F. Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Mol Endocrinol. 2000;14:1674–1681. doi: 10.1210/mend.14.10.0533. [DOI] [PubMed] [Google Scholar]
- Tosi M, Rodriguez-Estrada M, Lercker G, Poerio A, Trinchero A, Reggiani A, et al. Magnetic resonance spectroscopy and chromatographic methods identify altered lipid composition in human renal neoplasms. Int J Mol Med. 2004;14:93–100. doi: 10.3892/ijmm.14.1.93. [DOI] [PubMed] [Google Scholar]
- Wakil S, Abu-Elheiga L. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. 2009;(50):S138–143. doi: 10.1194/jlr.R800079-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C, Berkowitz B, Hatchell D. Oxygen kinetics in preretinal perfluorotributylamine. Exp Eye Res. 1992;55:119–126. doi: 10.1016/0014-4835(92)90099-e. [DOI] [PubMed] [Google Scholar]
- Wood P. Defects in mitochondrial beta-oxidation of fatty acids. Curr Opin Lipidol. 1999;10:107–112. doi: 10.1097/00041433-199904000-00004. [DOI] [PubMed] [Google Scholar]
- Yu F, White S, Zhao Q, Lee F. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano N, Lubensky I, Merino M, Linehan W, Walther M. Histopathology and molecular genetics of renal tumors toward unification of a classification system. J Urol. 1999;162:1246–1258. [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller K, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch-Marce M, Shimoda L, Tan Y, Baek J, Wesley J, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.