

Abstract
The p53 tumor suppressor potently limits the growth of immature and mature neurons under conditions of cellular stress. Although loss of p53 function contributes to the pathogenesis of central nervous system (CNS) tumors, excessive p53 function is implicated in neural tube defects, embryonic lethality, and neuronal degeneration. Thus, p53 function must be tightly controlled. The anti-proliferative properties of p53 are mediated, in part, through the induction of apoptosis, cell cycle arrest, and senescence. Although there is still much to be learned about the role of p53 in these processes, recent evidence supports exciting new roles for p53 in a wide range of processes, including neural precursor cell self-renewal, differentiation, and cell fate decisions. Understanding the full repertoire of p53 function in CNS development and tumorigenesis may provide us with novel points of therapeutic intervention for human diseases of the CNS.
Keywords: p53, MDM2, MDM4, apoptosis, nervous system, neural stem cell, cancer stem cell, medulloblastoma, glioblastoma, birth defects
Introduction
The p53 family (p53, p63, p73) of transcription factors occupies an important role in the cellular response to stress in developing and homeostatic tissues. In greater than 50% of human cancers, the p53 protein is mutated or functionally inactivated. The ability of p53 to suppress tumorigenesis is fundamentally linked to its central role in the cellular response to stress. Although normally low or undetectable, p53 levels and activity are rapidly increased in response to a wide range of stresses, including DNA damage, hypoxia, oncogene activation, microtubule disruption, and oxidative damage.1 Activation of p53 induces cellular protective processes such as apoptosis, cell cycle arrest, and senescence largely through the transcriptional regulation of p53 target genes.2 p53 potently limits the growth of damaged, potentially neoplastic cells in both embryonic and adult tissues, thus earning its moniker “guardian of the genome.”3 The importance of p53 in the apoptosis of mature neurons in response to DNA damage, excitotoxicity, or ischemia has been well established and is the topic of several recent reviews.4,5 This review will focus predominantly on the role of p53 in regulating neural precursor cell apoptosis during central nervous system (CNS) development and discuss emerging evidence that p53 also plays an important role in the regulation of neural precursor cell self-renewal, differentiation, and cell fate decisions. We also discuss how perturbations in the p53 pathway may contribute to neural tube defects and to the pathogenesis of the CNS tumors, medulloblastoma, and glioblastoma.
p53 in CNS Development
p53 mRNA is ubiquitously expressed throughout the rodent brain in early embryogenesis up to embryonic (E) day 10.5.6,7 At mid-gestation, coincident with increased cell specification and differentiation that accompanies organogenesis, p53 expression becomes more heterogeneous. Abundant in the highly proliferative neuroepithelium of the ventricular zones of the cerebellum, telencephalon, and mesencephalon, p53 mRNA expression is reduced in the postmitotic mantle zone and cortical plate. Analyses of transgenic mice expressing lacZ under control of a p53-responsive promoter reflect robust p53 transactivation function at E10.5,8,9 at the mid–hind brain boundary, a region from which the midbrain and cerebellum originate. In prenatal and newborn mice, p53 transactivation function is limited to the surface of the thalamus and superficial regions of the cerebral cortex, cerebellum, and hypothalamus, with only sporadic p53 activity in the adult brain.8-10 Thus, the majority of p53 protein in the developing and adult CNS is likely to be inactive.8-10
As p53 potently inhibits cell growth, it was widely anticipated that p53 would occupy an essential role in development. Therefore, it was somewhat unexpected when p53−/− mice, albeit highly tumor prone, were initially described as developmentally normal, suggesting that p53 was not required in embryogenesis. It was later established that in fact a variable percentage (8%-16%) of p53−/− females exhibit exencephaly, a neural tube defect (NTD) in which the neural tube fails to close in the developing brain.11,12 Additional malformations observed in p53−/− mice include spina bifida, in which the lower spinal neural tube fails to close,13 and retinal dysplasia, in which the neuroepithelium of the optic vesicle, a structure formed from the out-pouching of the neural tube, is aberrant folded.14 Together, these observations suggest that p53 has a physiological role in neural tube closure, a multifactorial process in which cell proliferation, death, and migration must be tightly coordinated. Although the pattern of apoptosis in the developing neural tube appears to be unaltered by the lack of p53,12 subtle disturbances in any of these processes can lead to NTDs. If p53 has a physiological role in neural tube closure, a compensatory factor such as p63 or p73, both of which are expressed in the developing CNS, must be activated in the majority of p53-deficient embryos that develop normally. Alternatively, the incomplete penetrance of embryonic lethality in p53−/− mice may reflect the requirement for p53 in a developmental checkpoint that is activated in only a subset of cells under specific conditions.
Consistent with this latter idea, mouse models deficient in key components of DNA repair pathways support spontaneous DNA damage as a potent p53-activating signal in CNS development. Mice deficient in DNA ligase 4 (Lig4) or XRCC4, two components of the nonhomologous end-joining (NHEJ) pathway that repairs double-stranded DNA breaks, exhibit defects in lymphocyte development, growth retardation, and neuronal apoptosis and die in late gestation.15,16 In both Lig4−/− and XRCC4−/− mice, a high level of p53-dependent apoptosis is found in newly formed postmitotic neurons throughout the developing CNS. The onset of apoptosis in Lig4−/− and XRCC4−/− mice parallels neurogenesis, occurring in the ventral spinal cord at E10 and dorsally at E11, suggesting that apoptosis is triggered upon the cessation of neurogenesis. Nascent postmitotic neurons in the developing embryo are also susceptible to p53-dependent apoptosis in the absence of DNA polymerase-β (Pol-β),17 which functions in the base excision repair (BER) pathway that repairs DNA lesions such as apurinic/apyrimidinic sites and DNA base modifications. The neuronal apoptotic phenotype of Pol-β−/− embryos is less severe than that observed in Lig4−/− and XRCC4−/− mice, perhaps reflecting the relative level of single-versus double-strand breaks that occur spontaneously.
Cycle dysregulation has also been proposed to signal p53 activation in the developing CNS. Germline retinoblastoma (Rb) deficiency in mice elicits p53-dependent apoptosis in postmitotic regions of the CNS, peripheral nervous system (PNS), and ocular lens.18-20 Rb binds to and inactivates the E2F family of transcription factors that function to promote the G1-S transition. Loss of E2F1 or E2F3 in Rb−/− embryos rescues inappropriate S-phase entry and apoptosis,21,22 thereby suggesting that the apoptotic function of p53 is activated in Rb−/− postmitotic neurons in response to the inappropriate exit from the cell cycle. Overexpression of E2F can activate p53 through the transcriptional regulation of ARF (p19arf in mice, p14arf in humans), a negative regulator of the p53 inhibitor, MDM2. However, apoptosis in the Rb−/− CNS is not rescued by the loss of ARF,23 indicating that Rb deficiency activates p53 through an alternative pathway. Interestingly, expression of the hypoxia-related genes, VEGF and LDH-A, is increased in the developing CNS of germline Rb−/− mice.24 However, in mice in which Rb is conditionally inactivated in the CNS, inappropriate S-phase entry persists, but the level of apoptosis and hypoxia-inducible gene expression is normal.24 As Rb−/− mice exhibit impaired erythropoeisis, it has been proposed that the pro-apoptotic signal in Rb−/− postmitotic neurons originates not from cell cycle dysregulation but rather from hypoxia due to defective erythroid cells. Aberrant proliferation may not therefore be sufficient to activate p53 in nascent postmitotic neurons but may predispose these cells to collaborative pro-apoptotic signals such as hypoxia.
Keeping p53 under Control: The MDM2/MDM4/p53 Axis
The exquisite sensitivity of the developing CNS to p53-mediated apoptosis dictates that strict controls must be operational to prevent p53 activation in the absence of cellular stress. Foremost among the gatekeepers to p53 activation are MDM2 and the related protein, MDM4.
MDM2 binds to and inhibits the ability of p53 to stimulate transcription.25,26 In addition, MDM2 regulates p53 protein stability. MDM2 targets p53 for destruction by the ubiquitin-proteasome pathway by functioning as an E3 ubiquitin ligase and facilitating the covalent attachment of ubiquitin to p53,27-29 thus regulating the overall level of p53 protein in a cell. The Mdm2 gene is a target of p53 transcriptional activation, thereby forming an autoregulatory loop in which p53 regulates expression of it own inhibitor. MDM4 also inactivates p53 transcriptional activity30 but, unlike MDM2, lacks ubiquitin ligase activity,31 and Mdm4 expression is not regulated by p53. Both Mdm2−/− and Mdm4−/− mice die early in embryogenesis but survive to adulthood when p53 is deleted,32-35 thereby highlighting the critical roles of MDM2 and MDM4 in negatively regulating p53 during development.
MDM2 and MDM4 are expressed in the CNS.35,36 Conditional mouse models using nestin-driven Cre recombinase demonstrate that loss of MDM2 in the developing CNS elicits massive p53-dependent apoptosis in the ventricular zone and intermediate zones of the cerebral cortex, leading to the degeneration of the neuroepithelium and hydrocephalus and to perinatal lethality.37,38 Studies using a novel hypomorphic allele, Mdm2puro, support a threshold requirement for MDM2 in the developing CNS. Mdm2 puro/Δ7–9 mice express ~30% of the wild-type level of MDM2 and are viable but have small brains and an elevated level of p53-mediated apoptosis in granular neuronal precursors (GNPs) of the cerebellum.39,40 As the restoration of p53 function in the mature brain of adult MDM2/p53-deficient mice fails to induce neuronal apoptosis,41 there appears to be an increased need for MDM2 to buffer p53 from endogenous p53 activating signals in the developing CNS.
The effects of MDM4 loss in the CNS are less severe than those observed for MDM2. Mice with CNS-specific ablation of MDM4 are viable but manifest significant growth retardation and cerebellar ataxia.37 Loss of MDM4 impairs cell proliferation in the embryonic neuroepithelium, lateral ventricular zone of the cerebral cortex, and external granular layer of the cerebellum. However, in contrast to its anti-proliferative effect in neural precursors, MDM4 deficiency promotes apoptosis in postmitotic neurons.37,38 As with MDM2, the effects of MDM4 loss on cell proliferation and survival are dependent on the presence of p53. Thus, MDM2 and MDM4 have nonredundant roles in regulating p53 in the developing CNS.
Although MDM2 and MDM4 independently control p53 function, their activities are likely to synergize to maintain p53 at low or undetectable levels. Deletion of one Mdm4 allele exacerbates the CNS phenotype of Nes-Cre; Mdm2–null embryos, as does the deletion of one Mdm2 allele in Nes-Cre; Mdm4–null mice. On a Nes-Cre background, loss of MDM2 did not elicit p53-dependent apoptosis in postmitotic neurons. However, studies with Nex-cre mice that specifically express Cre in differentiated neurons reveal that deletion of even one Mdm2 allele in this cell type potentiates apoptosis when Mdm4 is co-deleted.37 Analyses of Nes-Cre and Nex-cre Mdm2/Mdm4 compound mutants support a dose-dependent, synergistic effect of these two critical regulators on the inhibition of p53 function. Therefore, MDM2 and MDM4 each have an important role in limiting p53 function in both proliferating neuronal precursors and differentiated postmitotic neurons of developing CNS.37,38
Multiple proteins, in addition to MDM2 and MDM4, have been shown to interact with and influence p53 function through either direct regulation of p53 stability and/or activity or through modulation of MDM2 and/or MDM4 activity. A detailed list of proteins known to interact with p53, MDM2, and/or MDM4 can be found in Table 3 of Toledo and Wahl.42 A discussion of how all these factors may contribute to the regulation of p53 function is beyond the scope of this review. However, as will be explored in the following sections, both the activation and loss of p53 contribute to diseases of the CNS. It will be important to gain an improved understanding of how known regulators of p53 function specifically impinge upon the MDM2/MDM4/p53 axis in the developing and adult CNS.
p53 in Neural Stem and Progenitor Cells
Neural stem cells (NSCs) are self-renewing cells in the nervous system that can generate both neurons and glia.43 Early in nervous system development, NSCs can be found throughout the embryonic brain. Initial symmetrical division of NSCs serves to expand the stem cell pool, whereas subsequent asymmetric divisions give rise to self-renewing neural stem cells and neuronal progenitors and, later, glial progenitors.44 Adult neural stem cells have also been found within two well-described neurogenic niches: the subgranular zone (SGZ) in the dentate gyrus of hippocampus and the subventricular zone (SVZ) lining the wall of the lateral ventricles.45-47 More recently, adult neural stem cells have been identified in nonneurogenic regions of the mature brain, including the cerebellum,48 substantia nigra,49 and retina.50
In contrast to the hematopoietic system wherein hemapoietic stem cells (HSCs) are required to fulfill a relatively similar function throughout the life of the organism, embryonic and adult neural stem and lineage-restricted progenitors (collectively precursor cells) differ in their self-renewal and differentiation characteristics. NSCs isolated from early stage embryonic rat forebrains, for example, exhibit a high self-renewal capacity compared to late-stage embryonic and adult NSC from the same tissue. Moreover, NSC cell fate determination is temporally regulated during development as the differentiation of NSC from early and late-stage forebrains occurs predominantly toward the neuronal and glial lineages, respectively.51 Spatial and temporal specification of neural precursors is achieved through a combination of intrinsic and extrinsic cues. Growth factor concentration is one environmental cue predicted to play an important role in establishing neural precursor cell potential and phenotype. Genetic mutations acquired by neural precursor cells that alter properties, such as their sensitivity to different growth factors, may therefore promote cellular transformation.
Recent studies have begun to shed light onto the important role(s) of p53 in the regulation of NSCs. p53 is expressed within the neurogenic niche of the lateral ventricles.52,53 Abundant nuclear p53 is evident in the ependymal cell lining of the lateral ventricle wall as well as most cells of the SVZ, including astrocytes and progenitors. In agreement with the down-regulation of p53 in differentiating cells observed during embryogenesis, nuclear p53 immunoreactivity is absent or found at low levels in the majority of the mature brain, including differentiating cells in the rostral migratory stream,52 suggesting that p53 is preferentially expressed in neural precursors.
In the absence of markers to definitively identify NSCs in vivo, NSC behavior, including self-renewal and multipotentiality, can be assessed using neurospheres, floating aggregates of heterogeneous cell types, including neural stem and progenitor cells.45 Neurospheres established from either the embryonic olfactory bulb (OB) or adult SVZ of p53−/− mice proliferate more rapidly, are less apoptotic, and have an increased capacity for self-renewal than their wild-type counterparts.52,54-56 Within the SVZ, loss of p53 promotes cell division in slow-dividing astrocyte-like cells (Nestin+/GFAP+ type B), residing between the ependymal lining and striatal parenchyma,57,58 without a concomitant increase in cell proliferation,54 consistent with an enhancement of the self-renewal properties of this putative NSC population.
NSC proliferation, survival, and self-renewal are tightly coordinated with differentiation. When cultured in differentiation medium (2% FCS, EGF/ bFGF-free), p53-deficient neurospheres generate an increased number of Tuj1+ neuronal progenitors and decreased number of glial fibrillary acid protein (GFAP)+ astrocytes as compared to neurospheres with wild-type p53.54,55 The altered differentiation pattern in p53−/− neurospheres suggests that p53 may play a direct role in the negative regulation of neurogenesis. Within the SVZ, type B cells give rise to highly proliferative transit-amplifying precursor cells (type C cells) that can generate migratory neuroblasts (type A cells) and oligodendrycte progenitors,59 which migrate to the OB along the rostral migratory stream where they differentiate to form olfactory granule cells and periglomerular interneurons.43 In the absence of p53, the proliferation rate of type C cells is enhanced, although the number of C cells in the p53−/− SVZ is not increased. In contrast, A cells are more abundant in the p53−/− SVZ without an apparent increase in their proliferation rate,54 suggesting that p53 deficiency promotes that proliferation and rapid differentiation of type C cells into type A cells. Together, these findings suggest p53 may selectively regulate NSC self-renewal, differentiation, and cell fate determination depending on cellular and developmental context.
p53 and Brain Tumor Stem Cells
A growing number of studies have revealed that a small fraction of cells in tumors is capable of tumor initiation. Similar to stem cells found in normal tissues, these select tumors cells have the capacity for self-renewal and for producing a diverse repertoire of differentiated progeny, suggesting that they may be cancer stem cells. Initially identified in hematopoietic malignancies,60 cancer stem cells have now been found in solid tumors, including those of the breast,61 pancreas,62 colon,63 and brain.64,65 For brain tumors, it has been demonstrated that as few as a hundred glioblastoma or medulloblastoma (MB) tumor cells expressing the stem cell marker CD133 (prominin-1) can give rise to primary and secondary tumors upon serial xenograft transplantation,64 thereby supporting the identity of a subset of these cells as brain tumor stem cells (BTSCs). The origin of a BTSC is unclear. It may arise from the transformation of an NSC or from a lineage-restricted precursor cell or mature differentiated cell that, through oncogenic mutations, has acquired stem cell–like properties. Many of the pathways known to regulate NSC stemness are active in brain cancers, suggesting that these same pathways control the self-renewal and cell fate potential of BTSCs.66 Importantly, a BTSC within a tumor may not necessarily be the cell of origin for that tumor but may arise through the accumulation of de-differentiating mutations.67 Slowly proliferating and highly resistant to most therapeutic regimens, BTSCs may contribute to tumor regrowth and patient relapse.
The role of p53 in BTSCs has not been well established. Based on our current limited understanding of p53 function in neural precursors, several hypotheses may be put forth. First, loss of p53 may enhance the proliferation and self-renewal of neural stem and/or lineage-restricted progenitors, thereby expanding the pool of cells available for additional oncogenic mutations. Second, as p53 can promote or inhibit cell differentiation depending on cellular context, as well as influence cell fate decisions, p53 mutation may alter the differentiation program of neural precursors. Last, although much focus on p53 is directed at its growth inhibitory properties, accumulating evidence supports a role for p53 in the suppression of cell migration.68 The neurogenic niche has been shown to be important for the maintenance of stem cells in an undifferentiated state,69 and in the absence of p53, the premature exit of NSCs from the neurogenic niche may alter their differentiation program or capacity for tissue invasion. A greater understanding of the molecular, cellular, and environmental factors that regulate p53 function in NSCs is predicted to provide new insights into the role of p53 in BTSCs.
p53 in CNS Tumors
The p53 tumor suppressor protein is a potent inhibitor of cell proliferation. Activated in response to a wide range of cellular stresses (e.g., DNA damage, hypoxia, oncogene-induced hyperproliferation, and impaired ribosome biogenesis),70-73 p53 induces anti-proliferative processes, including cell cycle arrest73 and apoptosis,74 thereby potently limiting the survival of potentially pre-neoplastic cells. p53 function is lost in the majority of human tumors due to either mutation of the p53 gene75 or inactivation of the p53 protein through alternative mechanisms, including mislocalization of the p53 protein76 and inhibition of p53 by viral (e.g., human papillomavirus E6 protein)77,78 or cellular (e.g., MDM2)79,80 proteins, thus underscoring the importance of p53 function for tumor suppression. Here we discuss the role of p53 in two tumors of the CNS, medulloblastoma and glioblastoma.
p53 in Medulloblastoma
Medulloblastoma, a primitive neuroectodermal tumor of the cerebellum, is the most common malignant brain tumor of childhood. It has long been the prevailing view that MB pathogenesis reflects the dysregulation of the normal developmental program of the cerebellum. MB tumors are highly heterogenous with regard to histopathology, age of patient, and stage of metastasis at time of diagnosis. Current treatments for MB are not very specific for tumor cells, resulting in devastating side effects, including reduced intellect. Despite aggressive treatment with surgical resection, radiation, and chemotherapy, only 60% of children are cured of MB.81
The role of p53 in MB pathogenesis has not yet been conclusively defined. Persons with Li-Fraumeni syndrome that is caused by a germline mutation in p53 develop MB at a higher incidence than the general population.82,83 Similarly, p53 deficiency in mice in combination with mutations in other genes, including poly (ADP-ribose) polymerase (PARP), the cell cycle regulatory protein retinoblastoma (Rb), or the Sonic hedgehog (Shh) receptor Patched1 (Ptch1), greatly increases tumor incidence,84-87 indicating that loss of p53 can promote MB tumorigenesis. However, despite the high incidence of p53 mutations in most human tumors, the p53 gene is altered in <10% of sporadic human MB.88-95 Chromosome 17p, where p53 is located, is lost in 40% to 50% of sporadic MB tumors. It has since been found that loss of 17p in MB and p53 status is unrelated.88,96
New support for a role for p53 in MB tumorigenesis comes from a greater understanding of heterogeneity underlying MB tumors. Through the integration of molecular and clinical markers, including genetic abnormalities, patient age at time of diagnosis, tumor histology, and overall and progression-free survival, 4 MB subgroups with distinct signaling pathway alterations and clinicopathological features have been identified (see Table 1). These subgroups are characterized, in part, by deregulated Wnt (A) or Sonic hedgehog (B) signaling or by distinct gene expression profiles (C and D).97 Examination of p53 gene status in 310 primary human MB tumors representing all 4 MB subgroups confirmed that p53 gene mutations in MB are rare and failed to correlate with chromosome 17p deletions.96 In addition, this study yielded several novel insights. First, p53 gene mutations are overrepresented in MB tumors that have a mutation in the CTNNB1 gene (encoding β-catenin), indicative of Wnt activation, or that have a high level of NMYC amplification. By comparison, p53 mutations did not correlate with dysregulation of the Shh pathway or with subgroup D and were not present in MB tumors from subgroup C. Second, MB tumors harboring p53 mutations typically had a classic or large cell/anaplastic histology, whereas desmoplastic MB tumors that typically exhibit Shh pathway activation did not, consistent with the lack of p53 mutations in the Shh subgroup of MB. Last, in contrast to an earlier study with a smaller cohort of MB tumors,98 p53 gene status did not correlate with unfavorable prognosis.
Table 1.
Schematic Overview of the 4 Subgroups of Medulloblastoma (MB) and the Putative Mechanisms for p53 Inactivation Discussed in This Review
| MB Subgroup | ||||
|---|---|---|---|---|
| Wnt | Shh | C | D | |
| Cell of origin | NSC? | GNP? | ? | ? |
| Frequent molecular characteristics | CTNNB1 mutation; elevated MYC | Ptch mutation | Elevated MYC | 17q gain |
| p53 inactivation | p53 mutation | Enhanced MDM2 function? | ? | Elevated WIP1 |
| Predominant histology | Classic | Desmoplastic | Classic | Classic |
GNP, granular neuronal precursor; NSC, neural stem cell.
From the association of p53 mutations with particular subgroups of MB, we may begin to infer how loss of p53 mechanistically contributes to MB tumorigenesis. CTNNB1 gene mutation and NMYC amplification are predicted to impose a strong selective pressure for tumor cells lacking wild-type p53. p53 and β-catenin have been proposed to form a negative feedback loop in which overexpression of β-catenin activates p53 transactivation function and, in turn, p53 down-regulates β-catenin.99-101 The proliferation of MB tumor cells with aberrant levels of β-catenin resulting from CTNNB1 gene mutation may be initially restrained by heightened levels of p53. Similarly, deregulated NMYC expression can induce apoptosis concomitant with the promotion of cell proliferation,102 and cooperative mutations that abrogate apoptotic pathways such as p53 are predicted to augment tumorigenesis. In addition, p53 limits genomic instability in NMYC-overexpressing tumor cells by suppressing NMyc-mediated centrosome amplification.103 For MB tumor cells with CTNNB1 gene mutation or NMYC amplification, p53 mutations may be a necessary event for MB tumor progression. Consistent with the idea that loss of p53 is a late event in this subset of MB, p53 mutations were more frequently present in MB tumors from children >7 years of age and only rarely present in MB tumors from infants.96
Given the prevalence of p53 inactivation in human tumors, p53 function in MB tumors lacking p53 gene mutations is likely to be abrogated through alternative mechanisms. Recent work in our lab, for example, supports MDM2 as an important contributor to the inhibition of p53 in Shh-driven MB tumorigenesis. In cerebellar development, MDM2 is required to inhibit p53-mediated apoptosis in GNPs,39 the presumed cell of origin for MB tumors of the Shh subgroup, and MDM2 deficiency potently restricts cerebellar tumorigenesis in Ptch1 +/− mice, a model of human Shh-induced MB.39,104 Importantly, we find that Shh stimulation in GNPs promotes MDM2 protein accumulation and phosphorylation at Ser166,39 a modification known to promote MDM2-mediated degradation of p53,105-107 suggesting that Shh signaling limits p53 activity by increasing the anti-p53 function of MDM2. Accumulating evidence supports this idea. In cultured mesenchymal cells and mouse embryonic fibroblasts (MEFs), Shh signaling enhances the level and p53 binding affinity of MDM2, thereby abrogating p53-mediated growth arrest and apoptosis in response to oncogene-induced DNA damage.108 In transgenic mice in which the Shh effector Gli1 is overexpressed in the CNS, MDM2 is found highly expressed in the neonatal brain, and overexpression of Gli1 enhances cell proliferation concomitant with a decrease in p53 protein levels in these mice.56 Conversely, pharmacological inhibition of Shh signaling increases p53-mediated growth inhibition in vitro in multiple breast cancer cell lines,108 and knockdown of Gli1 has stabilized p53 in a glioblastoma cell line.56 Likewise, Shh deficiency in zebrafish elicits heightened levels of p53-dependent apoptosis in the retina and developing CNS.109 A hypothesis drawn from these results is that in human MB tumors in which Shh signaling is the initiating event, p53 function will be automatically limited through the same mechanisms that serve to restrict p53 activity in the developing cerebellum. Furthermore, Shh-mediated enhancement of the anti-p53 function of MDM2 is predicted to obviate the selective pressure for p53 mutations as well as for Mdm2 gene amplification events, which, although present in ~7% of human tumors with wild-type p53,91 are rare in MB.88,91,110
Intriguingly, when the level of MDM2 is reduced in GNPs, Shh signaling is attenuated concomitant with p53 activation,39 suggesting a complex interplay between the p53 and Shh pathways. Recently, p53 has been shown to negatively regulate the level and subcellular localization of Gli1 in NSCs,56 and both Gli1 and Gli2 expression is reduced in Mdm2 puro/Δ7-9 GNPs. Knockdown of p53 promotes NSC renewal synergistically with Gli1 overexpression, suggesting that p53 constitutively attenuates Gli1 function.56 The negative regulation of Gli activity by p53 would provide a plausible explanation to account for the earlier observation that Gli1 is highly expressed in MB tumors developing in mice lacking functional p53,86 in which known components of the Shh pathway are wild-type. The coordinate regulation of the p53 and Shh pathways may therefore provide a mechanism to allow Shh to drive high levels of proliferation during important periods of development without necessarily activating the growth inhibitory properties of p53 and, intriguingly, to perhaps also maintain a constant level of Shh signaling.
Several p53-negative regulatory proteins, in addition to MDM2, have also been implicated in MB tumorigenesis. The gene encoding the serine/threonine phosphatase Wip1 (also known as PPM1D) maps to chromosomal region 17q22-q23 and is highly expressed in MB with gain of chromosome 17q or i17q,111 genetic alterations associated with MB subgroups C and D.97 Wip1-mediated dephosphorylation of MDM2 enhances MDM2 stability and p53 binding affinity, thereby promoting p53 degradation.112 Recently, the MAGE-A family of cancer/testis antigens was reported to be overexpressed in 60% of MB cell lines and tumors.113 These metastasis-associated transcriptional regulators have been proposed to limit the transactivation function of p53 by blocking the binding of p53 to chromatin.114 Interestingly, MAGE-A is one of the targets down-regulated by miR-34, a microRNA that is deleted in a subset of MB tumors.115
The integration of molecular and clinical features of MB has greatly advanced our understanding of the heterogeneity of MB tumors. Rare gene mutations in MB such as those in p53 gain new significance when analyzed in conjunction with tumor-specific gene expression profiles and clinicopathological features. New knowledge about the gene and signaling pathways involved in different subgroups of MB is predicted to provide insight into MB pathogenesis and foster the development of new, more selective therapies for the treatment of MB.
p53 in Glioblastoma
Gliomas, a large class of tumors that includes low-grade diffuse astrocytoma, oligodendroglioma, and oligoastrocytoma (mixed glioma), are the most common malignant brain tumors in adults, accounting for more than 70% of primary tumors of the CNS. Malignant World Health Organization (WHO) grade IV astrocytoma, glioblastoma multiforme (GBM), is the most aggressive of all brain cancers, resulting in diffuse, highly infiltrating tumors with areas of extensive necrosis and vasculature. GBM affects the cerebral hemispheres and, in rare cases, the cerebellum.116 GBM can be separated into two distinct diseases, which, although histologically similar, differ in their molecular alterations, age of patients at time of clinical presentation, and response to radio- and chemotherapy. Primary or de novo GBMs account for >90% of GBM tumors and typically occur in patients with a mean age of 65 years with little evidence of a precursor lesion. In contrast, secondary GBMs (~5%) are associated with younger patients (mean age 45 years) and develop from a previous low-grade diffuse astrocytoma or anaplastic astrocytoma.117 Despite recent advances in our understanding of GBM biology and treatment, median survival rate in patients presenting with this tumor type is less than 12 months, and treatments are largely palliative.
Common genetic alterations in primary GBM include epidermal growth factor receptor (EGFR) amplifications (~40%) and PTEN (phosphatase and tensin homolog deleted on chromosome 10) mutations (15%-40%), but these are rare events in secondary GBM.118 p53 pathway mutations, in contrast to MB, are common in GBM.119-121 p53 gene mutations are present in 68% of secondary GBM and typically occur in hotspot codons 248 and 272. Loss of p53 function is likely an early event in secondary GBM as two-thirds of low-grade astrocytomas, a precursor lesion to secondary GBM, exhibit p53 mutations,122 and in mouse models, p53 inactivation promotes astrocytoma tumorigenesis.123,124 In primary GBM, p53 mutations occur less frequently (28%) and are spread throughout the gene. It has been hypothesized that p53 mutations in these tumors may be a late event, resulting from increased genomic instability.118
The importance of p53 pathway inactivation in GBM is further illustrated by the alteration of upstream regulators. The MDM2 ubiquitin ligase and related protein, MDM4, inhibits p53 function.125 An alternatively spliced transcript from the CDKN2A locus encodes the protein ARF. Binding of ARF to the C terminus of MDM2 results in relocalization of MDM2 to the nucleolus, thereby inhibiting the nucleoplasmic shuttling function of MDM2 required for MDM2-mediated degradation of p53.126-128 Gene amplifications in Mdm2 and Mdm4 occur in 10% and 4%, respectively, of GBMs and exclusively in GBMs that lack p53 mutations.121,129,130 Expression of ARF is lost in 76% of GBMs due to promoter methylation, homozygous deletion, or mutation.121,131 Recently, an integrative analysis of copy number and somatic cell alterations showed that the ARF/MDM2/MDM4/p53 pathway is disrupted in 78% of GBMs.121
Additional genomic alterations may also contribute to the neutralization of p53 activity in GBM. A novel member of the Bcl2 family, Bcl2-like 12 (Bcl2L12), is overexpressed in almost all primary GBMs and has been found to inhibit p53-mediated cellular senescence and cell death by binding to and impairing p53 transactivation potential.132,133 Genomic loss of ATM and CHEK2, two components of the DNA damage response required for p53-mediated apoptosis in response to radiation, is found in 13.3% and 22% of GBMs, respectively.134 The tumor suppressor CHD5, located at 1p36 that is deleted in GBM with 1p loss, has been shown to modulate p53 function by increasing expression of ARF.135 Considering the large number of factors that can regulate p53 activity,42 it is likely that upon further examination, additional genomic alterations in GBM will be shown to neutralize p53 function.
Similar to MB, intense effort is under way to generate a comprehensive molecular classification of GBM with the hope that this may ultimately lead to improved clinical outcome. A pilot study by the Cancer Genome Atlas (TCGA) Research Network has catalogued genomic alterations and gene expression profiles from a large cohort of morphologically similar GBMs. This work has led to the identification of 4 subgroups of GBM: proneural, neural, classical, and mesenchymal.136 Although present in 3 of the 4 subgroups, p53 mutations were predominantly found in the proneural subgroup of GBM. Significantly, 3 of the 4 known secondary GBMs included in the study were of the proneural subgroup, consistent with the idea that p53 loss is an early event in this group of GBMs. It has yet to be established whether GBM subgroups originate from NSCs that pursue different differentiation pathways or whether they reflect tumor initiation in different stem-like precursor cells. In light of the recently identified roles for p53 in NSC self-renewal and cell fate decisions, loss of p53 in NSCs may serve to increase NSC proliferation and self-renewal, leading to an expanded pool of precursor cells in which differentiation is compromised, thereby fostering their transformation into BTSCs for GBM. Supporting this idea, prenatal treatment with ENU results in the formation of glioblastoma-like tumors in 60% of p53−/− mice.54
GBM has proven to be highly resistant to most conventional therapeutic approaches, and for the majority of cases, surgery followed by chemo- and radiotherapy is largely palliative. Our improved understanding of GBM pathogenesis has, to date, failed to translate into more effective targeted therapies, and there has been little improvement in mean survival time for the past 10 years.137 Treatment difficulties include intratumoral heterogeneity as well as putative BTSC subpopulations. New strategies to specifically target BTSCs are particularly needed as these cells are highly efficient at DNA repair and typically express elevated levels of multiple-drug-resistant transporter genes, thereby causing them to be highly resistant to radiation and chemotherapeutic agents. In human GBM xenografts, CD133+ tumor cells were found enriched after ionizing radiation treatment.138 BTSCs are therefore likely to play an important role in GBM multidrug resistance and tumor regrowth. Efforts to develop therapeutics specifically targeted to the known genetic lesions of GBM have, to date, been largely unsuccessful. Thus, there is an immediate need for the identification of new drug targets and for the development of multimodality approaches to treat this devastating disease.
Strategies to increase the function of p53 in tumor cells are currently being explored and may improve the treatment of GBM. As a critical component of the cellular response to stress, p53 function is required for the cytotoxic effects of many commonly used cancer drugs and radiation. Loss of p53 function correlates with a decrease in sensitivity to these same genotoxic agents and an increase in tumor cell survival.139 The p53 status of individual tumor types is frequently an important prognostic indicator for the likelihood of successful treatment with radiation and some chemotherapeutics. Adjuvant therapeutics that augment p53 function are predicted to sensitize tumor cells to cancer therapies that rely on p53 for their efficacy.140 Different therapeutic strategies will be needed depending on whether p53 is mutated, deleted, or wild-type but functionally inactivated by viral or cellular p53 regulatory proteins.141 In this regard, pharmacological compounds (e.g., CP-31398, APR-246) have been identified that promote the correct folding and reactivation of p53 mutant proteins.142,143 An adenoviral-mediated gene transfer strategy is being explored to reintroduce wild-type p53 function in p53-null tumor cells. Yet another strategy is to use MDM2 antagonists (e.g., nutlin-3, RITA) that reduce the MDM2-p53 interaction and impair MDM2-mediated proteolytic degradation of p53. These agents have been demonstrated to potentiate p53 function in cultured tumor cells and in vivo xenograft models and enhance the chemosensitivity of tumor cells to DNA damaging agents.144-146 Importantly, in mice, even a moderate ~20% reduction in the level of MDM2 is sufficient to sensitize mice to the lethal effects of ionizing radiation due to the enhancement of p53 function,40 suggesting that even modest gains in p53 function may sensitize tumor cells to radiation and chemotherapeutic agents. More work is required to evaluate whether enhanced p53 activity in GBM tumor cells, particularly BTSCs, can potentiate cell death in response to current treatment modalities.
Contribution of p53 to CNS Pathology
Although important for tumor suppression, p53-mediated neuronal apoptosis is associated with neurological damage that occurs following acute insults such as ischemia, traumatic brain injury, or exposure to neurotoxins. Sustained p53-mediated apoptosis has also been implicated in chronic neurodegenerative pathologies, including Parkinson, Alzheimer, and Huntington diseases.147,148 Neuronal cells of the developing CNS, in particular, are highly sensitive to genotoxic stress, and the transactivation and apoptotic functions of p53 are rapidly increased throughout the embryonic brain following exposure to genotoxic agents such as gamma irradiation9,149 or the xenobiotic, TCDD.150 Mouse embryos lacking p53 treated with these same genotoxic agents exhibit an increased incidence of teratological malformations.149,150 In the embryo, p53 may suppress teratogenesis by limiting the propagation of severely damaged cells in the developing CNS and promote death of the organism in cases where the genotoxic damage is too great (Figure 1).
Figure 1.
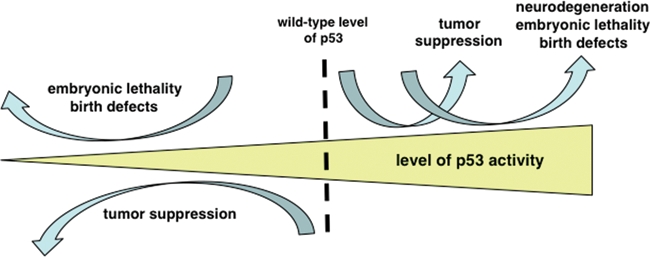
The level of p53 as a determinant of life and death. Elevating basal p53 function beyond its normal wild-type level can enhance tumor suppression. However, as p53 levels continue to rise, the benefits of enhanced tumor suppression become increasingly negated by the deleterious effects of the antigrowth properties of p53 in developing and homeostatic tissues. Chronic or aberrantly elevated p53 function is implicated in the neurodegeneration associated with several central nervous system pathologies, including Alzheimer and Parkinson diseases in adults and congenital birth defects and lethality in developing embryos. Conversely, loss of p53 function increases lifelong susceptibility to cancer and may abrogate a critical developmental checkpoint in the embryo.
As a suppressor of teratogenesis, p53 may reduce the incidence of congenital defects in surviving embryos. However, in some cases, perhaps more than are currently appreciated, the potent growth inhibitory properties of p53 may also underlie the CNS pathology of human congenital disorders. Evidence for a key role of p53 in congenital birth defects is provided by data from in vivo models. In Tcof1 +/− mice, a model of Treacher Collins syndrome, elevated apoptosis in the neuroepithelium is rescued by the loss of p53,151 thus ameliorating the craniofacial abnormalities typically associated with this disorder. A point mutant in the human Pax3 gene is associated with Waardenburg syndrome.152 The Pax3 transcription factor is highly expressed in the neuroepithelium and neural crest, and Pax3-deficient Splotch (Sp/Sp) mice exhibit neural tube defects, including exencephaly, spina bifida aperta, or both, which coincides with increased p53-dependent apoptosis in the neuroepithelium.153-155 Intriguingly, it has been recently demonstrated that within a panel of zebrafish carrying mutations in essential housekeeping genes, all had up-regulation of multiple p53 target genes. These mutants all exhibited increased neuronal apoptosis, which could be inhibited by the injection of anti-p53 morpholinos.156 Together, these studies in mice and zebrafish illustrate the sensitivity of the neuroepithelium and neural crest cells to p53-mediated apoptosis induced in response to mutations in genes with roles in a diverse range of essential processes.
How do so many gene mutations lead to p53 activation? As discussed previously, in some cases, an impaired ability to repair spontaneous DNA breaks due to loss of DNA repair pathway proteins (e.g., Lig4, XRCC4, Polβ) may result in an elevated level of endogenous genotoxic stress that is sufficient to trigger a p53-dependent DNA damage response. However, in many cases, the gene mutation is not predicted to directly lead to an increase in genotoxic stress. We propose that many of these gene mutations may perturb the network of negative regulators that normally limit p53 function, thereby lowering the threshold for p53 activation in response to spontaneous genotoxic stress or suboptimal trophic support.
For example, constitutive expression of the Notch1 intracellular domain (NICD) in the developing mammalian brain selectively promotes p53-dependent apoptosis in a subset of neural precursor cells but not in immature or mature postmitotic cells.157 Although the mechanism by which constitutive Notch1 signaling triggers p53 activation in neural progenitor cells is not yet well understood, the suppression of PI3K/AKT-mediated enhancement of MDM2 function may contribute. Upon growth factor stimulation, activated AKT phosphorylates MDM2 at Ser166 and Ser186 through both direct and indirect mechanisms, thereby enhancing MDM2-mediated inhibition of p53.105-108 In cultured cells, NICD signaling through activation of the CBF1 transcription factor regulates expression of PTEN,158 which encodes a lipid phosphatase that dephosphorylates and inactivates PI3K. The direct regulation of PTEN expression by Notch signaling is consistent with the finding that NICD overexpression reduces the level of total and phosphorylated AKT hepatocellular carcinoma cells (HCC), thereby enhancing p53 stability through the attenuation of MDM2-mediated proteasomal degradation of p53. Active Notch1 signaling sensitizes HCC cells to p53-mediated apoptosis in response to tumor necrosis factor–related apoptosis-inducing ligand (TRAIL).159 Taken together, these observations suggest that by inhibiting the AKT-mediated modulation of MDM2 function, Notch signaling may lower the threshold for p53 activation, thus sensitizing neural precursor cells to apoptotic stimuli.
In the aforementioned Splotch (Sp/Sp) mice, loss of Pax3 results in a posttranscriptional increase in p53 protein levels.153,160 Recent studies demonstrate that Pax3 enhances the rate of p53 degradation. Pax3 can down-regulate a p53 mutant incapable of binding MDM2,161 thereby implicating Pax3 in the regulation of other negative regulators of p53 stability (e.g., PirH2 or Cop1). The chromatin silencing factor, Sirt1, antagonizes p53 function by promoting its deacetylation,162 and mice deficient in Sirt1 also exhibit exencephaly that correlates with p53 hyperacetylation and enhanced p53-dependent apoptosis.163 Mutations in more than 150 genes, encoding proteins involved in a wide variety of biochemical pathways and cellular processes, result in NTDs in mice.164 To date, only a few NTD mutants have been tested for an interaction with p53. It is tempting to speculate that, as with zebrafish,156 many of these NTD mutations canalize on the MDM2/MDM4/p53 axis.
Together, these findings suggest that p53 functions in a developmental checkpoint in the CNS by integrating environmental and genetic information. Further understanding of the contribution of active p53 to human disease may yield new points of therapeutic intervention.
Concluding Remarks
Our knowledge of p53 function is immense, but the continued discovery of new roles for p53 in diverse cellular processes suggests that the majority of what we need to learn is still hidden from us. In particular, the recent demonstration that p53-mediated neuronal apoptosis is a major contributing factor to the lethality of mice and zebrafish that carry mutations in a wide range of housekeeping genes invites the further investigation of p53 pathway modifications in human congenital birth defects. Therapeutic strategies to inactivate p53 have not been very actively pursued, but such strategies do exist, and it remains to be determined whether there are windows of opportunity in which their application may mitigate some developmental malformations. In contrast, numerous therapeutic strategies to activate p53 are being explored. Although initial studies in animal models show that enhancing p53 function can be effective at de-bulking xenograft tumors, it remains to be determined whether this approach is effective at eliminating rare brain tumor stem cells. Moreover, issues regarding the delivery of p53 enhancing agents to tumors in the brain will need to be addressed. The decision as to whether to use a p53-targeted therapeutic approach in a tumor is predicted to be guided, in part, by advances in the classification of tumors, such as those described for medulloblastoma and glioblastoma. Among the recently identified functions for p53, perhaps the most exciting is the role of p53 in the regulation of neural precursor cell self-renewal, differentiation, and cell fate. New mouse models and tools to identify and track neural precursor cells in vivo will be required to further elucidate the role of p53 in this cell type. New information about the molecular and environmental factors that regulate p53 function in neural precursor cells may provide novel insight into how the loss of p53 promotes CNS tumorigenesis.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1. Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem. 1999;274:36031-4 [DOI] [PubMed] [Google Scholar]
- 2. Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003;10:431-42 [DOI] [PubMed] [Google Scholar]
- 3. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15-6 [DOI] [PubMed] [Google Scholar]
- 4. Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochem Res. 2003;28:15-27 [DOI] [PubMed] [Google Scholar]
- 5. Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761-77 [DOI] [PubMed] [Google Scholar]
- 6. Schmid P, Lorenz A, Hameister H, Montenarh M. Expression of p53 during mouse embryogenesis. Development. 1991;113:857-65 [DOI] [PubMed] [Google Scholar]
- 7. Rogel A, Popliker M, Webb CG, Oren M. p53 cellular tumor antigen: analysis of mRNA levels in normal adult tissues, embryos, and tumors. Mol Cell Biol. 1985;5:2851-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gottlieb E, Haffner R, King A, et al. Transgenic mouse model for studying the transcriptional activity of the p53 protein: age- and tissue-dependent changes in radiation-induced activation during embryogenesis. EMBO J. 1997;16:1381-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Komarova EA, Chernov MV, Franks R, et al. Transgenic mice with p53-responsive lacZ: p53 activity varies dramatically during normal development and determines radiation and drug sensitivity in vivo. EMBO J. 1997;16:1391-400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. MacCallum DE, Hupp TR, Midgley CA, et al. The p53 response to ionising radiation in adult and developing murine tissues. Oncogene. 1996;13:2575-87 [PubMed] [Google Scholar]
- 11. Armstrong JF, Kaufman MH, Harrison DJ, Clarke AR. High-frequency developmental abnormalities in p53-deficient mice. Curr Biol. 1995;5:931-6 [DOI] [PubMed] [Google Scholar]
- 12. Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, Jacks T. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet. 1995;10:175-80 [DOI] [PubMed] [Google Scholar]
- 13. Hosako H, Francisco LE, Martin GS, Mirkes PE. The roles of p53 and p21 in normal development and hyperthermia-induced malformations. Birth Defects Res B Dev Reprod Toxicol. 2009;86:40-7 [DOI] [PubMed] [Google Scholar]
- 14. Ikeda S, Hawes NL, Chang B, Avery CS, Smith RS, Nishina PM. Severe ocular abnormalities in C57BL/6 but not in 129/Sv p53-deficient mice. Invest Ophthalmol Vis Sci. 1999;40:1874-8 [PubMed] [Google Scholar]
- 15. Gao Y, Sun Y, Frank KM, et al. A critical role for DNA end-joining proteins in both lymphogenesis and neurogenesis. Cell. 1998;95:891-902 [DOI] [PubMed] [Google Scholar]
- 16. Frank KM, Sharpless NE, Gao Y, et al. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell. 2000;5:993-1002 [DOI] [PubMed] [Google Scholar]
- 17. Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J. 2000;19:1397-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clarke AR, Maandag ER, van Roon M, et al. Requirement for a functional Rb-1 gene in murine development. Nature. 1992;359:328-30 [DOI] [PubMed] [Google Scholar]
- 19. Lee EY, Chang CY, Hu N, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288-94 [DOI] [PubMed] [Google Scholar]
- 20. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295-300 [DOI] [PubMed] [Google Scholar]
- 21. Tsai KY, Hu Y, Macleod KF, Crowley D, Yamasaki L, Jacks T. Mutation of E2f-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol Cell. 1998;2:293-304 [DOI] [PubMed] [Google Scholar]
- 22. Ziebold U, Reza T, Caron A, Lees JA. E2F3 contributes both to the inappropriate proliferation and to the apoptosis arising in Rb mutant embryos. Genes Dev. 2001;15:386-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsai KY, MacPherson D, Rubinson DA, Crowley D, Jacks T. ARF is not required for apoptosis in Rb mutant mouse embryos. Curr Biol. 2002;12:159-63 [DOI] [PubMed] [Google Scholar]
- 24. MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol. 2003;23:1044-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237-45 [DOI] [PubMed] [Google Scholar]
- 26. Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857-60 [DOI] [PubMed] [Google Scholar]
- 27. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296-9 [DOI] [PubMed] [Google Scholar]
- 28. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25-7 [DOI] [PubMed] [Google Scholar]
- 29. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299-303 [DOI] [PubMed] [Google Scholar]
- 30. Shvarts A, Steegenga WT, Riteco N, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996;15:5349-57 [PMC free article] [PubMed] [Google Scholar]
- 31. Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol. 2000;20:1001-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206-8 [DOI] [PubMed] [Google Scholar]
- 33. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203-6 [DOI] [PubMed] [Google Scholar]
- 34. Parant J, Chavez-Reyes A, Little NA, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92-5 [DOI] [PubMed] [Google Scholar]
- 35. Migliorini D, Lazzerini Denchi E, Danovi D, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daujat S, Neel H, Piette J. Preferential expression of Mdm2 oncogene during the development of neural crest and its derivatives in mouse early embryogenesis. Mech Dev. 2001;103:163-5 [DOI] [PubMed] [Google Scholar]
- 37. Francoz S, Froment P, Bogaerts S, et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc Natl Acad Sci U S A. 2006;103:3232-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci U S A. 2006;103:3226-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Malek R, Matta J, Taylor N, Perry ME, Mendrysa SM. The p53 inhibitor MDM2 facilitates sonic hedgehog–mediated tumorigenesis and influences cerebellar foliation. PLoS ONE. 2011;6:e17884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mendrysa SM, McElwee MK, Michalowski J, O’Leary KA, Young KM, Perry ME. mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. 2003;23:462-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ringshausen I, O’Shea CC, Finch AJ, Swigart LB, Evan GI. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell. 2006;10:501-14 [DOI] [PubMed] [Google Scholar]
- 42. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev. 2006;6:909-23 [DOI] [PubMed] [Google Scholar]
- 43. Sohur US, Emsley JG, Mitchell BD, Macklis JD. Adult neurogenesis and cellular brain repair with neural progenitors, precursors and stem cells. Philos Trans R Soc Lond B Biol Sci. 2006;361:1477-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Temple S. The development of neural stem cells. Nature. 2001;414:112-7 [DOI] [PubMed] [Google Scholar]
- 45. Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255:1707-10 [DOI] [PubMed] [Google Scholar]
- 46. Richards LJ, Kilpatrick TJ, Bartlett PF. De novo generation of neuronal cells from the adult mouse brain. Proc Natl Acad Sci U S A. 1992;89:8591-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kempermann G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci. 1998;18:3206-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee A, Kessler JD, Read TA, et al. Isolation of neural stem cells from the postnatal cerebellum. Nat Neurosci. 2005;8:723-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843-50 [DOI] [PubMed] [Google Scholar]
- 50. Tropepe V, Coles BL, Chiasson BJ, et al. Retinal stem cells in the adult mammalian eye. Science. 2000;287:2032-6 [DOI] [PubMed] [Google Scholar]
- 51. Nagao M, Campbell K, Burns K, Kuan CY, Trumpp A, Nakafuku M. Coordinated control of self-renewal and differentiation of neural stem cells by Myc and the p19ARF-p53 pathway. J Cell Biol. 2008;183:1243-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meletis K, Wirta V, Hede SM, Nister M, Lundeberg J, Frisen J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363-9 [DOI] [PubMed] [Google Scholar]
- 53. van Lookeren Campagne M, Gill R. Tumor-suppressor p53 is expressed in proliferating and newly formed neurons of the embryonic and postnatal rat brain: comparison with expression of the cell cycle regulators p21Waf1/Cip1, p27Kip1, p57Kip2, p16Ink4a, cyclin G1, and the proto-oncogene Bax. J Comp Neurol. 1998;397:181-98 [DOI] [PubMed] [Google Scholar]
- 54. Gil-Perotin S, Marin-Husstege M, Li J, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Armesilla-Diaz A, Bragado P, Del Valle I, et al. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378-89 [DOI] [PubMed] [Google Scholar]
- 56. Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28:663-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97:703-16 [DOI] [PubMed] [Google Scholar]
- 58. Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17:5046-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alvarez-Buylla A, Kohwi M, Nguyen TM, Merkle FT. The heterogeneity of adult neural stem cells and the emerging complexity of their niche. Cold Spring Harb Symp Quant Biol. 2008;73:357-65 [DOI] [PubMed] [Google Scholar]
- 60. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730-7 [DOI] [PubMed] [Google Scholar]
- 61. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030-7 [DOI] [PubMed] [Google Scholar]
- 63. O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10 [DOI] [PubMed] [Google Scholar]
- 64. Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401 [DOI] [PubMed] [Google Scholar]
- 65. Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178-83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fomchenko EI, Holland EC. Stem cells and brain cancer. Exp Cell Res. 2005;306:323-9 [DOI] [PubMed] [Google Scholar]
- 67. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11 [DOI] [PubMed] [Google Scholar]
- 68. Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol. 2011;192:209-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Burness ML, Sipkins DA. The stem cell niche in health and malignancy. Semin Cancer Biol. 2010;20:107-15 [DOI] [PubMed] [Google Scholar]
- 70. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593-602 [DOI] [PubMed] [Google Scholar]
- 71. Graeber TG, Osmanian C, Jacks T, et al. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88-91 [DOI] [PubMed] [Google Scholar]
- 72. Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol. 1984;4:1689-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kastan MB, Zhan Q, el-Deiry WS, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587-97 [DOI] [PubMed] [Google Scholar]
- 74. Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345-7 [DOI] [PubMed] [Google Scholar]
- 75. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49-53 [DOI] [PubMed] [Google Scholar]
- 76. Moll UM, Riou G, Levine AJ. Two distinct mechanisms alter p53 in breast cancer: mutation and nuclear exclusion. Proc Natl Acad Sci U S A. 1992;89:7262-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Crook T, Tidy JA, Vousden KH. Degradation of p53 can be targeted by HPV E6 sequences distinct from those required for p53 binding and trans-activation. Cell. 1991;67:547-56 [DOI] [PubMed] [Google Scholar]
- 78. Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci U S A. 1991;88:5523-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Florenes VA, Maelandsmo GM, Forus A, Andreassen A, Myklebost O, Fodstad O. MDM2 gene amplification and transcript levels in human sarcomas: relationship to TP53 gene status. J Natl Cancer Inst. 1994;86:1297-302 [DOI] [PubMed] [Google Scholar]
- 80. Oliner JD, Kinzler KW, Meltzer PS, George DL, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80-3 [DOI] [PubMed] [Google Scholar]
- 81. Polkinghorn WR, Tarbell NJ. Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nat Clin Pract Oncol. 2007;4:295-304 [DOI] [PubMed] [Google Scholar]
- 82. Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1-13 [PMC free article] [PubMed] [Google Scholar]
- 83. Barel D, Avigad S, Mor C, Fogel M, Cohen IJ, Zaizov R. A novel germ-line mutation in the noncoding region of the p53 gene in a Li-Fraumeni family. Cancer Genet Cytogenet. 1998;103:1-6 [DOI] [PubMed] [Google Scholar]
- 84. Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994-1004 [PMC free article] [PubMed] [Google Scholar]
- 85. Shakhova O, Leung C, van Montfort E, Berns A, Marino S. Lack of Rb and p53 delays cerebellar development and predisposes to large cell anaplastic medulloblastoma through amplification of N-Myc and Ptch2. Cancer Res. 2006;66:5190-200 [DOI] [PubMed] [Google Scholar]
- 86. Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ. Null mutation of DNA strand break-binding molecule poly(ADP-ribose) polymerase causes medulloblastomas in p53(−/−) mice. Am J Pathol. 2003;162:343-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61:513-6 [PubMed] [Google Scholar]
- 88. Adesina AM, Nalbantoglu J, Cavenee WK. p53 gene mutation and mdm2 gene amplification are uncommon in medulloblastoma. Cancer Res. 1994;54:5649-51 [PubMed] [Google Scholar]
- 89. Ferretti E, De Smaele E, Di Marcotullio L, Screpanti I, Gulino A. Hedgehog checkpoints in medulloblastoma: the chromosome 17p deletion paradigm. Trends Mol Med. 2005;11:537-45 [DOI] [PubMed] [Google Scholar]
- 90. Saylors RL, III, Sidransky D, Friedman HS, et al. Infrequent p53 gene mutations in medulloblastomas. Cancer Res. 1991;51:4721-3 [PubMed] [Google Scholar]
- 91. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998;26:3453-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Raffel C, Thomas GA, Tishler DM, Lassoff S, Allen JC. Absence of p53 mutations in childhood central nervous system primitive neuroectodermal tumors. Neurosurgery. 1993;33:301-5; discussion 305-6 [DOI] [PubMed] [Google Scholar]
- 93. Badiali M, Iolascon A, Loda M, et al. p53 gene mutations in medulloblastoma: immunohistochemistry, gel shift analysis, and sequencing. Diagn Mol Pathol. 1993;2:23-8 [PubMed] [Google Scholar]
- 94. Wang W, Kumar P, Whalley J, et al. The mutation status of PAX3 and p53 genes in medulloblastoma. Anticancer Res. 1998;18:849-53 [PubMed] [Google Scholar]
- 95. Pazzaglia S, Mancuso M, Tanori M, et al. Modulation of patched-associated susceptibility to radiation induced tumorigenesis by genetic background. Cancer Res. 2004;64:3798-806 [DOI] [PubMed] [Google Scholar]
- 96. Pfaff E, Remke M, Sturm D, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010;28:5188-96 [DOI] [PubMed] [Google Scholar]
- 97. Northcott PA, Korshunov A, Witt H, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29:1408-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28:1345-50 [DOI] [PubMed] [Google Scholar]
- 99. Sadot E, Geiger B, Oren M, Ben-Ze’ev A. Down-regulation of beta-catenin by activated p53. Mol Cell Biol. 2001;21:6768-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Levina E, Oren M, Ben-Ze’ev A. Downregulation of beta-catenin by p53 involves changes in the rate of beta-catenin phosphorylation and Axin dynamics. Oncogene. 2004;23:4444-53 [DOI] [PubMed] [Google Scholar]
- 101. Damalas A, Ben-Ze’ev A, Simcha I, et al. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999;18:3054-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hogarty MD. The requirement for evasion of programmed cell death in neuroblastomas with MYCN amplification. Cancer Lett. 2003;197:173-9 [DOI] [PubMed] [Google Scholar]
- 103. Slack AD, Chen Z, Ludwig AD, Hicks J, Shohet JM. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007;67:2448-55 [DOI] [PubMed] [Google Scholar]
- 104. Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109-13 [DOI] [PubMed] [Google Scholar]
- 105. Ashcroft M, Ludwig RL, Woods DB, et al. Phosphorylation of HDM2 by Akt. Oncogene. 2002;21:1955-62 [DOI] [PubMed] [Google Scholar]
- 106. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598-603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ogawara Y, Kishishita S, Obata T, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843-50 [DOI] [PubMed] [Google Scholar]
- 108. Abe Y, Oda-Sato E, Tobiume K, et al. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc Natl Acad Sci U S A. 2008;105:4838-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Prykhozhij SV. In the absence of Sonic hedgehog, p53 induces apoptosis and inhibits retinal cell proliferation, cell cycle exit and differentiation in zebrafish. PLoS ONE. 2010;5:e13549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Giordana MT, Duo D, Gasverde S, et al. MDM2 overexpression is associated with short survival in adults with medulloblastoma. Neuro Oncol. 2002;4:115-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Castellino RC, De Bortoli M, Lu X, et al. Medulloblastomas overexpress the p53-inactivating oncogene WIP1/PPM1D. J Neurooncol. 2008;86:245-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lu X, Ma O, Nguyen TA, Jones SN, Oren M, Donehower LA. The Wip1 phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell. 2007;12:342-54 [DOI] [PubMed] [Google Scholar]
- 113. Kasuga C, Nakahara Y, Ueda S, et al. Expression of MAGE and GAGE genes in medulloblastoma and modulation of resistance to chemotherapy: laboratory investigation. J Neurosurg Pediatr. 2008;1:305-13 [DOI] [PubMed] [Google Scholar]
- 114. Marcar L, Maclaine NJ, Hupp TR, Meek DW. Mage-A cancer/testis antigens inhibit p53 function by blocking its interaction with chromatin. Cancer Res. 2010;70:10362-70 [DOI] [PubMed] [Google Scholar]
- 115. Ferretti E, De Smaele E, Po A, et al. MicroRNA profiling in human medulloblastoma. Int J Cancer. 2009;124:568-77 [DOI] [PubMed] [Google Scholar]
- 116. Grahovac G, Tomac D, Lambasa S, Zoric A, Habek M. Cerebellar glioblastomas: pathophysiology, clinical presentation and management. Acta Neurochir (Wien). 2009;151:653-7 [DOI] [PubMed] [Google Scholar]
- 117. Ohgaki H, Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009;100:2235-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170:1445-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64:6892-9 [DOI] [PubMed] [Google Scholar]
- 120. Zheng H, Ying H, Yan H, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455:1129-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. (TCGA) CGARN Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Okamoto Y, Di Patre PL, Burkhard C, et al. Population-based study on incidence, survival rates, and genetic alterations of low-grade diffuse astrocytomas and oligodendrogliomas. Acta Neuropathol. 2004;108:49-56 [DOI] [PubMed] [Google Scholar]
- 123. Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109-13 [DOI] [PubMed] [Google Scholar]
- 124. Zhu Y, Guignard F, Zhao D, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8:119-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wade M, Wang YV, Wahl GM. The p53 orchestra: Mdm2 and Mdmx set the tone. Trends Cell Biol. 2010;20:299-309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Tao W, Levine AJ. P19(ARF) stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci U S A. 1999;96:6937-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tao W, Levine AJ. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc Natl Acad Sci U S A. 1999;96:3077-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20-6 [DOI] [PubMed] [Google Scholar]
- 129. Reifenberger G, Liu L, Ichimura K, Schmidt EE, Collins VP. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993;53:2736-9 [PubMed] [Google Scholar]
- 130. Riemenschneider MJ, Buschges R, Wolter M, et al. Amplification and overexpression of the MDM4 (MDMX) gene from 1q32 in a subset of malignant gliomas without TP53 mutation or MDM2 amplification. Cancer Res. 1999;59:6091-6 [PubMed] [Google Scholar]
- 131. Nakamura M, Watanabe T, Klangby U, et al. p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol. 2001;11:159-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Stegh AH, Brennan C, Mahoney JA, et al. Glioma oncoprotein Bcl2L12 inhibits the p53 tumor suppressor. Genes Dev. 2010;24:2194-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Stegh AH, Kim H, Bachoo RM, et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007;21:98-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010;18:619-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bagchi A, Papazoglu C, Wu Y, et al. CHD5 is a tumor suppressor at human 1p36. Cell. 2007;128:459-75 [DOI] [PubMed] [Google Scholar]
- 136. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Stupp R, Hegi ME, van den Bent MJ, et al. Changing paradigms: an update on the multidisciplinary management of malignant glioma. Oncologist. 2006;11:165-80 [DOI] [PubMed] [Google Scholar]
- 138. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60 [DOI] [PubMed] [Google Scholar]
- 139. Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957-67 [DOI] [PubMed] [Google Scholar]
- 140. Lane DP, Fischer PM. Turning the key on p53. Nature. 2004;427:789-90 [DOI] [PubMed] [Google Scholar]
- 141. Brown CJ, Cheok CF, Verma CS, Lane DP. Reactivation of p53: from peptides to small molecules. Trends Pharmacol Sci. 2011;32:53-62 [DOI] [PubMed] [Google Scholar]
- 142. Foster BA, Coffey HA, Morin MJ, Rastinejad F. Pharmacological rescue of mutant p53 conformation and function. Science. 1999;286:2507-10 [DOI] [PubMed] [Google Scholar]
- 143. Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282-8 [DOI] [PubMed] [Google Scholar]
- 144. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8 [DOI] [PubMed] [Google Scholar]
- 145. Chen L, Agrawal S, Zhou W, Zhang R, Chen J. Synergistic activation of p53 by inhibition of MDM2 expression and DNA damage. Proc Natl Acad Sci U S A. 1998;95:195-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321-8 [DOI] [PubMed] [Google Scholar]
- 147. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275-83 [DOI] [PubMed] [Google Scholar]
- 148. Jacobs WB, Kaplan DR, Miller FD. The p53 family in nervous system development and disease. J Neurochem. 2006;97:1571-84 [DOI] [PubMed] [Google Scholar]
- 149. Norimura T, Nomoto S, Katsuki M, Gondo Y, Kondo S. p53-dependent apoptosis suppresses radiation-induced teratogenesis. Nat Med. 1996;2:577-80 [DOI] [PubMed] [Google Scholar]
- 150. Nicol CJ, Harrison ML, Laposa RR, Gimelshtein IL, Wells PG. A teratologic suppressor role for p53 in benzo[a]pyrene-treated transgenic p53-deficient mice. Nat Genet. 1995;10:181-7 [DOI] [PubMed] [Google Scholar]
- 151. Jones NC, Lynn ML, Gaudenz K, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14:125-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Chalepakis G, Goulding M, Read A, Strachan T, Gruss P. Molecular basis of splotch and Waardenburg Pax-3 mutations. Proc Natl Acad Sci U S A. 1994;91:3685-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev. 2002;16:676-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Epstein DJ, Vogan KJ, Trasler DG, Gros P. A mutation within intron 3 of the Pax-3 gene produces aberrantly spliced mRNA transcripts in the splotch (Sp) mouse mutant. Proc Natl Acad Sci U S A. 1993;90:532-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Auerbach R. Analysis of the developmental effects of a lethal mutation in the house mouse. J Exp Zool. 1954;127:305-29 [Google Scholar]
- 156. Danilova N, Kumagai A, Lin J. p53 upregulation is a frequent response to deficiency of cell-essential genes. PLoS ONE. 2010;5:e15938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Yang X, Klein R, Tian X, Cheng HT, Kopan R, Shen J. Notch activation induces apoptosis in neural progenitor cells through a p53-dependent pathway. Dev Biol. 2004;269:81-94 [DOI] [PubMed] [Google Scholar]
- 158. Whelan JT, Forbes SL, Bertrand FE. CBF-1 (RBP-J kappa) binds to the PTEN promoter and regulates PTEN gene expression. Cell Cycle. 2007;6:80-4 [DOI] [PubMed] [Google Scholar]
- 159. Wang C, Qi R, Li N, et al. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284:16183-90 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 160. He SJ, Stevens G, Braithwaite AW, Eccles MR. Transfection of melanoma cells with antisense PAX3 oligonucleotides additively complements cisplatin-induced cytotoxicity. Mol Cancer Ther. 2005;4:996-1003 [DOI] [PubMed] [Google Scholar]
- 161. Underwood TJ, Amin J, Lillycrop KA, Blaydes JP. Dissection of the functional interaction between p53 and the embryonic proto-oncoprotein PAX3. FEBS Lett. 2007;581:5831-5 [DOI] [PubMed] [Google Scholar]
- 162. Langley E, Pearson M, Faretta M, et al. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Cheng HL, Mostoslavsky R, Saito S, et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)–deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A Clin Mol Teratol. 2010;88:653-69 [DOI] [PubMed] [Google Scholar]