

Abstract
Idiopathic nephrotic syndrome (INS) is a genetically heterogeneous group of disorders characterized by proteinuria, hypoalbuminemia, and edema. Because it typically results in end-stage kidney disease, the steroid-resistant subtype (SRNS) of INS is especially important when it occurs in children. The present study included 29 affected and 22 normal individuals from 17 SRNS families; genome-wide analysis was performed with Affymetrix 250K SNP arrays followed by homozygosity mapping. A large homozygous stretch on chromosomal region 12p12 was identified in one consanguineous family with two affected siblings. Direct sequencing of protein tyrosine phosphatase receptor type O (PTPRO; also known as glomerular epithelial protein-1 [GLEPP1]) showed homozygous c.2627+1G>T donor splice-site mutation. This mutation causes skipping of the evolutionarily conserved exon 16 (p.Glu854_Trp876del) at the RNA level. Immunohistochemistry with GLEPP1 antibody showed a similar staining pattern in the podocytes of the diseased and control kidney tissues. We used a highly polymorphic intragenic DNA marker—D12S1303—to search for homozygosity in 120 Turkish and 13 non-Turkish individuals in the PodoNet registry. This analysis yielded 17 candidate families, and a distinct homozygous c.2745+1G>A donor splice-site mutation in PTPRO was further identified via DNA sequencing in a second Turkish family. This mutation causes skipping of exon 19, and this introduces a premature stop codon at the very beginning of exon 20 (p.Asn888Lysfs∗3) and causes degradation of mRNA via nonsense-mediated decay. Immunohistochemical analysis showed complete absence of immunoreactive PTPRO. Ultrastructural alterations, such as diffuse foot process fusion and extensive microvillus transformation of podocytes, were observed via electron microscopy in both families. The present study introduces mutations in PTPRO as another cause of autosomal-recessive nephrotic syndrome.
Main Text
Idiopathic nephrotic syndrome (INS), a disorder characterized by massive proteinuria due to loss of glomerular permselectivity resulting from podocyte functional alterations, affects 16 children out of 100,000.1 Whereas in the majority of cases the disorder is caused by immunological alterations readily responsive to glucocorticoid treatment, and individuals have an excellent long-term prognosis, 10%–20% of individuals are steroid resistant (SRNS) and typically progress to end-stage kidney disease (ESKD).2 In recent years, inherited defects in podocyte structure and function were observed in a fraction of children with the steroid-resistant subtype (SRNS). To date, abnormalities in seven genes expressed in the podocyte have been reported to cause nonsyndromic SRNS in humans.3–9
LAMB2 (MIM 150325) mutations cause Pierson syndrome (MIM 609049), which is characterized by congenital nephrotic syndrome with distinct eye abnormalities.10 Mutations in exons 8 and 9 of WT1 (MIM 607102) have been observed in individuals with isolated SRNS and in those with SRNS associated with Wilms' tumor or urogenital malformations (i.e., Denys-Drash syndrome [DDS] [MIM 194080] and Frasier syndrome [MIM 136680]).11 Together, these individuals account for approximately 15%–20% of SRNS cases, suggesting marked genetic heterogeneity of the disorder.
In the present study, we performed genome-wide analysis followed by homozygosity mapping to identify mutations in additional genes in a large cohort of SRNS individuals. The study included 17 autosomal-recessive SRNS families, defined according to standard criteria.12 Mutations in NPHS2 (MIM 604766) and WT1 (MIM 607102) in all cases and in NPHS1 (MIM 602716), LAMB2 (MIM 150325), and PLCE1 (MIM 608414) in cases with congenital nephrotic syndrome were ruled out via direct sequencing. In all, 29 affected and 22 normal individuals from the 17 families were included in the genome-wide analysis, which was performed with GeneChip mapping 250K NspI SNP arrays from Affymetrix according to the manufacturer's recommendations. Genotype files (CHP files) were generated with Affymetrix GTYPE software and were transferred to the VIGENOS (Visual Genome Studio) program (Hemosoft, Ankara), which facilitates visualization of a large quantity of genomic data in comprehensible visual screens.13
In affected cases, large homozygous segments overlapping within and between families were scored. This analysis, which did not show any shared regions of homozygosity among the 17 families, indicated extensive genetic heterogeneity of nephrotic syndrome. Each family was then individually analyzed; overlapping homozygous haplotype stretches in affected family members were recorded, and functional candidate genes positioned in these regions were identified. This approach showed that there was one consanguineous family (family A, Figures 1A and 1B) that contained two affected siblings and a large homozygous stretch spanning approximately 14 Mb between markers rs16928082 (SNP_A-2293685) and rs10841414 (SNP_A-2047221) on chromosome 12. This region harbored 182 genes. In this particular family, four additional loci on chromosomal regions 8qtel, 11p15, 17ptel, and 18q11 (Figure S1, available online) were also observed; however, the chromosomal region 12p was highlighted because of the presence of a highly relevant candidate gene—protein tyrosine phosphatase receptor type O (PTPRO), also known as GLEPP1 (glomerular epithelial protein-1) (MIM 600579).
Figure 1.
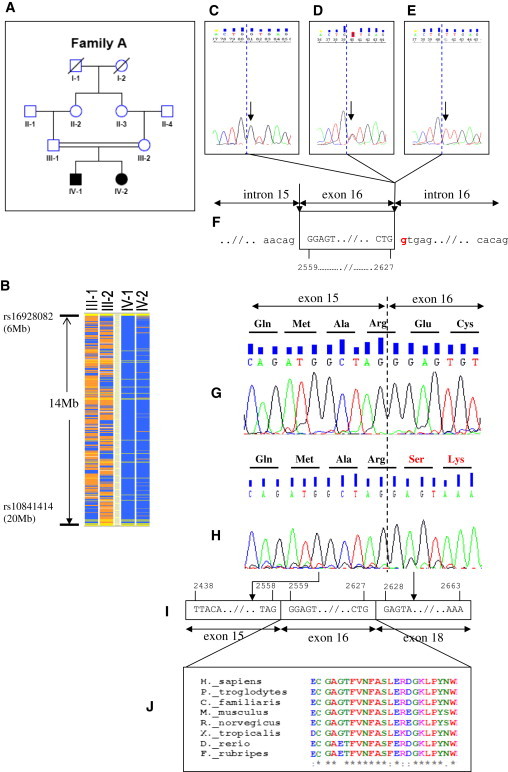
Positional Cloning of PTPRO Mutations in Family A
(A) The pedigree of the index family. Squares indicate males and circles indicate females. Solid symbols indicate affected individuals. Double-horizontal bars indicate consanguinity.
(B) The schematic representation of homozygosity data. Genotype files (CHP files) were generated with Affymetrix GTYPE software and were transferred to the VIGENOS (Visual Genome Studio) program (Hemosoft, Ankara),13 which facilitates visualization of a large quantity of genomic data in comprehensible visual screens. Homozygous genotypes identical to the genotype data obtained from the index case (IV-1) are shown in blue. Contrasting homozygote genotypes are shown in white, whereas heterozygous SNPs appear in orange. Noninformativeness as a result of heterozygous genotypes in parent-child trios is indicated in yellow. No calls are indicated in gray. The overlapping homozygous stretches in two affected individuals are approximately 14 Mb in size between markers rs16928082 and rs10841414 on chromosome 12.
(C–E) The sequence electropherograms of exon 16 of PTPRO. The wild-type sequence in a healthy control is shown in the left electropherogram (C, arrow). Whereas the healthy parents were heterozygous for the mutation (D, arrow), both affected siblings had a homozygous donor splice-site c.2627+1G>T (p.Glu854_Trp876del) mutation (E, arrow); dashed blue lines show exon-intron boundaries.
(F) The position of the mutation is shown as red in the genomic sequence. Horizontal arrows indicate subsequent introns and exons. Vertical arrows show intron-exon boundaries. Four-digit numbers (i.e., 2559 and 2627) show starting and ending nucleotides of exon 16.
(G) Sequencing of PTPRO cDNA of both peripheral blood lymphoctes from healthy individuals and nephrectomy specimens showed that exon 15 was followed by exon 16 (normal splicing). Dashed line shows exon-exon boundaries.
(H–J) c.2627+1G>T donor splice-site mutation (NM_002848.2) in Family A results in skipping of exon 16, which is conserved during evolution (see also Figure S3). Four-digit numbers show start and end positions of the respective exons.
The role of PTPRO in regulating glomerular pressure and permselectivity was previously demonstrated by the phenotype of the knockout model and antibody treatment studies.14,15 PTPRO primers (Table S1) were designed according to the NCBI genomic reference sequence along contig NC_000012.11 (National Center for Biotechnology Information [NCBI] RefSeq accession numbers NM_030667.1 [for the mRNA] and NP_109592.1 [for the protein]). The entire coding region and the exon-intron boundaries of PTPRO were sequenced directly from fragments amplified by PCR with an Applied Biosystems 3130 genetic analyzer according to the manufacturer's protocol.
In the present study, a homozygous c.2627+1G>T (p.Glu854_Trp876del) donor splice-site mutation in PTPRO in family A (in which both parents were heterozygous) was observed (Figures 1C–1F and Table 1).
Table 1.
Clinical Features of the Affected Individuals with PTPRO Mutations
| Family and Individual | Consang uinity | Nucleotide Alterationa | Segregation | Age at Onset (years) | Initial Up/Uc (mg/mg) |
Serum Albumin at Onset (g dL−1) |
Age at ESKD (years) | Histology | Last Up/Uc (mg/mg) |
Last serum albumin (g dL−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| A | ||||||||||
| IV-1 | yes | c.2627+1G>T | H, M, P | 7 | 9.9 | 2.5 | none at 11 years | ND | 3.8 | 4 |
| IV-2 | yes | c.2627+1G>T | H, M, P | 5 | 16.2 | 1.9 | none at 7 years | FSGS | 1.18 | 3.6 |
| B | ||||||||||
| IV-1 | yes | c.2745+1G>A | H, M, P | 14 | 14.1 | 2.2 | 18 (transplanted) | ND | 0.12 | 4 |
| IV-2 | yes | c.2745+1G>A | H, M, P | 11 | 7 | 4 | none at 15 years | MCD | 3.2 | 4.2 |
| IV-3 | yes | c.2745+1G>A | H, M, P | 9 | 6.1 | 4 | none at 12 years | ND | 2.9 | 3.6 |
| IV-4 | yes | WT | healthy | 0.1 | ||||||
The following abbreviations are used: WT, wild-type; H, Homozygous in affected individual; M, heterozygous mutation identified in mother; P, heterozygous mutation identified in father; Up/Uc: urinary protein/creatinine ratio; FSGS: Focal segmental glomerulosclerosis; MCD; minimal change disease; ND: not done. ESKD: End-stage kidney disease.
The nucleotide positions are numbered according to variant 2 of PTPRO (NM_002848.2). All mutations were absent in 180 healthy Turkish children.
In this family, the index individual (Figure 1A, IV-1) presented with SRNS at age 9. He was treated with prednisone and cyclophosphamide without effect. A partial reduction of proteinuria was noted in response to combined therapy with cyclosporin A, methylprednisolone pulses, enalapril, and losartan (Table 1). The second affected sibling (Figure 1A, IV-2) was diagnosed at age 5. Although cyclophosphamide and cyclosporin A failed to induce remission, a partial response to combined pulse methylprednisolone and cyclosporin A therapy was observed (Table 1).
We expanded this study to include other individuals with the identical phenotype in order to identify additional families. Screening for a highly polymorphic intragenic DNA marker (D12S1303) in 120 Turkish and 13 non-Turkish individuals in the PodoNet registry for homozygosity yielded 17 candidate families for direct sequencing. DNA-sequencing analysis identified a second Turkish family (family B) with a distinct homozygous c.2745+1G>A (p.Asn888Lysfs∗3) donor splice-site mutation in PTPRO (Figures 2A–2E and Table 1). This family included three affected siblings and one healthy sibling; the parents were heterozygous, and the healthy sibling did not carry the mutation (Figures 2B and 2C and Table 1).
Figure 2.
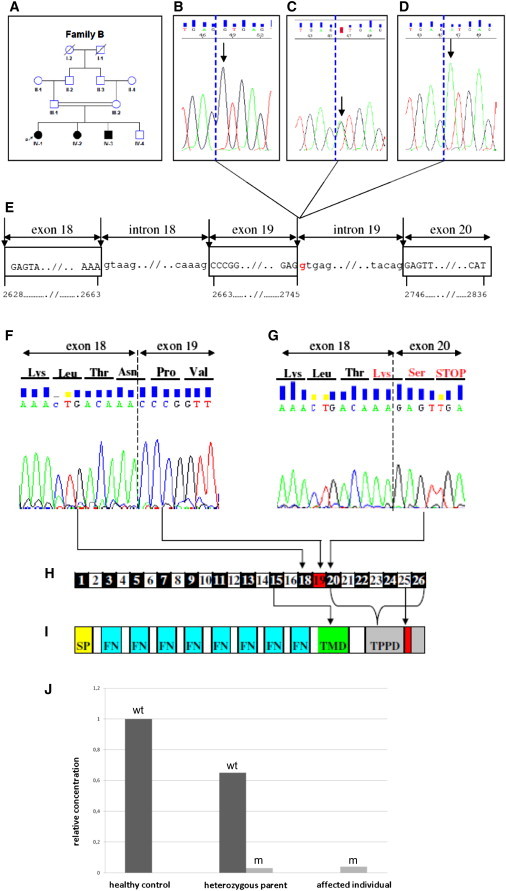
Identification of Family B with a Different PTPRO Mutation than Family A
(A) Pedigree of the second family. Squares indicate males and circles indicate females. Solid symbols indicate affected individuals. Double-horizontal bars indicate consanguinity. P indicates the proband.
(B–D) Sequence electropherograms of exon 19 of PTPRO. The wild-type sequence in both healthy offspring in the family and a healthy control (B, arrow). Whereas the unaffected parents were heterozygous for the mutation (C, arrow), all affected siblings had a homozygous donor splice-site c.2745+1G>A (p.Asn888Lysfs∗3) mutation (D, arrow). Dashed blue lines show exon-intron boundaries.
(E) Position of the mutation is shown as red in the genomic sequence. Horizontal arrows indicate subsequent introns and exons. Vertical arrows show intron-exon boundaries. Four-digit numbers show starting and ending nucleotides of the respective exons.
(F) Sequencing of PTPRO cDNA of both peripheral blood lymphocytes from healthy individuals and nephrectomy specimens showed that exon 18 was followed by exon 19 (normal splicing).
(G–I) NM_002848.2: c.2745+1G>A donor splice-site mutation results in skipping of evolutionary conserved exon 19, which introduces a premature stop codon at the very beginning of exon 20. Dashed line shows exon-exon boundaries. Organization of exons and respective domains in variant 2 of PTPRO is schematically shown (H and I). The following abbreviations are used: SP, signal peptide; FN, fibronectin type III; TMD, transmembrane domain; TPPD, tyrosine protein phosphatase domain.
(J) Allelic expression analysis of wild-type and mutant transcripts of PTPRO in a healthy sibling (IV-4), a heterozygous parent (III-2), and an affected individual (IV-2). According to comparative quantification analysis, the mutant transcript was decreased nearly 30-fold in cDNA samples from both a heterozygous parent and the affected individual (wt: wild-type allele, m: mutant allele).
The first sibling in family B (Figure 2A, IV-1) presented with SRNS at age 14; cyclophosphamide, pulse methylprednisolone, and cyclosporin A failed to induce remission. Renal function progressively deteriorated and ESKD occurred at age 18. She received a living-related kidney transplant, and there was no disease recurrence. This sibling also carried a heterozygous p.Arg229Gln (c.686G>A) variation in the podocin. The second and third siblings (Figure 2A, IV-2 and IV-3) were diagnosed by family screening while they were clinically asymptomatic after screening for proteinuria because of family history. The second sibling (Figure 2A, IV-2) was resistant to oral steroid treatment; however, partial remission with intensified immunosuppression and a renin-angiotensin system blockade (i.e., oral and pulse steroid, cyclophosphamide, cyclosporin A, enalapril, and losartan) was achieved. The third sibling (Figure 2A, IV-3) initially achieved complete remission with oral prednisone treatment but then developed a steroid-dependent clinical course. A course of oral cyclophosphamide and a maintenance course of low-dose alternate-day oral steroids yielded remission for 1 year. Subsequently, this individual experienced another relapse, which was completely resistant to oral steroids but partially responsive to monthly intravenous steroid pulses. At the time this report was written, when the siblings were ages 15 and 12, their glomerular filtration rate was normal; however, they both had proteinuria. The study protocol was approved by the Hacettepe University Ethics Committee (TBK08/1-57). Participants or their guardians provided written informed consent.
PTPRO extends over 51 kb and contains 26 exons. This gene encodes the receptor-type protein tyrosine phosphatase that contains a single intracellular catalytic domain with a characteristic signature motif. The gene product is an integral membrane protein. Several alternatively spliced transcript variants that exhibit tissue-specific expression have been described; among them, only variants 1 and 2 are expressed in the glomerulus. Variant 1 (NM_030667.1) encodes the longest isoform. The encoded protein has a large extracellular domain containing eight repeats of a fibronectin-type-III-like motif. Variant 2 (NM_002848.2) lacks exon 17, resulting in a missing juxtamembrane region (Figure S2).
We analyzed the effects of the identified mutations on RNA splicing in both families.
We first predicted the consequences of the mutations via in silico analysis by using automated splice-site analysis software.16,17 For the c.2745+1G>A mutation the software predicted an abolished site, whereas for the c.2627+1G>T mutation a leaky site was predicted. We then used cDNA analysis to observe these changes. We collected fresh heparinized blood from all the individuals, their parents, and healthy controls. In addition, a kidney biopsy was performed in one affected individual from each family (Figure 1A, IV-2, and Figure 2A, IV-2).
RNA was obtained from lymphocytes of affected individual and controls with the QIAGEN RNeasy Mini Kit and from kidney biopsy specimens as well as nephrectomized kidney tissues removed for unrelated causes with TRIzol reagent (Invitrogen) according to the manufacturer's recommendations. cDNA was generated by reverse transcription with the QIAGEN QuantiTect Reverse Transcription Kit. After cDNA synthesis, PTPRO transcripts were amplified with gene-specific primers (see Table S1), and then PCR products were sequenced.
Expression of variant 2 (NM_002848.2) was observed in both lymphocytes and kidney tissues. The results unequivocally showed the skipping of exon 16 in family A (Figures 1G–1I) and of exon 19 in family B (Figures 2F–2H) in both lymphocyte and kidney tissue cDNA in all the affected individuals.
In family A, the skipped exon resulted in a protein that lacked 23 amino acids (p.Glu854_Trp876del) (Figure 1J). These amino acids are not within a recognizable domain or motif of the protein; however, they are highly conserved in PTPRO orthologs in evolutionarily distant organisms such as Danio rerio and Fugu rubripes, suggesting a conserved function of the domain assembly within PTPRO (Figure 1J and Figure S3). In family B, the skipped exon introduced a premature stop codon at the very beginning of exon 20 (Figure 2G). This change could lead to a protein lacking the tyrosine protein phosphatase domain and the substrate-binding regions (Figure 2I) or nonsense-mediated decay of mRNA. Thus, we further analyzed allelic expression of wild-type and mutant PTPRO alleles in lymphocyte RNA samples from a heterozygous parent (Figure 2A, III-2), a healthy sibling (Figure 2A, IV-4), and an affected individual (Figure 2A, IV-2). Quantitative real-time PCR analysis showed a dramatic decrease in the mutant allele compared to the wild-type allele, suggesting that mRNA was degraded by nonsense-mediated decay (Figure 2J).
Because both families carrying PTPRO mutations were of Turkish origin, we excluded the presence of both mutations in 180 healthy Turkish controls by using an independent method—restriction fragment length polymorphism analysis (RFLP) (Figure 3). Using genomic DNA from family members, we assessed the presence of G→T donor splice-site mutations in intron 16 via amplification by PCR followed by BsrI digestion, and we assessed the presence of G→A donor splice-site mutations in intron 19 via amplification by PCR followed by HphI restriction digestion. We visualized 182 bp fragments for the mutated allele, and 116 and 66 bp fragments for the normal allele for BsrI digestion by using agarose gel electrophoresis. HphI digestion generated fragments of 294 bp for the mutated allele and 214, 66, and 14 bp for the normal allele; the expected fragments, except for 14 bp, were visualized. In addition, 15 individuals who were from the SRNS cohort from the Mario Negri Institute in Bergamo, Italy, and whose SNRS was unexplained were independently sequenced, and no mutations were identified.
Figure 3.
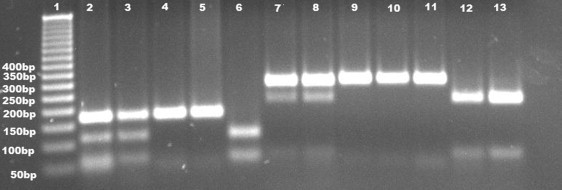
Restriction Fragment Length Polymorphism Analysis Visualized with Ethidium-Bromide Staining after Agarose Gel Electrophoresis
Lane 1 shows size marker with corresponding bands as base pairs. Lanes 2–5 show BsrI digestion in Family A with fragments of 182 bp for the mutated allele, and 116 and 66 bp, respectively, for the normal allele. Lanes 2 and 3 show heterozygous parents (III-1 and III-2, respectively, in Figure 1). Lanes 4 and 5 belong to affected siblings (IV-1 and IV2, respectively, in Figure 1A). Healthy control (as shown in lane 6) shows 2 bands (116 and 66 bp, respectively). Lanes 7-13 show HphI digestion in Family B with fragments of 294 bp for the mutated allele and 214 and 66 bp, respectively, for the normal allele. Lanes 7 and 8 depict heterozygous parents (III-1 and III-2, respectively, in Figure 2A) with three bands (294, 214, and 66 bp). Homozygous siblings (IV-1, IV-2, and IV-3 in Figure 2A), as shown in lanes 9, 10, and 11, give one band with 294 bp. Lanes 12 and 13 correspond to a healthy sibling (IV-4 in Figure 2A) and a healthy control, respectively, and are associated with two bands of 214 and 66 bp.
Kidney biopsies from each family were treated according to standard methods. For immunohistochemical analysis, antibody against PTPRO (anti-GLEPP1 5C11, BioGenex catalog number MU336-UC) at a dilution of 1:50 in antibody diluents buffer (ScyTek ABB125) was used. Specific immunoperoxidase staining was developed with the ScyTek UltraTek HRP Antipolyvalent Staining System (AMF080 horseradish peroxidase and goat 3,3′-diaminobenzidine kit, ScyTek) with 3,3′-diaminobenzidine (DAB) as the chromogen. DAB-stained slides were counterstained with hematoxylin. The kidney biopsy specimen of the second sibling in family A (Figure 1A, IV-2) showed two globally and three segmentally sclerosed glomeruli (among the 25 glomeruli retrieved) located in the juxtamedullary cortex (Figure 4A). Adhesion between Bowman capsules and capillary tufts was noted in another glomerulus. Cellular proliferation and matrix expansion were not observed, whereas mild focal tubulointerstitial fibrosis and atrophy was noted. Immunofluorescence analysis showed weak to moderate perimesangial and rare mesangial IgM but no other immunoglobulin or complement deposition. Immunohistochemical analysis with a monoclonal antibody against PTPRO showed diffuse and strong staining in the podocytes, and this staining was similar to that observed in control kidney tissue (Figure 4C). Electron microscopy showed diffuse foot process effacement and widespread attenuation of the glomerular basement membranes (<150 nm), which were thought to be constitutional because of the absence of hematuria. The outer and inner contours of the basement membrane were smooth and without signs of irregular thickening, lamellation, or splitting. Podocytes appeared swollen and vacuolated. Immune-type dense deposits were not present. Endothelial cells showed slight swelling and vacuolization (Figure 4E). The disease was diagnosed as focal segmental glomerulosclerosis. A kidney biopsy of the second sibling in family B (Figure 2A, IV-2) showed that of the 38 glomeruli, three were globally sclerosed; the remaining glomeruli were normal or showed slight mesangial prominence without hypercellularity or segmental sclerosis. The tubulointerstitial compartment was unremarkable except for narrow zones of interstitial fibrosis and tubular atrophy around the globally sclerosed glomeruli (Figure 4B). Immunofluorescence showed that there was not any immunoglobulin or complement deposition. In contrast to family A, immunohistochemical analysis in family B did not show immunostaining with monoclonal antibody against PTPRO in podocytes (Figure 4D). Electron microscopy showed diffuse foot process fusion and extensive microvillus transformation of the podocytes (Figure 4F). The pathological diagnosis was minimal change disease.
Figure 4.
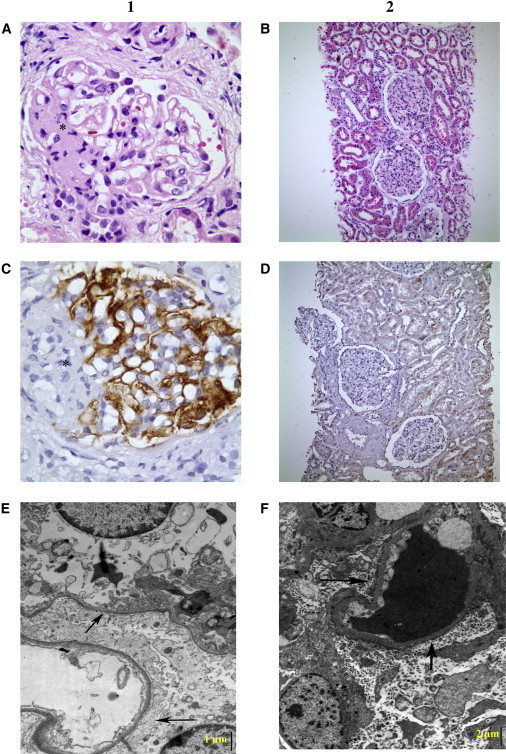
Histopathological Assessment of the Kidney Biopsies
(A and B) Light microscopy, (C and D) immunohistochemical, and (E and F) electron microscopy findings in biopsied affected individuals from families A and B are shown. The first and second columns are related to individual IV-2 in family A and individual IV-2 in family B, respectively. (A) One of the segmentally sclerosed glomeruli. The sclerosed segment is depicted by an asterisk (hematoxylin and eosin staining [H&E], 500×). (C) Podocytes showed strong staining with monoclonal antibody against PTPRO (immunohistochemistry, anti-GLEPP1 Ab, 500×). (E) Generalized foot process fusion (arrows) and widespread thinning of glomerular basement membranes, with smooth outer and inner contours (electron microscopy, lead citrate and uranyl acetate staining, 15000×). Bar indicates 1 μm. (B) Essentially normal glomeruli with open capillary lumina light microscopically (H&E, 100×). (D) Complete lack of PTPRO (immunohistochemistry, anti-GLEPP1 Ab, 100×). (F) Diffuse foot process effacement (arrows) and extensive microvillus transformation of podocytes (electron microscopy, lead citrate and uranyl acetate staining, 8000×). The scale bar indicates 2 μm.
PTPRO is a tyrosine phosphatase expressed at the apical membrane of the podocyte foot processes. Tyrosine phosphorylation of tight junction proteins plays a major role in controlling paracellular permeability, cell signaling, and actin cytoskeleton remodeling. Tyrosine phosphorylation of nephrin and ZO-1, key components of the slit diaphragm, alters their association with other slit pore proteins in the membrane.18,19 Abolished PTPRO expression has been observed in various proteinuric nephropathies, such as primary focal segmental glomerulosclerosis and severe IgA nephropathy, as well as in experimental podocyte damage leading to foot process effacement.20–22 In puromycin aminonucleoside (PAN) nephropathy, PTPro downregulation preceded the onset of proteinuria.22 The blockade of PTPro by specific antibodies increased albumin permeability in isolated rat and rabbit glomeruli, further supporting the essential role of this protein in maintaining glomerular permselectivity.15 In the present study, we observed that both mutations caused aberrant RNA splicing; in one case it led to a premature stop codon (p.Asn888Lysfs∗3) and degradation of the mRNA via nonsense-mediated decay and in another case to a protein lacking 23 amino acids (p.Glu854_Trp876del). Whereas nonsense-mediated decay of the mRNA resulting from the skipped exon 19 caused complete deficiency of the immunoreactive protein, the expression, and membrane location of the shortened protein resulting from skipped transcription of exon 16 appeared to be preserved. We think that the latter mutation might still be functionally relevant because the 23 missing amino acids constitute an evolutionarily highly preserved region of the protein.
In the present study, histopathological findings in one member of each family showed relatively mild lesions and focal segmental or global sclerosis of a small fraction of glomeruli. Ultrastructural analysis showed that the profound foot process effacement and altered phenotypic appearance of the podocytes resembled the anomalies observed in Ptpro−/− mice.14 Ptpro-deficient podocytes exhibit an amoeboid shape with blunted and widened foot processes rather than the typical octopoid shape, despite the normal glomerular structure observed with light microscopy.14 Although Ptpro−/− mice have shortened foot processes and a reduced total slit diaphragm length, they do not appear to develop major proteinuria. This apparent discrepancy with the increased albumin permeability induced by blocking PTPro activity in rat and rabbit glomeruli15 and with the nephrotic state observed in children with genetically altered PTPRO might be due to organism-specific differences in functional redundancy by other phosphatases.
Notably, four of the five affected children in the present study were partially responsive to intensified immunosuppressive treatment. In keeping with this observation, steroid treatment selectively attenuated downregulation of PTPro in the PAN model.22 Regulation of phosphatases is an important mechanism of glucocorticoid action in many biological systems;23 hence, PTPRO mutations may represent another hereditary disorder of podocyte function with partial susceptibility to immunosuppressive therapy, as previously observed in individuals with abnormalities in phospholipase C epsilon-1, another protein involved in podocyte cell signaling.8
The oldest sibling in family B (Figure 2A, IV-1) had a more severe disease course than the other affected siblings and had complete resistance to intensified immunosuppressive therapy and progressed to ESKD. Of note, the p.Arg229Gln polymorphism in the gene encoding podocin—the most commonly mutated protein in childhood-onset SRNS—was observed in this individual but not in the other siblings. p.Arg229Gln, which is the most frequent podocin variant and which occurs in 3%–13% of Europeans, is considered a nonneutral polymorphism, because the mutant protein exhibits decreased nephrin binding in vitro.24 Late-onset SRNS has been described in individuals compound heterozygous for the p.Arg229Gln variant together with a pathogenic NPHS2 mutation.25 An interesting possibility is that heterozygosity for podocin p.Arg229Gln modified the onset and course of the disease in family B.
In conclusion, we identified recessive mutations in PTPRO as another cause of childhood nephrotic syndrome with reduced steroid responsiveness and therefore propose use of the denomination NPHS6 as a synonym. While expanding the spectrum of hereditary nephrotic syndromes, PTPRO will also provide a better understanding of the pathogenetic mechanisms underlying proteinuric diseases and the clinical management of nephrotic children.
Acknowledgments
We thank the families for their participation in the study. The Department of Pediatrics, Nephrogenetics Laboratory, Hacettepe University Faculty of Medicine was established by the Hacettepe University Infrastructure Project (grant number 06A 101 008). This study was supported by the Scientific and Technological Research Council of Turkey (TÜBİTAK) (grant number 108S417), and the consortium (PodoNet) was supported by the European Research Area Network (E-RARE). P. Iatropoulos was partially supported by the PodoNet grant from the Istituto Superiore di Sanità (E-RARE 11). Human Genome Organization approval of the name NPHS6 was obtained. Members of the PodoNet consortium (in alphabetical order by institution): Hacettepe University Faculty of Medicine, Ankara, Turkey: F. Ozaltin and A.Bakkaloglu; Mario Negri Institute for Pharmacological Research, Bergamo, Italy: M. Noris; Instituto Giannina Gaslini, Genova, Italy: G.M. Ghiggeri; University of Heidelberg, Heidelberg, Germany: F. Schaefer; Hôpital Necker, Paris, France: C. Antignac; Ospedale Pediatrico Bambino Gesù, Rome, Italy: F. Emma.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Automated splice-site analysis, https://splice.uwo.ca
Ensembl, http://www.ensembl.org/Homo_sapiens/Gene/Summary?g=ENSG00000151490;r=12:15475451-15750335
National Center for Biotechnology Information (NCBI), http://www.ncbi.nlm.nih.gov
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser, February 2009 (GRCh37/hg19) freeze, http://genome.ucsc.edu
References
- 1.International Study on Kidney Disease in Children Nephrotic syndrome in children: prediction of histopathology from clinical and laboratory characteristics at time of diagnosis. A report of the International Study of Kidney Disease in Children. Kidney Int. 1978;13:159–165. doi: 10.1038/ki.1978.23. [DOI] [PubMed] [Google Scholar]
- 2.International Study on Kidney Disease in Children The primary nephrotic syndrome in children. Identification of patients with minimal change nephrotic syndrome from initial response to prednisone. A report of the International Study of Kidney Disease in Children. J. Pediatr. 1981;98:561–564. doi: 10.1016/s0022-3476(81)80760-3. [DOI] [PubMed] [Google Scholar]
- 3.Kestilä M., Lenkkeri U., Männikkö M., Lamerdin J., McCready P., Putaala H., Ruotsalainen V., Morita T., Nissinen M., Herva R. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell. 1998;1:575–582. doi: 10.1016/s1097-2765(00)80057-x. [DOI] [PubMed] [Google Scholar]
- 4.Boute N., Gribouval O., Roselli S., Benessy F., Lee H., Fuchshuber A., Dahan K., Gubler M.C., Niaudet P., Antignac C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000;24:349–354. doi: 10.1038/74166. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan J.M., Kim S.H., North K.N., Rennke H., Correia L.A., Tong H.Q., Mathis B.J., Rodríguez-Pérez J.C., Allen P.G., Beggs A.H., Pollak M.R. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000;24:251–256. doi: 10.1038/73456. [DOI] [PubMed] [Google Scholar]
- 6.Kim J.M., Wu H., Green G., Winkler C.A., Kopp J.B., Miner J.H., Unanue E.R., Shaw A.S. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science. 2003;300:1298–1300. doi: 10.1126/science.1081068. [DOI] [PubMed] [Google Scholar]
- 7.Winn M.P., Conlon P.J., Lynn K.L., Farrington M.K., Creazzo T., Hawkins A.F., Daskalakis N., Kwan S.Y., Ebersviller S., Burchette J.L. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- 8.Hinkes B., Wiggins R.C., Gbadegesin R., Vlangos C.N., Seelow D., Nürnberg G., Garg P., Verma R., Chaib H., Hoskins B.E. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006;38:1397–1405. doi: 10.1038/ng1918. [DOI] [PubMed] [Google Scholar]
- 9.Brown E.J., Schlöndorff J.S., Becker D.J., Tsukaguchi H., Tonna S.J., Uscinski A.L., Higgs H.N., Henderson J.M., Pollak M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010;42:72–76. doi: 10.1038/ng.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zenker M., Aigner T., Wendler O., Tralau T., Müntefering H., Fenski R., Pitz S., Schumacher V., Royer-Pokora B., Wühl E. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004;13:2625–2632. doi: 10.1093/hmg/ddh284. [DOI] [PubMed] [Google Scholar]
- 11.Mucha B., Ozaltin F., Hinkes B.G., Hasselbacher K., Ruf R.G., Schultheiss M., Hangan D., Hoskins B.E., Everding A.S., Bogdanovic R., Members of the APN Study Group Mutations in the Wilms' tumor 1 gene cause isolated steroid resistant nephrotic syndrome and occur in exons 8 and 9. Pediatr. Res. 2006;59:325–331. doi: 10.1203/01.pdr.0000196717.94518.f0. [DOI] [PubMed] [Google Scholar]
- 12.Arbeitsgemeinschaft für Pädiatrische Nephrologie Short versus standard prednisone therapy for initial treatment of idiopathic nephrotic syndrome in children. Lancet. 1988;1:380–383. [PubMed] [Google Scholar]
- 13.Uz E., Alanay Y., Aktas D., Vargel I., Gucer S., Tuncbilek G., von Eggeling F., Yilmaz E., Deren O., Posorski N. Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am. J. Hum. Genet. 2010;86:789–796. doi: 10.1016/j.ajhg.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wharram B.L., Goyal M., Gillespie P.J., Wiggins J.E., Kershaw D.B., Holzman L.B., Dysko R.C., Saunders T.L., Samuelson L.C., Wiggins R.C. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Invest. 2000;106:1281–1290. doi: 10.1172/JCI7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Charba D.S., Wiggins R.C., Goyal M., Wharram B.L., Wiggins J.E., McCarthy E.T., Sharma R., Sharma M., Savin V.J. Antibodies to protein tyrosine phosphatase receptor type O (PTPro) increase glomerular albumin permeability (P(alb)) Am. J. Physiol. Renal Physiol. 2009;297:F138–F144. doi: 10.1152/ajprenal.00122.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogan P.K., Faux B.M., Schneider T.D. Information analysis of human splice site mutations. Hum. Mutat. 1998;12:153–171. doi: 10.1002/(SICI)1098-1004(1998)12:3<153::AID-HUMU3>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 17.Nalla V.K., Rogan P.K. Automated splicing mutation analysis by information theory. Hum. Mutat. 2005;25:334–342. doi: 10.1002/humu.20151. [DOI] [PubMed] [Google Scholar]
- 18.Kurihara H., Anderson J.M., Farquhar M.G. Increased Tyr phosphorylation of ZO-1 during modification of tight junctions between glomerular foot processes. Am. J. Physiol. 1995;268:F514–F524. doi: 10.1152/ajprenal.1995.268.3.F514. [DOI] [PubMed] [Google Scholar]
- 19.Verma R., Kovari I., Soofi A., Nihalani D., Patrie K., Holzman L.B. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Invest. 2006;116:1346–1359. doi: 10.1172/JCI27414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sharif K., Goyal M., Kershaw D., Kunkel R., Wiggins R. Podocyte phenotypes as defined by expression and distribution of GLEPP1 in the developing glomerulus and in nephrotic glomeruli from MCD, CNF, and FSGS. A dedifferentiation hypothesis for the nephrotic syndrome. Exp. Nephrol. 1998;6:234–244. doi: 10.1159/000020528. [DOI] [PubMed] [Google Scholar]
- 21.Tian J., Wang H.P., Mao Y.Y., Jin J., Chen J.H. Reduced glomerular epithelial protein 1 expression and podocyte injury in immunoglobulin A nephropathy. J. Int. Med. Res. 2007;35:338–345. doi: 10.1177/147323000703500308. [DOI] [PubMed] [Google Scholar]
- 22.Clement L.C., Liu G., Perez-Torres I., Kanwar Y.S., Avila-Casado C., Chugh S.S. Early changes in gene expression that influence the course of primary glomerular disease. Kidney Int. 2007;72:337–347. doi: 10.1038/sj.ki.5002302. [DOI] [PubMed] [Google Scholar]
- 23.Beck I.M., Vanden Berghe W., Vermeulen L., Yamamoto K.R., Haegeman G., De Bosscher K. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr. Rev. 2009;30:830–882. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukaguchi H., Sudhakar A., Le T.C., Nguyen T., Yao J., Schwimmer J.A., Schachter A.D., Poch E., Abreu P.F., Appel G.B. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J. Clin. Invest. 2002;110:1659–1666. doi: 10.1172/JCI16242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machuca E., Hummel A., Nevo F., Dantal J., Martinez F., Al-Sabban E., Baudouin V., Abel L., Grünfeld J.P., Antignac C. Clinical and epidemiological assessment of steroid-resistant nephrotic syndrome associated with the NPHS2 R229Q variant. Kidney Int. 2009;75:727–735. doi: 10.1038/ki.2008.650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.