

Abstract
Organocatalytic domino oxa-Michael/aldol reactions between salicylaldehyde with electron deficient olefins are presented. We screened guanidine, 1,1,3,3-tetramethylguanidine (TMG) and L-pipecolinic acid as organocatalysts for this transformation. 3-Substituted 2-phenyl-2H-chromene derivatives are synthesized with high yields and with poor enantioselectivity (5–17% ee) using L-pipecolinic acid while TMG works well with cinnamaldehyde without using co-catalyst. These 3-substituted-2-phenyl-2H-chromene derivatives are further derivatized to synthesize triazole and biotin-containing chromene derivatives, to facilitate purification of protein targets.
For our ongoing chemical biology projects we are interested in developing new compounds that interact with developmentally important receptor-mediated pathways such as the TGF-β pathway, acting as antagonist or agonist.1,2 For this purpose, we described a chemical genetic approach by testing a library of 2-substituted-2H-chromene derivatives for their bioactivity on developing zebrafish embryos. The zebrafish embryo provides an ideal vertebrate model system for in vivo small molecule screens because of its optical transparency, accessibility during embryonic development, and permeability to small molecules. We synthesized a small library of 2-substituted-2H-chromene derivatives that were screened for bioactivity using the zebrafish model.3 Compound BT7 (1) (Fig. 1) was found to modulate a specific relevant pathway, namely p-SAPK/JNK, which is known to be downstream of TGF-β, and can mediate Smad-independent signaling.3
Figure 1.

Our next aim is to isolate BT7-interacting protein(s) by attaching a biotin moiety, which would facilitate purification of the protein target.
For this purpose, we sought to develop function-oriented BT7 analogues and, finally, to attach biotin using functional group transformations. Domino or cascade reactions that involve the formation of multiple stereo centers comprise one-pot a rapidly growing research field with respect to the synthesis of small molecules with complex architectures.4 However, the development of catalytic diastero- and enantioselective domino reactions is still a challenging task.5a–c In this context, the development of organocatalytic asymmetric domino reactions has been pursued. Nitrochromenes are versatile synthetic intermediates in organic synthesis owing to the various possible transformations of the nitro group into other useful functional groups. Useful synthetic methods reported for the preparation of 3-nitro- and 3-formyl-2-phenyl-2H-chromene are the domino Michael/aldol reactions of salicylaldehyde with β-nitrostyrene and cinnamaldehyde.5d–g Basically this involves L-proline and L-proline-based catalysts, along with a co-catalyst used for the above-mentioned purpose. However, this gives moderate to good yields and poor enantioselectivity. 6 Based on the development of these reactions and our research interest of finding catalytic domino reactions that give our function-oriented BT7 analogues (2, 3, and 4), we envisioned a reaction between differently substituted salicylaldehyde with β-nitrostyrene and cinnamaldehyde using simple catalyst like guanidine, 1,1,3,3-tetramethylguanidine (TMG)7 and pipecolinic acid. We choose these catalysts to improve selectivity, yield, reaction time, and to use an alternative protocol in which the absence of co-catalyst can provide a better yield.8
In an initial catalyst screen for the reaction between salicylaldehyde 5a and β-nitrostyrene 6, we found to our delight that amines 7, 8, and 9 were catalysts for the domino Michael/aldol reaction. In further experiments, other factors that influence the reaction were thoroughly investigated such as solvent, catalyst loading, and reaction temperature. In the beginning of our study β-nitrostyrene 6 and salicylaldehyde 5a were taken as the model substrates. The results are listed in Table 1. Catalyst 9 gave 81% yield of 2a at 80 °C in toluene with poor enantioselectivity (5% ee).
Table 1.
Catalyst screen for the amine-catalyzed enantioselective domino reactions between 5a and 6
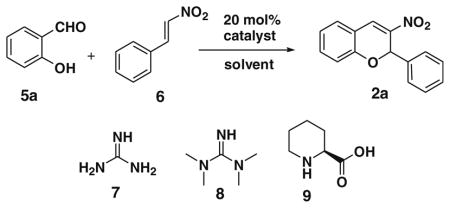 | |||
|---|---|---|---|
| Entry | Catalyst | Reaction condition | Yielda,b (%) |
| 1 | 7 | Toluene, rt, 5 d | 11 |
| 2 | 7 | Toluene, 80 °C, 48 h | 68 |
| 3 | 8 | Toluene, rt, 5 d | 30 |
| 4 | 8 | Toluene, 80 °C, 48 h | 75 |
| 5 | 9 | Toluene, 80°C, 24 h | 81 (5%, ee)b |
Reaction with 5 (1 mmol), 6 (1.2 mmol) in 2 ml toluene, isolated yield after column purified.
Determined by chiral-HPLC analyses using chiralpak AD.
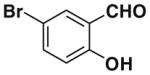
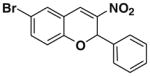
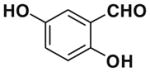
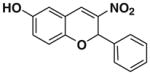
After successfully optimizing the catalyst 9, solvent (toluene), temperature (80 °C) and reaction time (24 h), we further explored to generalize this reaction. For this purpose we used substituted salicylaldehyde as substrates. Table 2 shows a number of examples of this chemistry. Significant variation in the electronic and steric features of salicylaldehydes is tolerated for the L-pipecolinic acid 9 catalyzed cascade process. The reaction between 3,5-dichlorosalicylaldehyde 5b with β-nitrostyrene 6 in the presence of 20 mol % of L-pipecolinic acid 9 furnished our target BT7 nitro chromene derivative 2b in 76% yield with poor enantioselectivity ee 2% (Table 2, entry 2). We then screened the 5-substituted salicylaldehydes, 5-chloro (5c), 5-bromo (5d) and β-nitrostyrene 6 under optimized conditions, which gave the corresponding chromene derivatives 9c and 9d in 79%, and 80% yields with poor enantioselectivity 17% and 18% ee, respectively (Table 2, entries 3 and 4). 5-hydroxy salicylaldehyde 5e gave the corresponding nitrochromene derivative in moderate yield 60% with 13% ee (Table 2, entry 5).
Table 2.
Scope of the domino Michael/aldol reaction between various salicylaldehydes with Eβ-
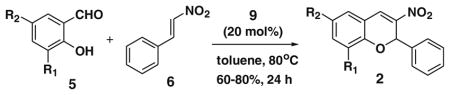
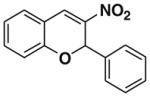
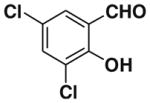
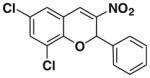
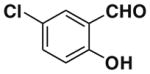
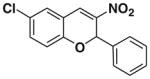
Reaction with 5 (1 mmol), 6 (1.2 mmol) and catalyst (20 mol %) in toluene.
Isolated yield of pure copmpound.
Determined by chiral-HPLC analyses using chiralpak AD.
The catalytic activity of 9 was further examined with the 2-amino-benzaldehyde 10 and β-nitrostyrene 6 under the same reaction conditions and gave the 3-nitro-2-phenyl-1,2-dihydro-quinoline10 11 in 85% yield and 26% ee (Scheme 1).
Scheme 1.

Having succeeded in synthesizing the BT7 nitrochromene derivative 2b, our attention was next focused on the attachment of biotin. Yao and co-workers reported the [3+2] cycloaddition of 3-nitro-2-phenyl-2H-chromene with sodium azide under catalyst free conditions at 80 °C in DMSO.11 Following this procedure we synthesized the BT7 triazole 12 by using 6,8-dichloro-3-nitro-2-phenyl-2H-chromene 2b and sodium azide at 80 °C in DMSO in 82% yield. We next examined the feasibility of attaching biotin to the BT7 triazole analogue 12 via triazole moiety using DCC under various reaction conditions, however, we did not observe the expected product formation (Scheme 2).
Scheme 2.

As we failed to make amide linkage of d-biotin with 12, we then decided instead of acid–amine coupling, alkyne-azide click chemistry could be implemented. Keeping this idea in mind we planned to synthesize 14. According to Scheme 3 and 14 could be synthesized from Michael adduct 3.
Scheme 3.

To synthesize compound 3, we screened the catalyst, 7, 8, 9, and L-proline with 3,5-dichlorosalicylaldehyde 5b and cinnamaldehyde 13 as substrates. To our surprise, the reaction proceeded only with 8 and gave 3 in 78% yield.12 After 72 h with L-proline only 10% conversion was observed. Reaction of 5b with 13 in the presence of L-pipecolinic acid and 7 was carried out but this failed to proceed at all even after 72 h. It is very interesting to note that in the case of β-nitrostyrene, all four catalysts gave 3-nitro-substituted chromene derivatives in moderate to good yields, but in the case of cinnamaldehyde, only catalyst 8 (TMG) gave 3-formyl substituted chromene derivatives. The detailed mechanism of this transformation is currently being investigated in our laboratory. Aldehyde 3 was further derivatized to 14. Compound 3 was treated with triphenylphosphine, CBr4 in Zn, resulting in dibromo derivative, which was further treated with n-BuLi in −78 °C to furnish alkyne 14.13 Before doing click chemistry with d-biotin azide, we initially attempted a model reaction, using simple benzyl azide in the presence of CuI and DIPEA, as base and successfully achieved the triazole 15 (Scheme 3).14 After obtaining the triazole derivative we also planned to synthesize ester and amide linkage of d-biotin. Thus, the aldehyde 3 was reduced by sodium borohydride in methanol, which produced the corresponding alcohol 16 in 92% yield.15
We then esterified this alcoholic derivative of BT7 16 with d-biotin using DCC, DMAP in dichloromethane and produced the d-biotin-attached BT7 analogue 17 in 74% yield (Scheme 4).15 After synthesizing 17, we focused to synthesize d-biotin-attached amide (Scheme 5).16 It is interesting to note that we tried with many reducing agents (but not by enzymatic process) to reduce only nitro group to amine keeping the vinylic double bond intact in the compound 2b but failed. However, the nitro compound 2b was reduced with BH3/THF to form the amine 18 which was isolated by crystallization using ether–hexane system in 82% yield. We observed both double bond and nitro group were reduced.8e The amine 18 was coupled with biotin using EDCI and DMAP in DMF to give BT7 biotin amide 19, which was purified by crystallization using ether and hexanes in 78% yield.16
Scheme 4.

Scheme 5.

After synthesizing of BT7 nitro 2b and aldehyde 3, we next turned our attention to functionalize BT7-carbonitrile 4. For this purpose the reaction between 5b and phenyl-acrylonitrile was executed with catalysts 7, 8, 9, and L-proline, to our surprise in all cases we failed to achieve the synthesis of compound 4. From this observation, it might be possible that, Michael acceptors playing crucial role in this domino oxa-Michael/aldol reaction, because in the case of β-nitrostyrene, all four catalysts worked well, for cinnamaldehyde only catalyst 8 worked, for acrylonitrile, L-proline worked (data not shown), but in the case of cinnamonitrile none of the four catalysts worked.
In summary, we report an organocatalytic domino oxa-Michael/aldol reaction that gives chromenes in high yields without using co-catalyst and in less reaction time. Hence, the reaction constitutes a simple catalytic high yield entry to pharmaceutically valuable 2H-chromene and 1,2-dihydro-quinoline derivatives. We successfully attached the biotin moiety to our lead compound BT7. For the first time, we observed the catalytic activity of L-pipecolinic acid to synthesize 3-substituted 2-phenyl chromene derivatives with poor enantioselectivities (5–117% ee). We are in the process of developing chiral L-pipecolinic acid-based and TMG-based catalysts to improve activity, improve stereoselectivity in various organic transformations and other enantioselective domino reactions. The detailed biological study of these compounds as possible TGF-β pathway modulators is ongoing in our laboratory.
Supplementary Material
Acknowledgments
The author B.C.D. is thankful to AECOM for start up funding. The instrumentation in the AECOM Structural NMR Resource is supported by the Albert Einstein College of Medicine and in part by Grants from the NSF (DBI9601607 and DBI0331934), the NIH (RR017998) and the HHMI Research Resources for Biomedical Sciences. We gratefully acknowledge the CRO laboratories inc. Dallas, TX, USA for providing HPLC data. Authors are thankful to reviewer for valuable suggestion.
Footnotes
Supplementary data (copies of 1H, 13C NMR and mass spectra) associated with this article can be found, in the online version, at doi:10.1016/j.tetlet.2010.02.143.
References and notes
- 1.MacRae CA, Peterson RT. Chem Biol. 2003;10:901–908. doi: 10.1016/j.chembiol.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 2.Peterson RT, Link BA, Dowling JE, Schreiber SL. Proc Natl Acad Sci USA. 2000;97:12965–12969. doi: 10.1073/pnas.97.24.12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torregroza I, Evans T, Das BC. Chem Biol Drug Des. 2009;73:339–345. doi: 10.1111/j.1747-0285.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Tietze LF, Brasche G, Gericke K. Domino Reactions in Organic Synthesis. Wiley-VCH; Weinheim: 2006. p. 672. [Google Scholar]; (b) Tietze LF. Chem Rev. 1996;96:115. doi: 10.1021/cr950027e. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Edmonds DJ, Bulger PG. Angew Chem, Int Ed. 2006;45:7134. doi: 10.1002/anie.200601872. [DOI] [PubMed] [Google Scholar]; (d) Guo H, Ma J. Angew Chem, Int Ed. 2006;45:354. doi: 10.1002/anie.200500195. [DOI] [PubMed] [Google Scholar]; (e) Pellieser H. Tetrahedron. 2006;62:2143. [Google Scholar]; (f) Ramon DJ, Yus M. Angew Chem, Int Ed. 2005;44:1602. doi: 10.1002/anie.200460548. [DOI] [PubMed] [Google Scholar]; (g) Enders D, Grondal C, Hüttl MRM. Angew Chem, Int Ed. 2007;46:1570–1581. doi: 10.1002/anie.200603129. [DOI] [PubMed] [Google Scholar]
- 5.(a) Rios R, Sundén H, Vesely J, Zhao GL, Dziedzic P, Córdova A. Adv Synth Catal. 2007;349:1028. [Google Scholar]; (b) Penon O, Carlone A, Mazzanati A, Locatelli M, Sambri L, Bartoli G, Melchiorre P. Chem Eur J. 2008;14:4788. doi: 10.1002/chem.200800440. [DOI] [PubMed] [Google Scholar]; (c) Enders D, Wang C, Bats DW. Angew Chem, Int Ed. 2008;47:3579. doi: 10.1002/anie.200802532. [DOI] [PubMed] [Google Scholar]; (d) Luo SP, Li ZB, Wang LP, Guo Y, Xia AB, Xu DQ. Org Biomol Chem. 2009;7:4539–4546. doi: 10.1039/b910835a. [DOI] [PubMed] [Google Scholar]; (e) Wang W, Li H, Wang J, Zu L. J Am Chem Soc. 2006;128:10354–10355. doi: 10.1021/ja063328m. [DOI] [PubMed] [Google Scholar]; (f) Li H, Wang J, E-Nunu T, Zu L, Jiang W, Wei S, Wang W. Chem Commun. 2007:507–509. doi: 10.1039/b611502k. [DOI] [PubMed] [Google Scholar]; (g) Sunden H, Rios R, Ibrahem I, Zhao GL, Eriksson L, Córdova A. Adv Synth Catal. 2007;349:827–832. [Google Scholar]; (h) Sundén H, Ibrahem I, Zhao GL, Eriksson L, Córdova A. Chem Eur J. 2007;13:574. doi: 10.1002/chem.200600572. [DOI] [PubMed] [Google Scholar]; (i) Ibrahem I, Sundén H, Rios R, Zhao GL, Córdova A. Chimia. 2007;61:219. [Google Scholar]
- 6.(a) Xu DQ, Wang YF, Luo SP, Zhang S, Zhong AG, Chen H, Xu ZY. Adv Synth Catal. 2008;350:2610–2616. [Google Scholar]; (b) Karthikeyan T, Sankararaman S. Tetrahedron: Asymmetry. 2008;19:2741–2745. [Google Scholar]
- 7.(a) Yanqing P, Gonghua S, Feifei H. Monatsh Chem. 2005;136:727–731. [Google Scholar]; (b) Nirmal KP, Hitoshi U, Masahiro T. Tetrahedron Lett. 2007;48:8700–8703. [Google Scholar]; (c) Junjie H, Yanfen X, Yingpeng S, Xuegong S, Xinfu P. Catal Commun. 2008;9:2077–2079. [Google Scholar]
- 8.(a) Carrie EA, Melissa MV, Scott JM. Org Lett. 2005;7:3849–3851. [Google Scholar]; (b) Zhouyu W, Xiaoxia Y, Siyu W, Pengcheng W, Anjiang Z, Jian S. Org Lett. 2006;8:999–1001. [Google Scholar]; (c) Fatome M, Andrieu L, Laval JD, Royer RL. Eur J Med Chem. 1976;11:81–82. [Google Scholar]; (d) Rene L, Royer R. Eur J Med Chem. 1975;10:72–78. [Google Scholar]; (e) Varma RS, Kadkhodayan M, Kabalka GW. Heterocycles. 1986;24:1647–1652. [Google Scholar]
- 9.General procedure for synthesis 3-nitro-2-phenyl-2H-chromenes: A mixture of salicylaldehyde (1 mmol) and β-nitrostyrene (1.2 mmol) in dry toluene under nitrogen atmosphere was added 20 mol % L-pipecolinic acid at room temperature. The reaction mixture was stirred at 80 °C under nitrogen atmosphere for 24 h. The reaction was quenched with saturated NH4Cl and extracted with ethyl acetate (3 × 10 mL). The extracts were washed with H2O and brine, then dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexanes/ethyl acetate (8:1) as the eluent to give 3-nitro-2-phenyl-2H-chromenes. The enantiomeric excess was determined by HPLC with chiralpak AD column.
- 10.(a) Ballini R, Bosica G, Fiorini D, Palmieri A. Green Chem. 2005;7:825–827. [Google Scholar]; (b) Yan MC, Tu Z, Lin C, Ko S, Hsu J, Yao CF. J Org Chem. 2004;69:1565–1570. doi: 10.1021/jo030070z. [DOI] [PubMed] [Google Scholar]
- 11.Pateliya MH, Rama Raju B, Kavala V, Kuo C-W, Yao C-F. Tetrahedron. 2009;65:5799–5804. [Google Scholar]
- 12.Synthesis of 6,8-dichloro-2-phenyl-2H-chromene-3-carbaldehyde (3): A mixture of 3,5-dichloro salicylaldehyde (1 mmol) and cinnamaldehyde (1.2 mmol) in dry toluene under nitrogen atmosphere was added 20 mol % 1,1,3,3-tetramethylguanidine at room temperature. The reaction mixture was stirred at 80 °C for 48 h under nitrogen atmosphere till the complete disappearance of starting material monitored by TLC. The reaction was quenched with saturated NH4Cl and extracted with ethyl acetate (3 × 10 mL). The extracts were washed with H2O (2 × 10 mL) and brine, then dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexanes/ethyl acetate (8:1) as the eluent to give 6,8-dichloro-2-phenyl-2H-chromene-3-carbaldehyde 3. 78% yield, 1H NMR (300 MHz, CDCl3): d 9.75 (s, 1H), 7.35–7.39 (m, 2H), 7.41–7.28 (m, 5H), 7.17–7.18 (d, 1H, J = 3 Hz), 6.49 (s, 1H); 13C NMR (75 MHz, CDCl3): d 189.9, 150.0, 139.4, 138.2, 136.1, 133.3, 129.4, 129.1, 127.4, 126.8, 124.0, 122.3, 75.1.
- 13.Alami M, Peyrat JF, Belachmi L, Brion JD. Eur J Org Chem. 2001:4207–4212. [Google Scholar]
- 14.Hotha S, Kashyap S. J Org Chem. 2006;71:364–367. doi: 10.1021/jo051731q. [DOI] [PubMed] [Google Scholar]
- 15.Synthesis of biotin attached BT7 analogue 17: To a solution of 6, 8-dichloro-2-phenyl-2H-chromene-3-carbaldehyde 3 (1 mmol) in methanol was added sodium borohydride (2 mmol). The mixture was then stirred at room temperature for 2 h and the reaction completion was indicated by TLC. After evaporation of the solvent, water was added and the product was extracted using ethyl acetate (3 × 10 mL). The extracts were washed with H2O (2 × 10 mL) and brine, then dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography using hexanes/ethyl acetate (8:2) as the eluent to give the alcohol 16 as an oil. Compound 16 was directly used for next step. To a stirred solution of d-biotin in DCM was added DCC under nitrogen atmosphere and stirred for 2 h at room temperature. To this stirred solution was added alcohol 16 in DCM and stirring continued for over night at room temperature under nitrogen atmosphere. After evaporation of the solvent, the product 17 was purified by silica gel chromatography (hexanes/ethyl acetate). 74% yield, 1H NMR (300 MHz, CDCl3): δ 7.95 (s, 1H), 7.35–7.38 (m, 5H), 7.31–7.34 (m, 1H), 6.82 (s, 1H), 6.43 (s, 1H), 6.37 (s, 1H), 6.10 (s, 1H), 4.59 (s, 2H), 4.25–4.32 (m, 1H), 4.12–4.18 (m, 1H), 3.04–3.13 (m, 1H), 2.78–2.87 (m, 1H), 2.56–2.60 (d, 1H, J = 12 Hz), 2.18–2.25 (t, 2H, J = 6 Hz), 1.43–1.48 (m, 4H), 1.27–1.29 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 173.2, 163.5, 163.2, 147.0, 138.4, 134.1, 130.0, 129.6, 128.3, 126.0, 124.8, 121.6, 120.9, 78.2, 63.8, 56.2, 48.3, 36.6, 33.8, 31.6, 28.8, 25.3, 25.0. ESI MS: [M+H] 533.1055, calcd 533.0990 for C26H26Cl2N2O4S.
- 16.Synthesis of biotin attached BT7 analogue 19: To a stirred solution of d-biotin (1.1 mmol) in DMF was added EDCI (3 mmol), DMAP (3 mmol) and compound 18 (1 mmol) under nitrogen atmosphere and stirred for overnight at room temperature. Reaction mixture was diluted with water and extracted with ethyl acetate (3 × 10 mL) and dried over Na2SO4. After evaporation of the solvent, the product was crystallized over ether–hexanes system resulted in compound 19 in 78% yield.1H NMR (300 MHz, CDCl3): δ 7.85 (m, 1H), 7.71–7.21 (m, 7H), 6.61 (br s, 1H), 6.35 (s, 2H), 5.41 (s, 1H), 4.71 (m, 1H), 4.35 (m, 1H), 4.21 (m, 1H), 3.32 (m, 2H), 2.92–2.51 (m, 2H), 2.22–1.91 (m, 2H), 0.9–1.71 (m, 6H); 13CNMR (75 MHz, CDCl3): 173.0, 163.6, 149.8, 138.8, 129.4, 128.5, 127.9, 127.0, 125.0, 124.7, 122.4, 79.1, 61.9, 60.0, 56.3, 45.0, 39.5, 35.5, 32.5, 28.8, 26.1 ESI MS:[M+H]+: 520.1151, calcd 519.1150 for C25H27Cl2N3O3S.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.