

Abstract
Cell–matrix adhesion depends on the collective behaviours of clusters of receptor–ligand bonds called focal contacts between cell and extracellular matrix. While the behaviour of a single molecular bond is governed by statistical mechanics at the molecular scale, continuum mechanics should be valid at a larger scale. This paper presents an overview of a series of recent theoretical studies aimed at probing the basic mechanical principles of focal contacts in cell–matrix adhesion via stochastic–elastic models in which stochastic descriptions of molecular bonds and elastic descriptions of interfacial traction–separation are unified in a single modelling framework. The intention here is to illustrate these principles using simple analytical and numerical models. The aim of the discussions is to provide possible clues to the following questions: why does the size of focal adhesions (FAs) fall into a narrow range around the micrometre scale? How can cells sense and respond to substrates of varied stiffness via FAs? How do the magnitude and orientation of mechanical forces affect the binding dynamics of FAs? The effects of cluster size, cell–matrix elastic modulus, loading direction and cytoskeletal pretension on the lifetime of FA clusters have been investigated by theoretical arguments as well as Monte Carlo numerical simulations, with results showing that intermediate adhesion size, stiff substrate, cytoskeleton stiffening, low-angle pulling and moderate cytoskeletal pretension are factors that contribute to stable FAs. From a mechanistic point of view, these results provide possible explanations for a wide range of experimental observations and suggest multiple mechanisms by which cells can actively control adhesion and de-adhesion via cytoskeletal contractile machinery in response to mechanical properties of their surroundings.
Keywords: cell adhesion, focal adhesion, receptor–ligand bond, adhesion lifetime, size effect, stiffness effect
1. Introduction
There have been growing research activities towards the understanding and control of cell adhesion in the past decades. The feature that distinguishes cell adhesion from conventional adhesive contact in engineering systems is the specific binding between cell surface proteins (receptors) and complementary molecules (ligands) on other cells or extracellular matrix (ECM). The specific receptor–ligand recognition is often considered a lock-and-key mechanism, while the various non-specific interactions including electrostatic and van der Waals forces are usually negligible owing to a layer of polymer brush surrounding the cell surface called glycocalyx [1]. There are literally hundreds of receptors that participate in cell adhesion, which are grouped into several main families: integrin, immunoglobulin, selectin and cadherin. Each of these families is accompanied by an equally diverse family of ligands [2,3]. This biological complexity is far beyond the scope of this paper and has been thoroughly reviewed elsewhere [2–6]. Receptor–ligand bonds function as transmembrane linkers between the cytoskeleton and ECM, where both mechanical cues and regulatory signals are transmitted in both directions across the plasma membrane for a wide range of cellular processes such as cell spreading, migration, proliferation and differentiation [7].
The aim of this paper is to outline some of the recent studies exploring the mechanical principles that may be possibly involved in cell–matrix adhesion. Only some of the key ideas and results are highlighted in the discussions given here. The reader is encouraged to consult various references given in the paper for more details.
2. Focal contacts in cell–matrix adhesion
The adhesion between cell and ECM is often localized to discrete contact regions called focal adhesions (FAs) [8], as illustrated in figure 1a. FAs usually evolve from small dot-like adhesions, commonly referred to as focal complexes (FXs), which are continuously formed and turned over under the protruding lamellipodia [9]. Mature and stable FAs depend on the clustering of molecular bonds, creating an adhesion plaque of complex macromolecular assemblies in which many cytoskeletal filaments are anchored (figure 1b). The recruitment of actin filaments and integrin receptors to the contact regime is essential to FAs, and artificially mutated integrins that lack an ability to connect with cytoskeletal filaments often fail to cluster and are unable to form stable adhesions [1]. Experimentally, the adhesion clusters between cell and matrix were found to have a characteristic length scale in the order of a few micrometres [10,11].
Figure 1.

A schematic of focal contacts in cell–ECM adhesion based on specific binding between receptors and complementary ligands. (a) Cell–ECM adhesion localized to discrete focal contacts. (b) Actin bundles anchored into an adhesion plaque that connects ECM through transmembrane molecular bonds. Focal adhesions can be exposed to cytoskeletally generated contractile forces in actin bundles, as well as externally applied loads outside of the cell.
The elastic properties of cytoskeleton and ECM play an essential role in the maintenance and development of FAs. One striking example is that stem cells will differentiate into different lineage cell types on substrates of different elastic rigidity [12]. Experiments have shown that many cell types spread and exhibit more polarized FAs on stiffer than on softer matrices [13–15], and cells tend to actively migrate towards stiffer regions when cultured on an elastically non-homogeneous substrate, a process known as durotaxis [16]. In vitro studies of cross-linked networks of actin bundles and other biopolymer networks showed that their elastic modulus can increase over several orders of magnitude in response to different levels of applied stress owing to the entropic elasticity of filaments [17,18]. Moreover, some cells such as fibroblasts can tune the stiffness of their cytoskeleton to match that of the substrate [19]. Other studies have shown that a contracting cytoskeleton can be used to sense mechanical properties of the ECM and in turn affect cell behaviours [20]; whether stiffness tuning of the cytoskeleton is the most important factor remains unclear.
Serving as the mechanical anchorage between cell and matrix, FAs are usually exposed to forces induced by external physical interactions such as blood flow, as well as those generated by the cell's own contractile machinery as stress fibres made of bundles of actin filaments and myosin II motors actively pull FAs towards the inside of the cell. The growth or shrinkage of FAs is strongly influenced by the magnitude of forces applied on it. Inhibition of the cytoskeletal contractility leads to dissolution of cytoskeleton and decrease of FAs [21], resulting in small, diffraction-limited spots with low traction stress near the cell periphery at the base of the lamellipodia. When myosin II activity is suppressed, application of an external force, irrespective of its physical origin, is found to stimulate growth of FAs in the direction of the force [22,23]. In the case of cell-generated tension, experiments have also shown that the size of mature FAs can reversibly increase or decrease in response to the magnitude of the applied force, with force per unit area (stress) maintained near a constant value around 5.5 kPa, which is remarkably similar for stationary cells of different types [24,25], although exceptional cases in which some small adhesions may produce non-proportionally high forces were also revealed [25,26]. Possible mechanisms by which externally applied forces and cytoskeleton-generated forces could exert changes in cell adhesion include force-induced protein unfolding with exposure or protection of binding sequences and conformational change of integrins (a more extended conformation is thought to enhance binding to ECM) [27–29]. Despite this line of studies demonstrating that forces affect protein structure and cellular function, it remains unclear whether mechanotransduction shares common mechanisms from physics or mechanics point of views.
3. Theoretical modelling of cell adhesion
Quantitative analysis and modelling of dynamic cell attachment are crucial for advancing conceptual insights along with technological applications. An early theory for describing cell adhesion was established by Bell [30] in a thermodynamic framework. The process of adhesion or de-adhesion of cells from substrates was subsequently modelled via peeling tests that are familiar in engineering design but is made more complicated by the biological interface and geometry involved [31,32]. More recent progresses have been made in modelling curved biological membranes spreading on a flat substrate mediated by binder diffusion [33,34], as well as receptor-mediated cellular uptake and release of viruses or nanoparticles [35]. Erdmann & Schwarz [36,37] studied the stochastic effects of a cluster of uniformly stressed molecular bonds transiting between open and closed states under the influence of thermal fluctuation. A common assumption of the existing models on cluster adhesion is ‘equal load sharing’, which means that the applied load is equally shared among all closed bonds. Based on this assumption, the deterministic equation of Bell's framework [30] predicted that molecular clusters remain stable and have infinite lifetime up to a critical load; in contrast, more recent work by Erdmann & Schwarz [36,37] indicated that the cluster lifetime is always finite and increases monotonically as the cluster size grows: the larger the cluster, the longer the cluster lifetime.
A number of theoretical studies have been performed to investigate how mechanical stimuli and cell–ECM properties affect the behaviours of FAs. Nicolas et al. [38] proposed that the FA mechanosensitivity can be enhanced by deformation-induced increase in the affinity of plaque proteins that form the adhesion. Shemesh et al. [39] considered FA growth as a consequence of enhanced aggregation of FA proteins in the direction of force application. Bruinsma [40] described the regulation of cytoskeletal forces generated along actin filaments and reinforcement of the FA–actin connection by force during the growth stage from initial contacts to FXs. Deshpande et al. [41] proposed a model of cellular contractility that accounts for dynamic reorganization of cytoskeleton. Smith et al. [42] showed force-induced adhesion strengthening by considering thermodynamic interplay between elastic response of membrane, entropy of free receptors and enthalpy of bond formation.
In spite of the tremendous progresses in understanding cell adhesion and dynamics of FAs over several decades, some fundamental questions still remain unanswered. In particular, physical and mechanical models have attempted to address several aspects of integrin-mediated cell–matrix adhesion: why does the size of FAs fall into a narrow range around the micrometre scale? How can cells sense and respond to substrates of varied stiffness via FAs? How do the magnitude and orientation of mechanical forces affect the binding dynamics of FAs? Can the binding dynamics of FAs be modelled taking cell–ECM elasticity into account? In the next few sections, we will discuss some recent attempts aimed to address these questions via a coupled stochastic–elastic modelling framework that unifies stochastic descriptions of individual molecular bonds and elastic descriptions of interfacial interactions (figure 2a,b,e,i,j).
Figure 2.

Schematic illustrations of the mechanical principles of focal contacts in cell–matrix adhesion. (a,b) Small adhesion size and stiff cell–matrix lead to uniform stress along the interface, whereas large adhesion size and soft cell–matrix lead to stress concentration near the adhesion edges [36,37,43,44]. (c) Schematic of a motor–clutch model where myosin motors pull an actin bundle against integrin adhesion [45]. (d) Schematic of a mechanical model of actin stress fibre formation and substrate elasticity sensing [46]. On one end, the actin bundle is anchored to an adhesion complex. At the other end, the bundle slides through the cytoskeleton. (e) Elastic recoil at open bonds that increases the surface separation at these bonds and kills rebinding events that are necessary for stable adhesion [47]. (f) Mechanosensitivity at single molecule level [27]. Application of force activates receptor–ligand bonds from inactive to active, and therefore induces FA development. (g) A force-driven mechanism for focal adhesion mechanosensitivity [38]. Force induces compression at FA front edge, increases the affinity of the adhesion molecules and leads to FA development. (h) A thermodynamic model of FA building proteins [39]. Under a pulling force, assembly of new FA proteins to the adhesion structure reduces the mechanical stress and is energetically favourable. (i) Low-angle pulling on stress fibres reduces the average interfacial traction in the focal contact, thereby prolonging the adhesion lifetime [44]. (j) Cytoskeletal pretension induced by myosin contractility tends to smooth out the distribution of forces on receptor–ligand bonds between individual stress fibres and matrix, thereby increasing the adhesion lifetime [48].
4. Stochastic dynamics of single receptor–ligand bonds
The specific receptor–ligand bonds are often considered a lock-and-key mechanism, which can transit stochastically between a closed (binding) state and an open (broken) state. A single closed receptor–ligand bond in cell adhesion has a binding energy of 10–25 kBT (kBT: the product of Boltzmann constant and absolute temperature) and can undergo a transition from the original closed state to an open state owing to thermally activated bond dissociation even in the absence of an external force [49]. In the past two decades, intensive studies, including experiments based on dynamic force spectroscopy [50–52] and theoretical work by Evans & Ritchie [53], and more recently by Freund [54], have been carried out to understand the dissociation of single molecular bonds under an applied force. The process of bond dissociation is often regarded as thermally assisted escape over a potential energy barrier [55,56]. Application of an external force changes the energy landscape and therefore influences the rupture process. For time-independent loading, both theories and experiments have indicated that the dissociation rate koff of a closed bond increases exponentially with a force F acting on the bond as [30]:
 |
4.1 |
where k0 is the spontaneous dissociation rate in the absence of the force, xb is the distance between the minimum of the binding potential and the transition state barrier and kBT is the unit of thermal energy. For molecular bonds in FAs, 1/k0 falls in the range from a fraction of a second to around 100 s [56]. It follows from equation (4.1) that Fb = kBT/xb can be recognized as an intrinsic force scale. Typically, xb ≈ 1 nm and Fb is estimated to be around 4 pN. The dimensionless lifetime τT = k0t, where t is the real time, of a single closed bond is then τT(1) = exp(−F/Fb). Therefore, a single molecular bond is unstable over time even without an applied force.
For failure of a multiple-bond adhesion, such as those in FAs, one must take into account the fact that individual bonds can rebind after they break until the whole adhesion is detached. The analysis of single bond force spectroscopy [53] did not consider such rebinding, but theoretical considerations by Erdmann & Schwarz [36,37] indicated that bond rebinding can greatly enhance the adhesion lifetime. Rebinding of an open bond can occur as long as a broken receptor and ligand comprising a pair are held in close proximity for a sufficient amount of time. In a recent model of bond rebinding, the whole process is regarded as consisting of two steps (figure 3): first, the receptor and complementary ligand have to come sufficiently close within a binding radius lbind to form a complex; second, the complex reacts at a rate  to form the final receptor–ligand bond [57,58]. The reaction rate kon between a ligand on a substrate surface and a receptor tethered to a cell wall by a linear spring with stiffness kLR and rest length lb is assumed to depend on the cell–substrate surface separation δ as [43,44,57,58]:
to form the final receptor–ligand bond [57,58]. The reaction rate kon between a ligand on a substrate surface and a receptor tethered to a cell wall by a linear spring with stiffness kLR and rest length lb is assumed to depend on the cell–substrate surface separation δ as [43,44,57,58]:
 |
4.2 |
where Z is the partition function for a receptor confined in a harmonic potential between −lb and δ − lb (figure 3).
Figure 3.

Dissociation–association transition between closed and open bonds. The dissociation of single closed bonds is assumed force-dependent as described by the classical Bell's equation. In modelling bond association, a tethered receptor is assumed to approach and react with an opposed ligand. The two surfaces are separated by a distance δ. The receptor is modelled as a binding site tethered to the cell by a linear spring with stiffness kLR and rest length lb.
The rebinding rate described above depends strongly on the surface separation. This strongly decaying behaviour of rebinding rate with increasing separation is expected to play a very important role in the dynamics and stability of molecular bond clusters when the elasticity of the system is considered. For example, the surface separation of an adhesion cluster under a remote tensile force is generally larger at the cluster edges than at the centre and, consequently, rebinding is less likely to occur at the edges. Once the opposing surfaces are separated by more than a critical distance, rebinding becomes impossible and the cluster is expected to undergo crack-like failure from its adhesion edges [43,44].
5. Stochastic dynamics of bond clusters
Although the statistical description of single molecular bonds is by now well accepted, the collective behaviour of multiple molecular bonds, such as the bond clusters in FAs, can be much more complex and less understood. A single molecular bond only has a limited lifetime while a cluster of bonds can survive for much longer owing to the cooperative effects in a stochastic ensemble [36,37]. How can this transition be modelled and can this tell us something about the mechanics of cell adhesion? Theoretical effort to address this question was pioneered by Bell [30] who applied the kinetic theory of chemical reactions to predict the thermodynamic competition between bond breaking and reforming. Subsequently, Seifert [59] studied the dynamic behaviour of a molecular bond cluster subjected to linearly ramping forces. Erdmann & Schwarz [36,37] developed a more rigorous theory of cluster lifetime based on the one-step master equation [60] in stochastic dynamics.
Experiments have shown that the size of large FAs in stationary cells can reversibly increase or decrease in response to cell-generated forces with force per unit area maintained near a constant value [24,25], although some small adhesions could produce non-proportionally high forces [25,26]. In this sense, it will be instructive to study the behaviour of molecular bond clusters subjected to a constant average force per bond, i.e. a total force proportional to the cluster size. Consider the case that a pulling force fNt is applied on a molecular cluster between two rigid bodies. The cluster consists of a total of Nt receptor–ligand bonds with stiffness kLR and rest length lb, as indicated in figure 4a. The nominal force sustained by individual bonds is f (normalized by the force scale Fb in §4). Suppose that k bonds are closed and (Nt–k) bonds are open at a given time τ (0 ≤ k ≤ Nt). The k closed bonds share the total applied force equally, so that the actual force acting on each closed bond is fNt/k. Each of the (Nt–k) open bonds is assumed to rebind at a separation-dependent rate described in equation (4.2).
Figure 4.

Effect of cluster size and applied force magnitude on the adhesion lifetime of a cluster of molecular bonds between two rigid bodies subjected to a force proportional to its size. (a) The system under study consists of Nt receptor–ligand bonds, k of which are closed and equally share the total load fNt. The Nt–k open bonds rebind at a separation-dependent rate g described in equation (5.3). (b) The lifetime τT of the molecular bond cluster as a function of the cluster size Nt at various load levels f (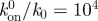 ).
).
For the given initial condition k(τ = 0) = Nt, the average lifetime of a molecular bond cluster, defined throughout this paper as the mean first passage time to reach the final failure state of k = 0, can be calculated analytically for the present rigid case as [61]:
 |
5.1 |
under the reflecting boundary condition at k = Nt and absorbing boundary condition at k = 0. The above equation can be derived by summing up the average time for all possible pathways transiting from the initial cluster size Nt towards the absorbing boundary k = 0 with their statistical weights. Under the present setting,
 |
5.2 |
 |
5.3 |
where β = kLRb2/(2kBT), and  is a prefactor for rebinding rate; Δ = δ/b and Lb = lb/b are the normalized surface separation and bond rest length, respectively. The surface separation Δ in equation (5.3) is also a function of k as Δ = Lb + (Nt/k) (fFb/kLRb).
is a prefactor for rebinding rate; Δ = δ/b and Lb = lb/b are the normalized surface separation and bond rest length, respectively. The surface separation Δ in equation (5.3) is also a function of k as Δ = Lb + (Nt/k) (fFb/kLRb).
If Nt = 1, equation (5.1) is reduced to τT(1) = exp(−f), which is just the lifetime of a single molecular bond. In the case of zero rebinding, the second term of equation (5.1) vanishes and the cluster lifetime becomes  , which, in the absence of an applied force, is further reduced to
, which, in the absence of an applied force, is further reduced to  , corresponding to the Ntth harmonic number.
, corresponding to the Ntth harmonic number.
For typical values kLR = 0.25 pN nm−1, b = 32 nm, lb = 11 nm and Fb = 4 pN (used throughout the discussions in this paper unless stated otherwise), the lifetime τT of a molecular bond cluster is plotted as a function of its size for various load levels in figure 4b. The rebinding prefactor,  is taken to be 104. An immediate observation is that a single bond is always unstable, and bond clustering prolongs the lifetime by many orders of magnitude through rebinding. In other words, individual molecular bonds are very weak but many bonds together collectively result in long-term stability. Moreover, the lifetime τT increases monotonically with growing cluster size Nt under the assumption of equal load sharing, which is similar to the results of Erdmann & Schwarz [36,37] based on a separation-independent rebinding rate.
is taken to be 104. An immediate observation is that a single bond is always unstable, and bond clustering prolongs the lifetime by many orders of magnitude through rebinding. In other words, individual molecular bonds are very weak but many bonds together collectively result in long-term stability. Moreover, the lifetime τT increases monotonically with growing cluster size Nt under the assumption of equal load sharing, which is similar to the results of Erdmann & Schwarz [36,37] based on a separation-independent rebinding rate.
6. Role of cell–matrix elasticity in the dynamics of bond clusters
In the presence of elastic deformation of cell and ECM, the reverse and forward rates in equations (4.1) and (4.2) would also depend on the local force and surface separation at a bond location within the adhesion domain. The analytical solution to the original master equation is generally unavailable in the case of compliance-induced spatially dependent rupture and rebinding rates. The effects of cell–ECM stiffness on cluster lifetime have been incorporated into the stochastic dynamics of bond clusters through an elastic Green's function approach [43,44]. A Monte Carlo scheme has been developed based on Gillespie's algorithm [62,63] to numerically solve the spatio-temporal process governed by the master equation. The basic idea is to cast stochastic trajectories of cluster evolution in accordance with the above described reaction rates and then average over many independent trials to obtain useful statistical information. Qian et al. [43,44] have applied the so-called ‘first-reaction method’ [62,63], which was also adopted by Erdmann & Schwarz [36,37] in their simulations under the assumption of equal load sharing. In the simulations, each bond location (closed or open) is considered an independent reaction site. At any step during the cluster evolution, reactions were selected through random number sampling to determine whether the next event is bond breaking or rebinding, and when or where the next activity would occur. The ‘first-reaction method’ [62,63] is to generate a series of independent random numbers ξν, ν = 1, 2, …, Nt uniformly distributed over the interval [0, 1] and calculate the reaction time for individual reaction sites according to τν = −ln ξν/aν, aν referring to rupture–rebinding rates at individual bond locations. The time for the next reaction is chosen to be the smallest among τν, i.e. τμ = min (τν). At the same time, the location for the next event is identified to be the reaction site μ where τμ is chosen. The event type for the next reaction is ‘rupture’ if the bond at site μ is currently closed and ‘rebinding’ if it is currently open.
Any changes of bond state require an update of bond force and surface separation in the elasticity part of the model, which is then used to determine the subsequent reaction rates. This stochastic–elastic coupling usually starts with the initial condition that all bonds within the adhesion domain are closed, and proceeds until all bonds are open. The total elapsed time is recorded as the cluster lifetime τT.
7. Size window for stable focal adhesions
The experimental observation that focal contacts in cell–matrix adhesion lie within a narrow size range in the order of a few micrometres [10,11] has puzzled the community for a long time. Based on the solution to a one-step master equation, Erdmann & Schwarz [36,37] demonstrated that molecular clusters below a critical size behave like a single bond with a finite lifetime while those above the critical size survive over a much prolonged time owing to the collective effect of clustering (figure 2a). Therefore, adhesion size can play a very important role in the stability of a bond cluster: small clusters can easily switch between adhesion and de-adhesion, similar to FXs which are subjected to frequent turnover, while large clusters tend to have a much longer lifetime similar to stable FAs. Built upon the work by Erdmann & Schwarz [36,37] Qian et al. [43,44] included the effects of elasticity and non-uniform stress distribution on the stability of a single or a periodic array of adhesion clusters under normal or inclined loads, with results predicting a size-dependent transition between uniform and crack-like distributions of interfacial traction, a window of cluster size for relatively stable adhesion and an optimal size for maximum strength (figure 2b). Analysis by Lin & Freund [64] based on a direct analogy between FAs and periodic cracks led to similar conclusions. Here we discuss these size effects using a theoretical model involving a periodic array of adhesion clusters of molecular bonds between two dissimilar elastic media subjected to a tensile stress σ∞ applied at an inclined angle θ with respect to the cell–ECM interface (figure 5a), following the analysis by Qian et al. [44].
Figure 5.

A stochastic–elastic model of focal contacts illustrating the effect of adhesion size and cell–substrate elasticity on the adhesion lifetime. (a) Schematic illustration of a periodic array of adhesion clusters between two dissimilar elastic media under an inclined tensile stress σ∞. (b) The lifetime τT of the periodic adhesion clusters as a function of the cluster size Nt for different values of the reduced modulus E*. The pulling angle θ is fixed at 45°. The selected values of E* are 1, 10, 100 and 300 kPa. (c) The lifetime τT of the periodic adhesion clusters as a function of the pulling angle θ at various levels of applied stress when Nt = 40 and E* = 10 kPa. All plots are based on  and c/a = 2, and are adapted from Qian et al. [44].
and c/a = 2, and are adapted from Qian et al. [44].
Both cell and substrate are modelled as semi-infinite elastic media with Young's modulus and Poisson's ratio EC, νC and ES, νS, respectively. It will be convenient to define a reduced elastic modulus E* according to the convention of contact mechanics [65]:
 |
7.1 |
Consider the situation that interfacial adhesion arises solely from the receptor–ligand bonds modelled as Gaussian chains having a finite stiffness kLR and zero rest length. All bonds are assumed to be closed at the initial state and subsequently can transit statistically between open (broken) and closed (linked) states as described by Bell [30]. The bonds are grouped in adhesion clusters of size 2a which are periodically distributed at a distance of 2c along the interface. Within each cluster, the bonds are uniformly distributed at spacing b, corresponding to a bond density of ρLR = 1/b2. The average bond density along the interface is  . Owing to the periodic nature of the problem, only one cluster with a total number of bonds Nt = 2a/b is considered. A plane strain elasticity model is adopted to determine the distributions of interfacial traction and separation within the bond cluster.
. Owing to the periodic nature of the problem, only one cluster with a total number of bonds Nt = 2a/b is considered. A plane strain elasticity model is adopted to determine the distributions of interfacial traction and separation within the bond cluster.
The present setting can be viewed as a combination of the bond dynamics obeying one-step master equation [60] and the periodic crack model in interfacial fracture mechanics [66]. In the absence of molecular bonds, the model is reduced to a periodic array of interfacial cracks between two elastic media and in the limit of rigid elastic media, it is reduced to the type of cluster model discussed by Erdmann & Schwarz [36,37].
Using the elastic Green's functions for semi-infinite media [65], it can be shown that the normal and shear tractions along the interface, σ(x) and τ(x) obey the following integral equations [44]:
 |
7.2 |
where
 |
7.3 |
corresponds to one of the Dundurs constants [67] for elastic problems in a bimaterial system. Biological materials are often modelled to be incompressible with Poisson's ratio of nearly 1/2, in which case β ≈ 0. Thus, how the interfacial traction is distributed within the adhesion domain is found to be solely governed by a stress concentration index [44]
| 7.4 |
which is linearly proportional to the adhesion size, the bond stiffness and density, and inversely proportional to the reduced elastic modulus of cell and substrate. In the limit when α → 0, it is clear that both normal and shear interfacial tractions will be uniformly distributed within the adhesion domain. In the opposite limit when α → ∞, both σ(x) and τ(x) become crack-like with square root singularity near the adhesion edges. For intermediate values of α, numerical analysis of equation (7.2) indicates that the interfacial tractions are nearly uniform for α smaller than 0.1, while crack-like stress concentration emerges near the adhesion edges for α values larger than 1.0 [44]. In particular, the elastic modulus of both cell and substrate needs to be sufficiently large in order to keep α small enough for equal load sharing.
Guided by the scaling law in equation (7.4), a series of two-scale elastic-Monte Carlo simulations have been performed to investigate the dynamics of bond clusters coupling the elastic descriptions of adhesive contact at a large scale and stochastic breaking–rebinding of molecular bonds at a small scale [44]. The lifetime τT of the periodic clusters is shown in figure 5b as a function of the cluster size Nt for different values of the reduced elastic modulus E* between 1 and 300 kPa. The loading angle θ is fixed at 45°. The simulation results indicate that there exists a size window for relatively stable adhesion. In all cases, the traction distribution along the cell–ECM interface is non-uniform and the failure becomes increasingly crack-like at increasing cluster size. Very small clusters resemble single molecule behaviour with limited lifetime and large clusters fail by severe stress concentration near the adhesion edges. Increasing the reduced elastic modulus tends to stabilize and strengthen the adhesion by alleviating stress concentration within the FA domain. It is observed that the size window of stable adhesion shifts and broadens as the cell and substrate stiffen, which can be understood from the point of view that large values of E* decrease the stress concentration index α towards the regime of uniform interfacial traction. The concept of a size window for stable adhesion here is similar to another study on a single cluster under normal tensile load [43] and should be a general feature of molecular adhesion clusters between elastic media because stochastic effects are expected to dominate at small scales and crack-like failure dominates at large scales. Increasing adhesion size or decreasing cell–ECM modulus tends to increase α towards the regime of crack-like stress concentration, hence reducing the lifetime and stability of FAs.
The lifetime τT of the periodic cluster array is plotted as a function of the pulling angle θ at various stress levels in figure 5c. Here Nt = 40 and E* = 10 kPa are fixed in the calculation. For a given magnitude of the applied stress σ∞, decreasing θ tends to stabilize the adhesion. In fact, the adhesion lifetime asymptotically approaches infinity as the pulling angle is reduced to below a critical threshold. This is especially interesting in view of the fact that cells generally flatten when successfully adhered to a substrate and immediately suggests a regulation mechanism by which cells can switch between long- and short-lived adhesions by adjusting pulling direction around the critical angle (figure 2i).
Thus, a possible explanation for the characteristic micrometre-size scale of FAs is that small adhesion size leads to single-molecule-like behaviour of limited lifetime because of statistical effect, whereas large adhesion also leads to single-molecule-like behaviour because of the focusing effect of stress concentration that confines the bond dissociation events to a smaller number of bonds near the adhesion edges. Optimal adhesion is achieved only at intermediate adhesion sizes. Additionally, cell–matrix stiffening and low-angle pulling by cytoskeletal stress fibres help to stabilize FAs.
8. Effect of substrate–cytoskeleton stiffness on dynamics of focal contacts
A recent study by Chan & Odde [45] investigated ECM rigidity sensing of neuronal filopodia via a stochastic model of the ‘motor–clutch’ force transmission system shown in figure 2c, where integrin molecules function as mechanical clutches linking F-actin to the substrate and mechanically resisting myosin-driven F-actin retrograde flow. The model predicts two distinct regimes in retrograde flow speed and integrin traction force, and the predicted critical matrix stiffness leading to different regimes is of the same order as the brain tissue. Stiffer substrates result in a rapid build-up of tension within individual engaged integrins and an abrupt disengagement from substrates. In this case, the F-actin bundle is continuously slipping over the substrates. On softer substrates, in contrast, the rate of tension increase in the clutch is slow enough that a sufficient number of integrin bonds remain engaged at the early stage of adhesion. At a critical time, the failure of a single bond results in catastrophic failure of all bonds. This force transmission from myosin-driven F-actin retrograde flow to FAs has been experimentally identified by the clutch between integrins and ECM, with interesting results showing that the immobilization of FAs relative to the ECM occurred at a constant tension [68], and forces of similar magnitude were built up over similar timescales for FAs on soft (0.6 kPa) and stiff (2.8 kPa) matrices [69]. In other recent studies on clutch dynamics, a kinetic model has been introduced to simulate the turnover processes of actin filaments, integrin bonds and the involved FA proteins [70]; Sabass & Schwarz [71] considered the competition between the binding dynamics at the moving cell–substrate interface and the frictional dissipation inside the cell; Li et al. [72] explained the observed biphasic relationship between actin retrograde flow speed and traction force [73] from the perspective that force transmission is limited by bond breakage at high speeds and the build-up of force within FAs is limited at low speeds. A more sophisticated model of ECM rigidity sensing was proposed recently by Walcott & Sun [46], who coupled FA resistance against myosin-driven activity, a Hill-type of force–velocity relation of myosin pulling [74] and actin cytoskeleton reorganization under force to show that the stiffness of the substrate directly influences differential formation of stress fibres in the cytoskeleton and ultimately leads to changes in intracellular biochemistry (figure 2d). In this study, stiffer substrates are shown to be more favourable for stress fibre aggregation because of more rapidly built-up cytoskeletal force.
Qian et al. [43,44] have previously shown that the soft matrix-induced stress concentration at adhesion edges can decrease the lifetime of a molecular cluster, as reviewed in §7. However, the elasticity-induced stress concentration is only one of the devastating effects on the lifetime and stability of focal contacts. A more striking effect is that the surface separation between cell and substrate after a closed bond is released also depends sensitively on substrate stiffness, irrespective of the stress concentration effects at adhesion edges [47]. The local elastic recoil following a bond rupture can lead to large surface separation, thereby preventing future rebinding of the bonds (figure 2e). To demonstrate this additional effect of elasticity on adhesion lifetime, let us consider molecular bond clusters subjected to a uniform tension applied directly along the interface between cell and substrate [47]. For a single adhesion patch under a uniform tensile stress p over the adhesion domain −a ≤ x ≤ a, as indicated in figure 6a, the governing equation under plane strain situation is [65]
| 8.1 |
where σ(x) is the traction within the adhesion region sustained by molecular bonds, and E* has been defined in equation (7.1). The solution to equation (8.1) is simply σ(x) = p when all of the bonds are closed. It is seen that, instead of a remotely applied force which tends to induce a non-uniform distribution of bond forces within the cluster [43], the uniformly applied stress p at the interface ensures that all bonds are nominally subjected to an equal force. In this setting, the effects of an initial non-uniform stress distribution within the cluster are excluded from the analysis, but the cell–substrate stiffness can still strongly influence the adhesion lifetime because soft media substantially diminish the separation-dependent rebinding rate as a result of the elastic recoil at open bonds (figure 6a).
Figure 6.

A stochastic–elastic model of focal adhesion demonstrating the effect of cell–substrate compliance in suppressing the rebinding of bonds in the absence of initial stress concentration within the adhesion domain. (a) A single adhesion patch between two elastic media subjected to a uniform tensile stress p directly applied along the interface. In this case, the applied load is initially equally shared among all bonds, independent of the system elasticity. (b) The cluster lifetime τT as a function of the reduced modulus E* for different cluster sizes. The load level f is fixed at 0.5. (c) The ratio of total events between bond rebinding and bond rupture influenced by the reduced modulus E* of the cell and the substrate (f = 0.5). (d) Analytical results of the cluster lifetime τT for the cases of rigid media (upper) and zero rebinding (lower) compared with the Monte Carlo simulations for stiff (E* = 10 MPa) and soft (E* = 10 kPa) cell and substrate (f = 0.5). All plots are based on  , and are adapted from Qian & Gao [47]. (b,c) Filled squares, eight bonds; filled circles, 20 bonds; filled triangles, 40 bonds; filled inverted triangles, 100 bonds.
, and are adapted from Qian & Gao [47]. (b,c) Filled squares, eight bonds; filled circles, 20 bonds; filled triangles, 40 bonds; filled inverted triangles, 100 bonds.
In a cluster of molecular bonds at the cell–substrate interface, breaking one bond bears some resemblance to a microcrack of size 2b (b is the bond spacing) in an infinite elastic media. A rough estimate of the elastic recoil at the centre of the crack is δe = 4pb/E* [75]. In addition, the tensile stress p also induces an average separation between the cell and the substrate, which is equal to δu = pb2/kLR, where kLR is the bond stiffness. Equation (4.2) then suggests that the rebinding rate of the broken bond is governed by the non-dimensional parameter
 |
8.2 |
Since the rebinding rate is proportional to exp (−χ2), the larger the χ, the smaller the rebinding rate. The effect of soft elastic modulus on decreasing rebinding rate is clearly seen.
Figure 6b shows the cluster lifetime τT as a function of the reduced elastic modulus E* for different values of the cluster size Nt. The nominal load sustained by individual bonds, i.e. f as defined in §5, is identical in all cases. For cell–ECM with physiological E* value within 1–100 kPa, the cluster lifetime is reduced by two orders of magnitude from stiff to soft cases (figure 6b). For given parameters kLR = 0.25 pN nm−1 and b = 32 nm, δe and δu are actually comparable when E* is around 10 kPa. In this case, the elastic recoil causes large reductions in cluster lifetime by decreasing the probability of rebinding. This is confirmed by tracking the ratio of total events between bond rebinding and bond rupture during the cluster evolution, as indicated in figure 6c.
At a fixed cluster size, focal contacts become more and more stable as cell and ECM stiffen, approaching the behaviour of clusters between two rigid bodies, given by equation (5.1). For very soft cell–ECM, the surface separation at open bond locations is so large that rebinding becomes almost impossible. Removal of all rebinding terms in equation (5.1) gives
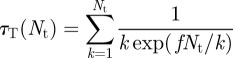 |
8.3 |
under the condition of equal load sharing. This result can serve as an estimate of lifetime for a molecular cluster between very soft cell and substrate. As shown in figure 6d, the Monte Carlo simulations of the cluster lifetime τT for stiff (E* = 10 MPa) and soft (E* = 10 kPa) cell–substrate agree well with the analytical predictions for the cases of rigid media and zero rebinding (equations (5.1) and (8.3)).
Guided by the scaling law in equation (8.2), the cooperation of molecular bonds in FAs is also strongly influenced by the spacing between neighbouring bonds: the larger the spacing b, the larger the parameter χ and the smaller the rebinding rate. This is qualitatively consistent with the experimental observation that FA is inhibited and cells do not spread for ligand spacing larger than 73 nm, while formation of focal contacts and cell spreading to a pancake-like shape can operate normally only for ligand spacing smaller than 58 nm [76].
Therefore, the way that cytoskeleton–ECM stiffness influences FA stability does not rely solely on how the load is transmitted in the adhesion region. Even for molecular clusters under initially uniform pulling forces, the cell–substrate elasticity can destabilize FAs by suppressing the rebinding of open bonds. While the effect of stress concentration in adhesive contact is well known in contact mechanics theory [77] as well as in applications such as gecko adhesion [78,79,80], the role of elasticity in suppressing bond rebinding is a unique feature of molecular adhesion. The sensitivity of FAs to cell–substrate stiffness cannot be alleviated simply by removing stress concentration in the system.
9. Effect of cytoskeleton pretension on cell–matrix adhesion
It is well known that the action of myosin motors induces pretension in the cytoskeleton [81,82]. There is ample experimental evidence that intracellular tension in cytoskeleton is crucial for FA stability [21]. One pathway for the cytoskeletal forces to the growth of FAs can be that these forces could switch the mechanosensing proteins within FAs from an inactive state to an active state by inducing protein conformational change, which could then up- or downregulate FA growth [40]. Nicolas et al. [38] proposed that the FAs mechanosensitivity can be enhanced by a deformation-induced increase in the affinity of plaque proteins that form the adhesion (figure 2g), which can account for observations of anisotropic FA growth with tension [22]. Alternatively, Shemesh et al. [39] suggested that the growth of FAs can be understood as a one-dimensional molecular aggregation process governed by thermodynamic principle (figure 2h), in which assembly of new plaque proteins to the pulled adhesion structure reduces the mechanical force and is energetically favourable. The pretension can also regulate the strength of FAs by altering the FA-associated protein interactions (figure 2f). For example, transmembrane protein α5β1 forms force-enhanced ‘catch bonds’ with the ECM protein fibronectin [27]. Such catch bonds were initially proposed by Dembo et al. [31], experimentally observed for the first time for leucocyte binding proteins [83], and have been thoroughly reviewed for molecular interactions in cell adhesion [84–86].
The basic mechanical principle of the effect of pretension on FA stability is still a subject of debate. A possible clue to this question has emerged from the recent finding that pretension in spatula hairs of geckos can dramatically enhance the critical pull-off force at small peeling angles [87]. In general, cytoskeletal contractility has at least two effects on FAs. The first effect is that it stiffens actin networks in the cytoskeleton, thereby increasing the adhesion lifetime by increasing the reduced elastic modulus of the system [43,44], as discussed in the previous two sections. The second effect is that the contractile force generates pretension in the stress fibres, which may also change the adhesion lifetime (figure 2j). Chen & Gao [48] investigated the effect of cytoskeletal pretension on focal contacts by considering an individual stress fibre attached to a rigid substrate via a cluster of molecular bonds subjected to a horizontal pulling force P, as illustrated in figure 7a. The stress fibre is modelled as an elastic beam with tension stiffness EA, where E is the Young modulus and A is the cross-sectional area. A cluster of receptor–ligand bonds with stiffness kLR is assumed to be equally spaced at a distance b along the interface between the fibre and the substrate. The bonds can break and reform stochastically according to reaction rates similar to those described in equations (4.1) and (4.2). Following Kendall's [88] elastic thin film peeling model, the ratio between the maximum bond force at the contact edge and the applied force can be estimated as [48]
| 9.1 |
where Λ = kLRb/(EA) is recognized as a force concentration index at the fibre–substrate interface. As the bond forces are generally concentrated in the vicinity of the contact edge, equation (9.1) indicates that the force concentration would decrease as the force concentration index Λ is reduced. Harder stress fibres and/or softer molecular bonds lead to smaller force concentration near the contact edge (figure 7b). The concept that the interfacial force distribution is governed by the force concentration index Λ is similar to that developed by Qian et al. [43,44] where interfacial traction distribution between two elastic media is governed by the stress concentration index α.
Figure 7.

A stochastic–elastic model of focal adhesion between an elastic fibre under pretension and a rigid substrate. (a) A stress fibre adhered to a substrate via molecular bonds. The fibre is subjected to a pretension before the fibre–substrate binding. (b) The force distribution along an array of molecular bonds. The smaller the force concentration index Λ, the more uniform the interfacial force distribution. The paramters for the calculation are kLR = 0.25 pN nm−1, Nt = 500 and P = 16 pN. (c) Effect of a stairwise increasing pattern of pretension, from 0 to 49P0/50 at uniform steps over 50 equal segments along the stress fibre, on the distribution of interfacial forces within the molecular bonds. The force concentration index Λ is fixed at 1.0 (kLR = 0.25 pN nm−1, Nt = 501, P = 16 pN). (d) The adhesion lifetime versus the magnitude of pretension in the stress fibre for various combinations of the force concentration index Λ and the adhesion size Nt (γ = 1.0, Fb = 1 pN and P = 36 pN). All plots are adapted from Chen & Gao [48].
In the presence of a uniform pretension P0 within the stress fibre, the force concentration factor near the contact edge is modified to
 |
9.2 |
where u is the maximum displacement of bond upon breaking. This result immediately shows that the pretension tends to lower the force concentration near the contact edge, and sufficiently large pretension can make the bond force distribution nearly uniform along the fibre–substrate interface, as indicated in figure 7c.
Systematic Monte Carlo simulations have been conducted to investigate the effect of pretension on the adhesion lifetime by varying the fibre stiffness, the adhesion size, and the magnitude and distribution of pretension in the stress fibre. The results show that the presence of pretension in the stress fibre tends to shift the interfacial failure mode from a crack-like propagation in the absence of pretension to almost uniformly distributed bond rupture, hence dramatically enhancing the adhesion lifetime [48]. This is in excellent agreement with the theoretical interpretation in equation (9.2). In a demonstrating example shown in figure 7d, the pretension is assumed to increase stairwise from 0 to P over four equal segments over the entire stress fibre. As the magnitude of pretension increases, the adhesion lifetime can be prolonged by several orders of magnitude. There exists an optimal magnitude of pretension for the maximum adhesion lifetime. Too small or too large contractile forces are both detrimental to the stability of molecular adhesion.
The above analysis suggests that pretension within cytoskeletal stress fibre tends to shift the interfacial failure mode from crack-like propagation towards uniform bond rupture within the adhesion domain, thereby greatly increasing the adhesion lifetime. These results provide possible clues to the molecular mechanisms by which cell adhesion can be actively controlled through the magnitude and pattern of myosin activities within the cytoskeleton.
10. Conclusive remarks
The studies discussed in this paper have been aimed at illustrating some of the basic mechanical principles of FAs between cell and ECM using simple analytical and numerical models. Various models differ in their assumptions concerning the physical mechanisms underlying the mechanical and dynamic behaviour of focal contacts in cell–matrix adhesion. Given the complexity of biological systems, it should not be surprising if in practice different mechanisms act in parallel. Although the existing experimental evidences are far from allowing definitive conclusions to be drawn regarding different theoretical models and simulations, these efforts do provide interesting insights into a number of physical and mechanical aspects of cell–matrix adhesion.
Theoretical models suggest that the reason for FAs to lie in a narrow size range from a few hundred nanometres to a few micrometres [10,11] might be that the growth of FAs eventually leads to crack-like delamination failure near the adhesion edges. From this point of view, the growth of FAs is self-limiting, and the optimal size window for stable adhesion is in the submicrometre to micrometre range, depending on the rigidity of the cell and the substrate. The effects of the reduced elastic modulus E* of cell and matrix on bond rebinding and adhesion lifetime, independent of how the load is distributed within FAs, imply that very soft substrates tend to diminish the adaptive capability of cells by suppressing bond rebinding irrespective of the cytoskeleton stiffness, which may prevent short-lived FXs from maturing into stable FAs. This is also in qualitative agreement with the experimental observations that stable and large FAs can only form on sufficiently rigid substrates [15,16]. The fact that FAs on stiff substrates are more stable provides a possible driving force for cells to migrate towards stiffer parts of the substrate. On hard substrates, the reduced elastic modulus E* tends to be dominated by the stiffness of the cytoskeleton. The cytoskeletal contractile forces can stiffen the cytoskeleton by decreasing entropic elasticity of the actin network, as suggested by the in vitro studies of cross-linked biopolymer networks [17,18], and therefore benefit the long-term stability of FAs. This is consistent with the experimental observations that cytoskeletal contractile forces are necessary to stabilize cell adhesion [21]. The role of cytoskeletal contractility is also demonstrated through an alternative view that pretension in the cytoskeletal stress fibres tends to shift the interfacial failure mode from crack-like propagation towards uniform bond breaking, thereby stabilizing FAs. The dependence of adhesion lifetime on the pulling angle of stress fibres has also been discussed: low-angle pulling dramatically increases the adhesion lifetime. Therefore, cell spreading and flattening over a substrate result in low-angle pulling on FAs and therefore benefit stable adhesion. All these results are consistent with related experimental observations and suggest multiple mechanisms by which cells can actively control adhesion and de-adhesion by modulating the cytoskeleton or adjusting the pulling force/angle of stress fibres. Generally, intermediate adhesion size, rigid substrate, cytoskeleton stiffening, cytoskeletal pretension and low-angle pulling are factors that contribute to stable FAs, whereas extreme adhesion size, soft substrate, cytoskeleton softening, dissolution of actin network and high-angle pulling are factors that tend to destabilize FAs.
Theoretical models described in the present paper suggest the following principles of focal contacts in cell–matrix adhesion:
— Principle of enhancing adhesion by bond clustering. Compared with isolated receptor–ligand bonds, clustering of multiple bonds into focal contacts greatly enhances the lifetime and stability of cell–matrix adhesion.
— Principle of enhancing adhesion by increasing the reduced elastic modulus of cell and matrix. The distributions of bond force and surface separation at the cell–matrix interface depend on the reduced elastic modulus of the cell–matrix system, as defined in equation (7.1). A small modulus, which could result from the presence of a soft matrix or dissolution of cytoskeleton, has two devastating effects on focal contacts. First, it induces severe stress concentration near the adhesion edges and crack-like failure around the rims of focal contacts (figure 2b). The stress concentration effect places an upper limit on the size of focal contacts. Second, small modulus tends to increase local surface separation at open bonds and make them difficult to reform the adhesion, effectively killing the possibility of bond rebinding (figure 2e). The second effect indicates that the role of elasticity is intrinsic in molecular adhesion between soft materials and cannot be alleviated simply by removing stress concentration in the system.
— Principle of enhancing adhesion by low-angle pulling. The adhesion lifetime depends on the direction of stress fibres that exert forces on the focal contacts, as shown in figure 5c. For a given magnitude of pulling force, there usually exists a critical angle below which the adhesion lifetime rapidly rises to long-term stability. This effect is analogous to a more inclined stress fibre exerting smaller average traction on the adhesive interface, as illustrated in figure 2i. This principle may have important implications on how cells control adhesion/de-adhesion during cell migration.
— Principle of enhancing adhesion by cytoskeletal pretension. Cytoskeletal pretension induced by myosin contractility tends to smooth out the distribution of forces on receptor–ligand bonds between individual stress fibres and matrix, thereby increasing adhesion lifetime by orders of magnitudes (figure 2j). This important effect of cytoskeleton contractility may provide a very effective control for cells to regulate adhesion with ECM through metabolic activities in the cytoskeleton. The basic principle can also be understood from the fact that the energy release rate of a thin film on a rigid substrate can be effectively reduced by a moderate pretension in the film. In the absence of any pretension in the film, the energy release rate for interfacial delamination is G = σ2h/(2E), where σ is the stress in the debonded part of the fibre induced by the applied force, h is the film thickness and E is the Young modulus of the film. In the presence of a pretension σ0 in the bonded part of the film, the energy release rate is reduced to G = (σ − σ0)2h/(2E), indicating that a moderate pretension can effectively reduce the driving force for interfacial delamination. The effect of pretension on interfacial adhesion can have general implications in the mechanical properties and functions of muscles and muscular–skeletal junctions.
11. Outlook
At this point of time, the biophysical or biomechanical modelling of FA mechanosensitivity is still at a very primitive stage of research. Increasingly sophisticated models may be necessary to take into account increasing biological complexities, e.g. inside-out and outside-in signalling in both directions across the cell membrane, because cell adhesion is believed to involve complexities far beyond simple passive mechanical attachments. Integrins, the principal receptors on animal cells for binding most ECM proteins, can switch between different conformations, rendering different levels of affinity to their ligands on the ECM side in response to intracellular proteins (e.g. talin), ligation on ECM or divalent cations [1]. All these structural changes and signalling events have yet to be integrated into any theoretical models. Nevertheless, as we discussed here, it seems useful to formulate some possible ground rules based on some basic physical and mechanical principles, which could assist in the integration of relevant biological aspects into a coherent theoretical framework. The mechanics of FAs provides an excellent opportunity for researchers with backgrounds in physical and biological sciences to work together. A major effort on the biological side will be to continue elucidating the relationship between molecular structures, functions and signalling pathways, which can be a critical input for appropriate modelling in the future.
The stochastic–elastic modelling framework [43,44,47,48] used to address the questions posed in §§7–9 of this paper has been built upon the classical model of Bell [30] and its recent extension by Erdmann & Schwarz [36,37]. This framework unifies stochastic descriptions of individual molecular bonds and elastic descriptions of interfacial adhesion. Of special interest has been how the spatially distributed interfacial traction and separation governed by the system elasticity influence the lifetime and strength of adhesion clusters. This paper has reviewed different aspects of the model including the size- and angle-dependent lifetime and stability of FAs [43,44], the role of cell–matrix stiffness in the rebinding kinetics of molecular adhesion [47] and the effect of cytoskeletal pretension in stabilizing cluster adhesion [48]. More generally, such an approach can be extended to a broad range of situations involving spatially dependent bond traction, bond separation and/or non-homogeneous bond density. For example, when leucocytes tether to and roll on vessel walls under blood flow, molecular bonds near the periphery of the contact region are expected to be more stretched than those at the centre [89,90]. In cell–cell adhesion, the formation of an immunological synapse involves multiple types of receptor–ligand bonds with different rest lengths, and the adhesion complex is often organized into patterns with spatially varying bond density [91,92]. The stochastic–elastic coupling approach could serve as a basis for further study of such problems.
An implicit assumption made in the stochastic–elastic models of FAs [43,44,47] is that these adhesions are operating near their rupture limit during FA assembly and growth. We caution that this assumption has not been experimentally proved and should be further investigated. Experimentally measured traction stress is only a direct measure of the tension that is transmitted from the contractile actin cytoskeleton to the ECM via FAs; there is currently no way of knowing whether this force transmission is limited by the amount of tension generated or by weak elastic elements within the actin cytoskeleton or FA. Before this issue is fully clarified by experiments, one way to proceed is to make some reasonable assumptions and then look for possible inconsistencies/contradictions in subsequent research. Much of the current modelling effort in cell–matrix adhesion has followed this approach and therefore requires further experimental validation over the coming years.
While the discussions in this paper are consistent with some of the findings that FAs alter their behaviour as a result of changes in cluster size, cell–matrix rigidity and cytoskeletal contractility, the precise mechanisms remain elusive. While a clear picture of FA mechanosensitivity is not yet available, there are new techniques that enable systematic and accurate in vitro or in vivo measurements of molecular bond kinetics [27], FA molecular architecture [93], cell-generated forces [94], FA protein recruitment [95], ECM deformation [96], etc., which would be important in establishing the validity of modelling efforts. The development of theoretical studies and a deeper understanding of the underlying physical principles on FAs are essential prerequisites for the design of more effective experiments. We believe that further communications between theoretical models and experimental investigations can significantly improve our understanding of the complex processes involved in the dynamics of focal contacts in cell–matrix adhesion.
References
- 1.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. 2002. Molecular biology of the cell. New York, NY: Garland Science [Google Scholar]
- 2.Burridge K., Chrzanowska-Wodnicka M. 1996. Focal adhesions, contractility, and signaling. Ann. Rev. Cell Dev. Biol. 12, 463–518 10.1146/annurev.cellbio.12.1.463 (doi:10.1146/annurev.cellbio.12.1.463) [DOI] [PubMed] [Google Scholar]
- 3.Burridge K., Fath K., Kelly T., Nuckolls G., Turner C. 1988. Focal adhesions: transmembrane junctions between the extracellular-matrix and the cytoskeleton. Ann. Rev. Cell Biol. 4, 487–525 10.1146/annurev.cb.04.110188.002415 (doi:10.1146/annurev.cb.04.110188.002415) [DOI] [PubMed] [Google Scholar]
- 4.Bershadsky A., Balaban N. Q., Geiger B. 2003. Adhesion-dependent cell mechanosensitivity. Ann. Rev. Cell Dev. Biol. 19, 677–695 10.1146/annurev.cellbio.19.111301.153011 (doi:10.1146/annurev.cellbio.19.111301.153011) [DOI] [PubMed] [Google Scholar]
- 5.Geiger B., Bershadsky A., Pankov R., Yamada K. M. 2001. Transmembrane extracellular matrix–cytoskeleton crosstalk. Nat. Rev. Mol. Cell Biol. 2, 793–805 10.1038/35099066 (doi:10.1038/35099066) [DOI] [PubMed] [Google Scholar]
- 6.Zaidel-Bar R., Itzkovitz S., Ma'ayan A., Iyengar R., Geiger B. 2007. Functional atlas of the integrin adhesome. Nat. Cell Biol. 9, 858–868 10.1038/ncb0807-858 (doi:10.1038/ncb0807-858) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoenwaelder S. M., Burridge K. 1999. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 11, 274–286 10.1016/S0955-0674(99)80037-4 (doi:10.1016/S0955-0674(99)80037-4) [DOI] [PubMed] [Google Scholar]
- 8.Geiger B., Bershadsky A. 2001. Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13, 584–592 10.1016/S0955-0674(00)00255-6 (doi:10.1016/S0955-0674(00)00255-6) [DOI] [PubMed] [Google Scholar]
- 9.Alexandrova A. Y., Arnold K., Schaub S., Vasiliev J. M., Meister J. J., Bershadsky A. D., Verkhovsky A. B. 2008. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS ONE 3, e3234. 10.1371/journal.pone.0003234 (doi:10.1371/journal.pone.0003234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaidel-Bar R., Ballestrem C., Kam Z., Geiger B. 2003. Early molecular events in the assembly of matrix adhesions at the leading edge of migrating cells. J. Cell Sci. 116, 4605–4613 10.1242/jcs.00792 (doi:10.1242/jcs.00792) [DOI] [PubMed] [Google Scholar]
- 11.Zamir E., et al. 2000. Dynamics and segregation of cell–matrix adhesions in cultured fibroblasts. Nat. Cell Biol. 2, 191–196 10.1038/35008607 (doi:10.1038/35008607) [DOI] [PubMed] [Google Scholar]
- 12.Engler A. J., Sen S., Sweeney H. L., Discher D. E. 2006. Matrix elasticity directs stem cell lineage specification. Cell 126, 677–689 10.1016/j.cell.2006.06.044 (doi:10.1016/j.cell.2006.06.044) [DOI] [PubMed] [Google Scholar]
- 13.Engler A., Bacakova L., Newman C., Hategan A., Griffin M., Discher D. 2004. Substrate compliance versus ligand density in cell on gel responses. Biophys. J. 86, 617–628 10.1016/S0006-3495(04)74140-5 (doi:10.1016/S0006-3495(04)74140-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geiger B., Spatz J. P., Bershadsky A. D. 2009. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 10, 21–33 10.1038/nrm2593 (doi:10.1038/nrm2593) [DOI] [PubMed] [Google Scholar]
- 15.Pelham R. J., Wang Y. L. 1997. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc. Natl Acad. Sci. USA 94, 13 661–13 665 10.1073/pnas.94.25.13661 (doi:10.1073/pnas.94.25.13661) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo C. M., Wang H. B., Dembo M., Wang Y. L. 2000. Cell movement is guided by the rigidity of the substrate. Biophys. J. 79, 144–152 10.1016/S0006-3495(00)76279-5 (doi:10.1016/S0006-3495(00)76279-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gardel M. L., Shin J. H., MacKintosh F. C., Mahadevan L., Matsudaira P., Weitz D. A. 2004. Elastic behavior of cross-linked and bundled actin networks. Science 304, 1301–1305 10.1126/science.1095087 (doi:10.1126/science.1095087) [DOI] [PubMed] [Google Scholar]
- 18.Storm C., Pastore J. J., MacKintosh F. C., Lubensky T. C., Janmey P. A. 2005. Nonlinear elasticity in biological gels. Nature 435, 191–194 10.1038/nature03521 (doi:10.1038/nature03521) [DOI] [PubMed] [Google Scholar]
- 19.Solon J., Levental I., Sengupta K., Georges P. C., Janmey P. A. 2007. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J. 93, 4453–4461 10.1529/biophysj.106.101386 (doi:10.1529/biophysj.106.101386) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Discher D. E., Janmey P., Wang Y. L. 2005. Tissue cells feel and respond to the stiffness of their substrate. Science 310, 1139–1143 10.1126/science.1116995 (doi:10.1126/science.1116995) [DOI] [PubMed] [Google Scholar]
- 21.Totsukawa G., Wu Y., Sasaki Y., Hartshorne D. J., Yamakita Y., Yamashiro S., Matsumura F. 2004. Distinct roles of MLCK and ROCK in the regulation of membrane protrusions and focal adhesion dynamics during cell migration of fibroblasts. J. Cell Biol. 164, 427–439 10.1083/jcb.200306172 (doi:10.1083/jcb.200306172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bershadsky A., Kozlov M., Geiger B. 2006. Adhesion-mediated mechanosensitivity: a time to experiment, and a time to theorize. Curr. Opin. Cell Biol. 18, 472–481 10.1016/j.ceb.2006.08.012 (doi:10.1016/j.ceb.2006.08.012) [DOI] [PubMed] [Google Scholar]
- 23.Riveline D., Zamir E., Balaban N. Q., Schwarz U. S., Ishizaki T., Narumiya S., Kam Z., Geiger B., Bershadsky A. D. 2001. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 153, 1175–1185 10.1083/jcb.153.6.1175 (doi:10.1083/jcb.153.6.1175) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Balaban N. Q., et al. 2001. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat. Cell Biol. 3, 466–472 10.1038/35074532 (doi:10.1038/35074532) [DOI] [PubMed] [Google Scholar]
- 25.Tan J. L., Tien J., Pirone D. M., Gray D. S., Bhadriraju K., Chen C. S. 2003. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc. Natl Acad. Sci. USA 100, 1484–1489 10.1073/pnas.0235407100 (doi:10.1073/pnas.0235407100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beningo K. A., Dembo M., Kaverina I., Small J. V., Wang Y. L. 2001. Nascent focal adhesions are responsible for the generation of strong propulsive forces in migrating fibroblasts. J. Cell Biol. 153, 881–887 10.1083/jcb.153.4.881 (doi:10.1083/jcb.153.4.881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong F., Garcia A. J., Mould A. P., Humphries M. J., Zhu C. 2009. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 185, 1275–1284 10.1083/jcb.200810002 (doi:10.1083/jcb.200810002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takagi J., Petre B. M., Walz T., Springer T. A. 2002. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell 110, 599–611 10.1016/S0092-8674(02)00935-2 (doi:10.1016/S0092-8674(02)00935-2) [DOI] [PubMed] [Google Scholar]
- 29.Xiao T., Takagi J., Coller B. S., Wang J. H., Springer T. A. 2004. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67 10.1038/nature02976 (doi:10.1038/nature02976) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bell G. I. 1978. Models for specific adhesion of cells to cells. Science 200, 618–627 10.1126/science.347575 (doi:10.1126/science.347575) [DOI] [PubMed] [Google Scholar]
- 31.Dembo M., Torney D. C., Saxman K., Hammer D. 1988. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B 234, 55–83 10.1098/rspb.1988.0038 (doi:10.1098/rspb.1988.0038) [DOI] [PubMed] [Google Scholar]
- 32.Evans E. 1985. Detailed mechanics of membrane-membrane adhesion and separation. I. Continuum of molecular cross-bridges. Biophys. J. 48, 175–183 10.1016/S0006-3495(85)83770-X (doi:10.1016/S0006-3495(85)83770-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Freund L. B., Lin Y. 2004. The role of binder mobility in spontaneous adhesive contact and implications for cell adhesion. J. Mech. Phys. Solids 52, 2455–2472 10.1016/j.jmps.2004.05.004 (doi:10.1016/j.jmps.2004.05.004) [DOI] [Google Scholar]
- 34.Shenoy V. B., Freund L. B. 2005. Growth and shape stabillity of a biological membrane adhesion complex in the diffusion-mediated regime. Proc. Natl Acad. Sci. USA 102, 3213–3218 10.1073/pnas.0500368102 (doi:10.1073/pnas.0500368102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao H. J., Shi W. D., Freund L. B. 2005. Mechanics of receptor-mediated endocytosis. Proc. Natl Acad. Sci. USA 102, 9469–9474 10.1073/pnas.0503879102 (doi:10.1073/pnas.0503879102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erdmann T., Schwarz U. S. 2004. Stability of adhesion clusters under constant force. Phys. Rev. Lett. 92, 108102. 10.1103/PhysRevLett.92.108102 (doi:10.1103/PhysRevLett.92.108102) [DOI] [PubMed] [Google Scholar]
- 37.Erdmann T., Schwarz U. S. 2004. Stochastic dynamics of adhesion clusters under shared constant force and with rebinding. J. Chem. Phys. 121, 8997–9017 10.1063/1.1805496 (doi:10.1063/1.1805496) [DOI] [PubMed] [Google Scholar]
- 38.Nicolas A., Geiger B., Safran S. A. 2004. Cell mechanosensitivity controls the anisotropy of focal adhesions. Proc. Natl Acad. Sci. USA 101, 12 520–12 525 10.1073/pnas.0403539101 (doi:10.1073/pnas.0403539101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shemesh T., Geiger B., Bershadsky A. D., Kozlov M. M. 2005. Focal adhesions as mechanosensors: a physical mechanism. Proc. Natl Acad. Sci. USA 102, 12 383–12 388 10.1073/pnas.0500254102 (doi:10.1073/pnas.0500254102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bruinsma R. 2005. Theory of force regulation by nascent adhesion sites. Biophys. J. 89, 87–94 10.1529/biophysj.104.048280 (doi:10.1529/biophysj.104.048280) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deshpande V. S., McMeeking R. M., Evans A. G. 2006. A bio-chemo-mechanical model for cell contractility. Proc. Natl Acad. Sci. USA 103, 14 015–14 020 10.1073/pnas.0605837103 (doi:10.1073/pnas.0605837103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith A. S., Sengupta K., Goennenwein S., Seifert U., Sackmann E. 2008. Force-induced growth of adhesion domains is controlled by receptor mobility. Proc. Natl Acad. Sci. USA 105, 6906–6911 10.1073/pnas.0801706105 (doi:10.1073/pnas.0801706105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian J., Wang J., Gao H. 2008. Lifetime and strength of adhesive molecular bond clusters between elastic media. Langmuir 24, 1262–1270 10.1021/la702401b (doi:10.1021/la702401b) [DOI] [PubMed] [Google Scholar]
- 44.Qian J., Wang J., Lin Y., Gao H. 2009. Lifetime and strength of periodic bond clusters between elastic media under inclined loading. Biophys. J. 97, 2438–2445 10.1016/j.bpj.2009.08.027 (doi:10.1016/j.bpj.2009.08.027) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan C. E., Odde D. J. 2008. Traction dynamics of filopodia on compliant substrates. Science 322, 1687–1691 10.1126/science.1163595 (doi:10.1126/science.1163595) [DOI] [PubMed] [Google Scholar]
- 46.Walcott S., Sun S. X. 2010. A mechanical model of actin stress fiber formation and substrate elasticity sensing in adherent cells. Proc. Natl Acad. Sci. USA 107, 7757–7762 10.1073/pnas.0912739107 (doi:10.1073/pnas.0912739107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qian J., Gao H. 2010. Soft matrices suppress cooperative behaviors among receptor-ligand bonds in cell adhesion. PLoS ONE 5, e12342. 10.1371/journal.pone.0012342 (doi:10.1371/journal.pone.0012342) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen B., Gao H. 2010. Mechanical principle of enhancing cell–substrate adhesion via pre-tension in the cytoskeleton. Biophys. J. 98, 2154–2162 10.1016/j.bpj.2010.02.007 (doi:10.1016/j.bpj.2010.02.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leckband D., Israelachvili J. 2001. Intermolecular forces in biology. Q. Rev. Biophys. 34, 105–267 10.1017/S0033583501003687 (doi:10.1017/S0033583501003687) [DOI] [PubMed] [Google Scholar]
- 50.Alon R., Hammer D. A., Springer T. A. 1995. Lifetime of the P-selectin–carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature 374, 539–542 10.1038/374539a0 (doi:10.1038/374539a0) [DOI] [PubMed] [Google Scholar]
- 51.Florin E. L., Moy V. T., Gaub H. E. 1994. Adhesion forces between individual ligand–receptor pairs. Science 264, 415–417 10.1126/science.8153628 (doi:10.1126/science.8153628) [DOI] [PubMed] [Google Scholar]
- 52.Merkel R., Nassoy P., Leung A., Ritchie K., Evans E. 1999. Energy landscapes of receptor–ligand bonds explored with dynamic force spectroscopy. Nature 397, 50–53 10.1038/16219 (doi:10.1038/16219) [DOI] [PubMed] [Google Scholar]
- 53.Evans E., Ritchie K. 1997. Dynamic strength of molecular adhesion bonds. Biophys. J. 72, 1541–1555 10.1016/S0006-3495(97)78802-7 (doi:10.1016/S0006-3495(97)78802-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freund L. B. 2009. Characterizing the resistance generated by a molecular bond as it is forcibly separated. Proc. Natl Acad. Sci. USA 106, 8818–8823 10.1073/pnas.0903003106 (doi:10.1073/pnas.0903003106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans E. 2001. Probing the relation between force–lifetime–and chemistry in single molecular bonds. Ann. Rev. Biophys. Biomol. Struct. 30, 105–128 10.1146/annurev.biophys.30.1.105 (doi:10.1146/annurev.biophys.30.1.105) [DOI] [PubMed] [Google Scholar]
- 56.Evans E., Calderwood D. A. 2007. Forces and bond dynamics in cell adhesion. Science 316, 1148–1153 10.1126/science.1137592 (doi:10.1126/science.1137592) [DOI] [PubMed] [Google Scholar]
- 57.Erdmann T., Schwarz U. S. 2006. Bistability of cell–matrix adhesions resulting from nonlinear receptor-ligand dynamics. Biophys. J. 91, L60–L62 10.1529/biophysj.106.090209 (doi:10.1529/biophysj.106.090209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erdmann T., Schwarz U. S. 2007. Impact of receptor–ligand distance on adhesion cluster stability. Eur. Phys. J. E 22, 123–137 10.1140/epje/e2007-00019-8 (doi:10.1140/epje/e2007-00019-8) [DOI] [PubMed] [Google Scholar]
- 59.Seifert U. 2000. Rupture of multiple parallel molecular bonds under dynamic loading. Phys. Rev. Lett. 84, 2750–2753 10.1103/PhysRevLett.84.2750 (doi:10.1103/PhysRevLett.84.2750) [DOI] [PubMed] [Google Scholar]
- 60.van Kampen N. G. 1992. Stochastic processes in physics and chemistry. Amsterdam, The Netherlands: Elsevier Science [Google Scholar]
- 61.Goel N. S., Richter-Dyn N. 1974. Stochastic models in biology. New York, NY: Academic Press [Google Scholar]
- 62.Gillespie D. T. 1976. General method for numerically simulating stochastic time evolution of coupled chemical-reactions. J. Comput. Phys. 22, 403–434 10.1016/0021-9991(76)90041-3 (doi:10.1016/0021-9991(76)90041-3) [DOI] [Google Scholar]
- 63.Gillespie D. T. 1977. Exact stochastic simulation of coupled chemical-reactions. J. Phys. Chem. 81, 2340–2361 10.1021/j100540a008 (doi:10.1021/j100540a008) [DOI] [Google Scholar]
- 64.Lin Y., Freund L. B. 2008. Optimum size of a molecular bond cluster in adhesion. Phys. Rev. E 78, 021909. [DOI] [PubMed] [Google Scholar]
- 65.Johnson K. L. 1985. Contact mechanics. Cambridge, UK: Cambridge University Press [Google Scholar]
- 66.Rice J. R. 1968. In fracture: an advanced treatise (ed. Liebowitz H.), pp. 191–311 New York, NY: Academic Press [Google Scholar]
- 67.Dundurs J. 1969. Mathematical theory of dislocations. New York, NY: American Society of Mechanical Engineers [Google Scholar]
- 68.Aratyn-Schaus Y., Gardel M. L. 2010. Transient frictional slip between integrin and the ECM in focal adhesions under myosin II tension. Curr. Biol. 20, 1145–1153 10.1016/j.cub.2010.05.049 (doi:10.1016/j.cub.2010.05.049) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aratyn-Schaus Y., Oakes P. W., Gardel M. L. 2011. Dynamic and structural signatures of lamellar actomyosin force generation. Mol. Biol. Cell 22, 1330–1339 10.1091/mbc.E10-11-0891 (doi:10.1091/mbc.E10-11-0891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Macdonald A., Horwitz A. R., Lauffenburger D. A. 2008. Kinetic model for lamellipodal actin–integrin ‘clutch’ dynamics. Cell Adhes. Migration 2, 95–105 10.4161/cam.2.2.6210 (doi:10.4161/cam.2.2.6210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sabass B., Schwarz U. S. 2010. Modeling cytoskeletal flow over adhesion sites: competition between stochastic bond dynamics and intracellular relaxation. J. Phys.-Condensed Matter 22, 194112. 10.1088/0953-8984/22/19/194112 (doi:10.1088/0953-8984/22/19/194112) [DOI] [PubMed] [Google Scholar]
- 72.Li Y., Bhimalapuram P., Dinner A. R. 2010. Model for how retrograde actin flow regulates adhesion traction stresses. J. Phys. Condensed Matter 22, 194113. 10.1088/0953-8984/22/19/194113 (doi:10.1088/0953-8984/22/19/194113) [DOI] [PubMed] [Google Scholar]
- 73.Gardel M. L., Sabass B., Ji L., Danuser G., Schwarz U. S., Waterman C. M. 2008. Traction stress in focal adhesions correlates biphasically with actin retrograde flow speed. J. Cell Biol. 183, 999–1005 10.1083/jcb.200810060 (doi:10.1083/jcb.200810060) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hill A. V. 1938. The heat of shortening and dynamic constants of muscle. Proc. R. Soc. Lond. B 126, 136–195 10.1098/rspb.1938.0050 (doi:10.1098/rspb.1938.0050) [DOI] [Google Scholar]
- 75.Tada H., Paris P. C., Irwin G. R. 2000. The stress analysis of cracks handbook. New York, NY: American Society of Mechanical Engineers [Google Scholar]
- 76.Arnold M., Cavalcanti-Adam E. A., Glass R., Blummel J., Eck W., Kantlehner M., Kessler H., Spatz J. P. 2004. Activation of integrin function by nanopatterned adhesive interfaces. Chem. Phys. Chem. 5, 383–388 10.1002/cphc.200301014 (doi:10.1002/cphc.200301014) [DOI] [PubMed] [Google Scholar]
- 77.Johnson K. L., Kendall K., Roberts A. D. 1971. Surface energy and contact of elastic solids. Proc. R. Soc. Lond. A 324, 301–313 10.1098/rspa.1971.0141 (doi:10.1098/rspa.1971.0141) [DOI] [Google Scholar]
- 78.Gao H. J., Yao H. M. 2004. Shape insensitive optimal adhesion of nanoscale fibrillar structures. Proc. Natl Acad. Sci. USA 101, 7851–7856 10.1073/pnas.0400757101 (doi:10.1073/pnas.0400757101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Buehler M. J., Yao H. M., Gao H. J., Ji B. H. 2006. Cracking and adhesion at small scales: atomistic and continuum studies of flaw tolerant nanostructures. Modell. Simul. Mater. Sci. Eng. 14, 799–816 10.1088/0965-0393/14/5/001 (doi:10.1088/0965-0393/14/5/001) [DOI] [Google Scholar]
- 80.Gao H. J., Wang X., Yao H. M., Gorb S., Arzt E. 2005. Mechanics of hierarchical adhesion structures of geckos. Mech. Mater. 37, 275–285 10.1016/j.mechmat.2004.03.008 (doi:10.1016/j.mechmat.2004.03.008) [DOI] [Google Scholar]
- 81.Polte T. R., Eichler G. S., Wang N., Ingber D. E. 2004. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am. J. Physiol.-Cell Physiol. 286, C518–C528 10.1152/ajpcell.00280.2003 (doi:10.1152/ajpcell.00280.2003) [DOI] [PubMed] [Google Scholar]
- 82.Pourati J., Maniotis A., Spiegel D., Schaffer J. L., Butler J. P., Fredberg J. J., Ingber D. E., Stamenovic D., Wang N. 1998. Is cytoskeletal tension a major determinant of cell deformability in adherent endothelial cells? Am. J. Physiol.-Cell Physiol. 274, C1283–C1289 [DOI] [PubMed] [Google Scholar]
- 83.Marshall B. T., Long M., Piper J. W., Yago T., McEver R. P., Zhu C. 2003. Direct observation of catch bonds involving cell-adhesion molecules. Nature 423, 190–193 10.1038/nature01605 (doi:10.1038/nature01605) [DOI] [PubMed] [Google Scholar]
- 84.McEver R. P., Zhu C. 2010. Rolling cell adhesion. Ann. Rev. Cell Dev. Biol. 26, 363–396 10.1146/annurev.cellbio.042308.113238 (doi:10.1146/annurev.cellbio.042308.113238) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu C. 2000. Kinetics and mechanics of cell adhesion. J. Biomech. 33, 23–33 10.1016/S0021-9290(99)00163-3 (doi:10.1016/S0021-9290(99)00163-3) [DOI] [PubMed] [Google Scholar]
- 86.Zhu C., Bao G., Wang N. 2000. Cell mechanics: mechanical response, cell adhesion, and molecular deformation. Ann. Rev. Biomed. Eng. 2, 189–226 10.1146/annurev.bioeng.2.1.189 (doi:10.1146/annurev.bioeng.2.1.189) [DOI] [PubMed] [Google Scholar]
- 87.Chen B., Wu P. D., Gao H. J. 2009. Pre-tension generates strongly reversible adhesion of a spatula pad on substrate. J. R. Soc. Interface 6, 529–537 10.1098/rsif.2008.0322 (doi:10.1098/rsif.2008.0322) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kendall K. 1975. Thin-film peeling: elastic term. J. Phys. D-Appl. Phys. 8, 1449–1452 10.1088/0022-3727/8/13/005 (doi:10.1088/0022-3727/8/13/005) [DOI] [Google Scholar]
- 89.Chang K. C., Tees D. F. J., Hammer D. A. 2000. The state diagram for cell adhesion under flow: leukocyte rolling and firm adhesion. Proc. Natl Acad. Sci. USA 97, 11 262–11 267 10.1073/pnas.200240897 (doi:10.1073/pnas.200240897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schwarz U. S., Alon R. 2004. L-selectin-mediated leukocyte tethering in shear flow is controlled by multiple contacts and cytoskeletal anchorage facilitating fast rebinding events. Proc. Natl Acad. Sci. USA 101, 6940–6945 10.1073/pnas.0305822101 (doi:10.1073/pnas.0305822101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qi S. Y., Groves J. T., Chakraborty A. K. 2001. Synaptic pattern formation during cellular recognition. Proc. Natl Acad. Sci. USA 98, 6548–6553 10.1073/pnas.111536798 (doi:10.1073/pnas.111536798) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weikl T. R., Lipowsky R. 2004. Pattern formation during T-cell adhesion. Biophys. J. 87, 3665–3678 10.1529/biophysj.104.045609 (doi:10.1529/biophysj.104.045609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kanchanawong P., Shtengel G., Pasapera A. M., Ramko E. B., Davidson M. W., Hess H. F., Waterman C. M. 2010. Nanoscale architecture of integrin-based cell adhesions. Nature 468, 580–584 10.1038/nature09621 (doi:10.1038/nature09621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Legant W. R., Miller J. S., Blakely B. L., Cohen D. M., Genin G. M., Chen C. S. 2010. Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nat. Methods 7, 969–971 10.1038/nmeth.1531 (doi:10.1038/nmeth.1531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.del Rio A., Perez-Jimenez R., Liu R. C., Roca-Cusachs P., Fernandez J. M., Sheetz M. P. 2009. Stretching single talin rod molecules activates vinculin binding. Science 323, 638–641 10.1126/science.1162912 (doi:10.1126/science.1162912) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maskarinec S. A., Franck C., Tirrell D. A., Ravichandran G. 2009. Quantifying cellular traction forces in three dimensions. Proc. Natl Acad. Sci. USA 106, 22108–22113 10.1073/pnas.0904565106 (doi:10.1073/pnas.0904565106) [DOI] [PMC free article] [PubMed] [Google Scholar]