

The ice-binding protein from Leucosporidium sp. AY30 was cloned, expressed, purified and crystallized. A complete data set was collected to 1.5 Å resolution.
Keywords: freezing, ice-binding proteins, cold-adaptation, Antarctic yeast
Abstract
Freezing is dangerous to cellular organisms because it causes an increase in the concentration of ions and other solutes in the plasma, denatures biomolecules and ruptures cell membranes. Some cold-adapted organisms can survive at subzero temperatures by producing proteins that bind to and inhibit the growth of ice crystals. To better understand the structure and function of these proteins, the ice-binding protein from Leucosporidium sp. AY30 (LeIBP) was overexpressed, purified and crystallized. The native crystal belonged to space group P43212, with unit-cell parameters a = b = 98.05, c = 106.13 Å. Since LeIBP lacks any cysteine or methionine residues, two leucine residues (Leu69 and Leu155) were substituted by methionine residues in order to obtain selenomethionine-substituted LeIBP for use in multiple-wavelength anomalous diffraction (MAD) phasing. The selenomethionine-substituted mutant crystallized in the same space group as the native protein.
1. Introduction
Many organisms are able to protect themselves from freezing at subzero temperatures at which intracellular fluids would normally freeze. One strategy that organisms have evolved in order to tolerate subzero temperatures is the expression of antifreeze proteins (AFPs) and ice-binding proteins (IBPs). Both AFPs and IBPs are able to bind to ice, which makes further growth of ice energetically unfavourable, and have been discovered in many cold-adapted organisms (Duman, 2001 ▶; Raymond & DeVries, 1977 ▶). AFPs have been identified in bacteria, plants, invertebrates and fish (Duman, 2001 ▶; Duman & Olsen, 1993 ▶; Jia & Davies, 2002 ▶) and have been characterized according to their structures and thermal hysteresis (TH) values; the TH is the difference between the freezing and melting points (Duman & Olsen, 1993 ▶). Fish AFPs have been categorized into five types (AFP I–IV and AFGP) and insect AFPs have been grouped into three types (those with right-handed and left-handed β-helices and those containing a glycine-rich repeat; Jia & Davies, 2002 ▶).
IBPs have also been found in many organisms and tend to have very weak TH relative to AFPs (Janech et al., 2006 ▶; Raymond & Janech, 2009 ▶). All of these IBPs have no features in common with the numerous AFPs that have been identified to date in fish, insects, plants and bacteria, and are therefore divided into separate groups. IBPs have recently been identified in bacteria, diatoms and fungi that were classified into a distinct cluster in a phylogenetic tree. The first ice-active fungal protein (∼25 kDa) was found in the snow mould Typhula ishikariensis (Hoshino, Kiriaki, Ohgiya et al., 2003 ▶) and a protein which has a similar N-terminal sequence was identified in another snow mould, Corpinus psychromobidus (Hoshino, Kiriaki & Nakajima, 2003 ▶). Similar ice-binding proteins have been identified in sea-ice diatoms (Janech et al., 2006 ▶), a sea-ice bacterium (Raymond et al., 2007 ▶) and a bacterium found in the deep ice core of the Antarctic ice sheet (Raymond et al., 2008 ▶). It has been suggested that IBPs limit cell damage by preventing the recrystallization of extracellular ice (Raymond & Janech, 2009 ▶).
In a recent report, the first yeast IBP was identified from Leucosporidium sp. AY30 (LeIBP; Lee et al., 2010 ▶). LeIBP was reported to have one N-glycosylation site at residue Asn185. The deduced amino-acid sequence has 55 and 48% sequence identity to the ice-binding proteins from T. ishikariensis (a snow mould) and Navicula glaciei (a sea-ice diatom), respectively. However, in the phylogenetic analysis the yeast IBP was grouped separately from other fungal proteins as well as from diatom and bacterial IBPs. Furthermore, it had no sequence similarity to AFPs with available crystal structures and no three-dimensional structures of IBPs are available. These findings suggest that LeIBP has unique structural features compared with previously available AFPs and may bind to the ice lattice in a different manner.
To better understand the structure and function of yeast ice-binding proteins and how they interact with bound ice, we overexpressed and crystallized LeIBP and obtained preliminary X-ray crystallographic data as a first step towards protein structure determination.
2. Materials and methods
2.1. Cloning, overexpression and purification
The gene encoding the LeIBP protein was amplified from a Leucosporidium sp. complementary DNA library by polymerase chain reaction (PCR) using the forward primer 5′-CATATGCAGCGCGACCTCTTCGT-3′ and the reverse primer 3′-TCTAGATTAAAGCCACTGGCGGGC-5′ (NdeI and XbaI restriction sites are indicated in bold). The PCR product was then digested and ligated into a pcoldI vector (Takara, Otsu, Shiga, Japan). The resulting plasmid (LeIBP/pcoldI) was transformed into Escherichia coli strain BL21 (DE3) (Novagen, San Diego, California, USA) and the cells were grown at 310 K in Luria–Bertani medium supplemented with ampicillin (50 µg ml−1). The transformants were grown in LB medium at 310 K until they reached an optical density of 0.6 at 600 nm and expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 12 h at 288 K. Cells were then harvested by centrifugation at 5000g for 30 min at 277 K. The pelleted cells were suspended in buffer A (20 mM potassium phosphate pH 7.4, 500 mM NaCl, 5 mM imidazole) and lysed by sonication. The crude lysate was centrifuged at 20 000g for 1 h at 277 K. The supernatant was mixed with His-Bind agarose resin (Elpis, Daejeon, Republic of Korea) and unbound proteins were washed out with buffer B (20 mM potassium phosphate pH 7.4, 500 mM NaCl and 60 mM imidazole). The LeIBP protein was recovered by elution with buffer C (20 mM potassium phosphate pH 7.4, 200 mM NaCl and 500 mM imidazole). The protein was further purified to its final state by gel filtration on a HiLoad 26/60 Superdex 200 column (GE Healthcare, Schenectady, New York, USA) previously equilibrated with buffer D (20 mM Tris–HCl pH 7.9, 1 mM DTT). The purified samples were concentrated to 10 mg ml−1 using an Amicon Ultra-15 ultrafiltration device (Millipore, Decatur, Illinois, USA). This procedure yielded approximately 25 mg LeIBP protein from a 1 l culture. For multiple-wavelength anomalous dispersion (MAD) phasing, selenomethionine-substituted protein was prepared as follows. Two leucine residues at positions 69 and 155 of the LeIBP protein were substituted by methionine residues using PCR-mediated site-directed mutagenesis. The LeIBP/pcoldI plasmid was used as the template, with L69M (5′-CAG GTC ACC GGT ATG ATC TAC GCT GCA GAC-3′) and L155M (5′-CTG GAG GCC TCA GCC ATG GCC GAC GGT GTT-3′) as the forward primers and L69M (5′-GTC CAG TGG CCA TAC TAG ATG CGA CGT CTG-3′) and L165M (5′-GAC CTC CGG AGT CGG TAC CGG CTG CCA CAA-3′) as the reverse primers. The L69M and L155M LeIBP double-mutant construct was confirmed by DNA-sequence analysis. The selenomethionine-substituted LeIBP mutant protein was purified as follows. After cell lysis by sonication in buffer E (20 mM Tris–HCl pH 7.9, 500 mM NaCl, 5 mM imidazole), the crude lysate was centrifuged at 20 000g for 1 h at 277 K. The supernatant was loaded onto an Ni2+-chelating HiTrap chelating HP column (GE Healthcare, Schenectady, New York, USA) equilibrated with buffer E. The protein was eluted with a linear gradient of buffer E containing 1 M imidazole. The protein was purified to its final state by gel filtration on a HiLoad 16/60 Superdex 200 column (GE Healthcare, Schenectady, New York, USA) previously equilibrated with buffer F (20 mM Tris–HCl pH 7.9, 200 mM NaCl, 2 mM DTT). The purified samples were concentrated to 10 mg ml−1 using an Amicon Ultra-15 ultrafiltration device (Millipore, Decatur, Illinois, USA).
2.2. Crystallization and X-ray analysis
Crystallization trials were performed using the sitting-drop vapour-diffusion method (0.5 µl protein solution and 0.5 µl reservoir solution equilibrated against 50 µl reservoir solution) to set up 96-well Intelli-Plates (Art Robbins Instruments, Sunnyvale, California, USA) at 295 K. The initial screening was set up by hand using an eLINE electronic pipette (Biohit, Helsinki, Finland). Commercial screening kits such as Crystal Screen, Crystal Screen 2, MembFac, PEG/Ion, PEG/Ion 2, PEGRx, PEGRx 2, Index and Natrix (Hampton Research, Aliso Viejo, California, USA) and Wizard I and II (Emerald BioSystems, Bainbridge Island, Washington, USA) were used. About 600 conditions were screened to find the initial crystallization conditions. Large single native and selenomethionine-substituted crystals were grown from Wizard I condition No. 17 [0.1 M sodium acetate pH 4.5, 0.2 M lithium sulfate, 30%(w/v) PEG 8000] and Crystal Screen 2 condition No. 13 [0.1 M sodium acetate pH 4.6, 0.2 M ammonium sulfate, 30%(w/v) PEG MME 2000], respectively (Fig. 1 ▶). Crystal growth was scaled up to the hanging-drop vapour-diffusion method in 24-well VDX plates (Hampton Research, Aliso Viejo, California, USA) at 295 K. Each hanging drop was prepared by mixing 1 µl protein solution and 1 µl reservoir solution and was equilibrated against 500 µl reservoir solution. Crystals appeared within 2 d and grew to full size within two weeks; the refined crystallization conditions for the native and the selenomethionine-substituted crystals were 0.1 M sodium acetate pH 4.5, 0.18 M lithium sulfate, 25%(w/v) PEG 8000 and 0.1 M sodium acetate pH 4.5, 0.22 M ammonium sulfate, 31%(w/v) PEG MME 2000, respectively. For data collection, crystals were first cryoprotected using Paratone-N (Hampton Research, Aliso Viejo, California, USA) and then flash-cooled at 100 K in a liquid-nitrogen stream. Data sets were collected to a resolution of 1.5 Å from a native crystal on beamline 4A and to a resolution of 2.0 Å from a selenomethionine-substituted crystal on beamline 6C at the Pohang Light Source (Pohang, Republic of Korea) at 100 K. A total of 360 images were collected with an oscillation angle of 1°. The following multiple-wavelength anomalous diffraction (MAD) data sets were collected: peak (0.97935 Å), inflection (0.97958 Å) and high-energy remote (0.97168 Å) from the selenium absorption edge. The data sets were indexed, processed and scaled using the HKL-2000 software package (Otwinowski & Minor, 1997 ▶).
Figure 1.

Crystals of the LeIBP protein from Leucosporidium sp. AY30. (a) Native crystal, (b) selenomethionine-substituted crystal. The crystal dimensions of the native and selenomethionine-substituted crystals were 0.3 × 0.2 × 0.1 and 0.2 × 0.4 × 0.15 mm, respectively.
3. Results and discussion
The gene encoding the LeIBP protein from Leucosporidium sp. was cloned. The protein was overexpressed in E. coli and purified for structural studies. The molecular weight of LeIBP was calculated to be 24.9 kDa, with 261 amino acids. The native crystal belonged to the tetragonal space group P43212, with unit-cell parameters a = b = 98.05, c = 106.13 Å. The selenomethionine-substituted crystal also belonged to space group P43212, with unit-cell parameters a = b = 97.77, c = 106.33 Å (Fig. 1 ▶). The data-collection statistics are summarized in Table 1 ▶. The V M value of the native crystal was 2.55 Å3 Da−1 (Matthews, 1968 ▶), which suggested that the crystal was most likely to contain two molecules per asymmetric unit, with a solvent content of 51.81%. For MAD phasing, we prepared crystals of a double mutant containing two selenomethionine residues. Two leucine residues at positions 69 and 155 were substituted by methionine and selenomethionine-substituted protein was overexpressed, purified and crystallized. Attempts to solve the selenium substructure using the MAD data sets did not lead to good phases. Using the peak data only, we performed single-wavelength anomalous diffraction (SAD) phasing. The location of the Se-atom positions, initial phase calculations, phase improvement by density modification and initial map calculations were performed using SOLVE/RESOLVE (Terwilliger & Berendzen, 1999 ▶; Terwilliger, 2000 ▶). A total of four selenium sites in the asymmetric unit were determined and the overall figure of merit (FOM) was 0.65; 394 residues (81%) out of a total of 484 residues were placed automatically. The final model building and crystallographic refinement are now in progress.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| SeMet-labelled | ||||
|---|---|---|---|---|
| Native | Peak | Inflection | Remote | |
| Space group | P43212 | P43212 | ||
| Wavelength (Å) | 1.0000 | 0.97935 | 0.97958 | 0.97168 |
| Unit-cell parameters (Å) | a = b = 98.05, c = 106.13 | a = b = 97.77, c = 106.33 | a = b = 97.76, c = 106.32 | a = b = 97.77, c = 106.33 |
| Resolution range (Å) | 50–1.5 (1.55–1.50) | 50–2.0 (2.07–2.00) | 50–2.0 (2.07–2.00) | 50–2.0 (2.07–2.00) |
| Total reflections | 1993499 | 1037163 | 1024513 | 1008845 |
| Unique reflections | 80774 | 35632 | 35509 | 35665 |
| Multiplicity | 24.7 (20.2) | 29.1 (29.1) | 28.9 (28.1) | 28.3 (24.3) |
| Completeness (%) | 97.0 (70.8) | 100 (100) | 100 (100) | 100 (100) |
| Rmerge† (%) | 5.5 (22.2) | 12.9 (41.6) | 13.3 (49.3) | 15.1 (56.2) |
| 〈I/σ(I)〉 | 92.7 (12.5) | 26.3 (6.7) | 24.8 (5.6) | 17.7 (5.0) |
| Anomalous statistics | ||||
| Completeness (%) | 100 (100) | 100 (100) | 100 (100) | |
| Multiplicity | 15.1 (14.3) | 15.1 (14.3) | 15.1 (14.3) | |
| Signal-to-noise ratio | 1.47 (0.27) | 0.74 (0.00) | 0.77 (0.18) | |
R
merge = 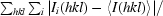
 , where I
i(hkl) represents the observed intensity, 〈I(hkl)〉 represents the average intensity and i counts through all symmetry-related reflections.
, where I
i(hkl) represents the observed intensity, 〈I(hkl)〉 represents the average intensity and i counts through all symmetry-related reflections.
Acknowledgments
We thank the staff at beamlines 4A and 6C of Pohang Light Source, Republic of Korea for assistance during data collection. This work was supported by a Korea University Grant and by KOPRI grant Nos. PE11100 and PG10010 (HJK).
References
- Duman, J. G. (2001). Annu. Rev. Physiol. 63, 327–357. [DOI] [PubMed]
- Duman, J. G. & Olsen, T. M. (1993). Cryobiology, 30, 322–328.
- Hoshino, T., Kiriaki, M. & Nakajima, T. (2003). Cryo Letters, 24, 135–142. [PubMed]
- Hoshino, T., Kiriaki, M., Ohgiya, S., Fujiwara, M., Kondo, H., Nishimiya, Y., Yumoto, I. & Tsuda, S. (2003). Can. J. Bot. 81, 1175–1181.
- Janech, M. G., Krell, A., Mock, T., Kang, J.-S. & Raymond, J. A. (2006). J. Phycol. 42, 410–416.
- Jia, Z. & Davies, P. L. (2002). Trends Biochem. Sci. 27, 101–106. [DOI] [PubMed]
- Lee, J. K., Park, K. S., Park, S., Park, H., Song, Y. H., Kang, S.-H. & Kim, H. J. (2010). Cryobiology, 60, 222–228. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Raymond, J. A., Christner, B. C. & Schuster, S. C. (2008). Extremophiles, 12, 713–717. [DOI] [PubMed]
- Raymond, J. A. & DeVries, A. L. (1977). Proc. Natl Acad. Sci. USA, 74, 2589–2593. [DOI] [PMC free article] [PubMed]
- Raymond, J. A., Fritsen, C. & Shen, K. (2007). FEMS Microbiol. Ecol. 61, 214–221. [DOI] [PubMed]
- Raymond, J. A. & Janech, M. G. (2009). Cryobiology, 58, 151–156. [DOI] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]