
Figure 1.
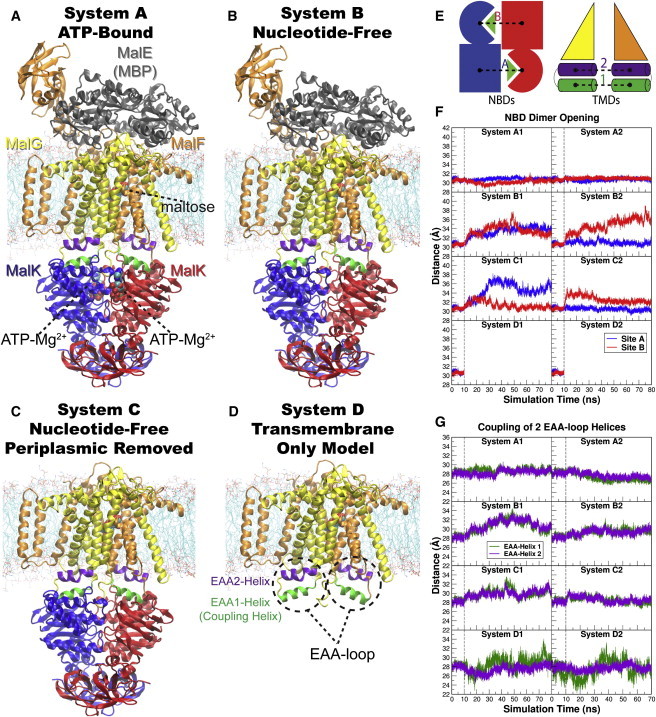
Overview of the equilibrium simulations. (A–D) Initial structure of the four simulation systems, showing proteins (in ribbons), bound substrates (including maltose and MgATP, shown as van der Waals spheres and labeled in panel A), and lipids (in line representations). The color scheme for this and all following figures is: MalK monomers in blue and red, MalF in orange, MalG in yellow, MalE in dark gray (labeled in panel A); the two EAA1 (the coupling helices) and EAA2 helices are highlighted in green and purple, respectively (labeled in panel D). (E) Schematic representation of the distances measured to quantify the NBD opening and the separation of the EAA helix pairs for panels F and G. Left: the NBD opening in each simulation system is measured as the center-of-mass distance between the helical subdomain of one MalK monomer (P88–E151, squares) and the RecA-like subdomain of the opposite MalK monomer (A2–Y87 and P152–G235, pac-man shaped); the bound nucleotides are shown as green triangles occupying the binding sites A and B. Right: the separation of the two sets of EAA helices are measured as the distances between their centers of masses (MalF:P396–G407 with MalG:D185–G196 as set 1, and MalF:F411–L422 with MalG:W200–S211 as set 2). (F) The conformational changes in the NBDs of each simulation system measured by the degree of NBD dimer opening. (G) The conformational changes of the EAA loops of the TMDs, measured as the separation of the two EAA helix pairs. The vertical dashed lines at 10 ns in panels F and G denote the time point at which all the simulations were branched out from their common parent equilibration simulation system.