
Table 3. Memory and CPU requirements.
| Memory (GB) | CPU time (Hours) | ||||
| Method | Phenotype | Null | Real | Null | Real |
| GWiS | PR | 1.2 | 1.2 | 9.4 | 43.1 |
| QRS | 1.2 | 1.2 | 11.0 | 31.9 | |
| QT | 1.2 | 1.2 | 11.2 | 67.0 | |
| minSNP | PR | 0.6 | 0.6 | 13.6 | 62.0 |
| QRS | 0.6 | 0.6 | 15.8 | 45.9 | |
| QT | 0.6 | 0.6 | 16.1 | 96.4 | |
| minSNP-P | PR | 0.6 | 0.6 | 11.9 | 54.2 |
| QRS | 0.6 | 0.6 | 13.8 | 40.1 | |
| QT | 0.6 | 0.6 | 14.0 | 84.3 | |
| BIMBAM | PR | 0.6 | 0.6 | 14.1 | 42.3 |
| QRS | 0.6 | 0.6 | 16.5 | 33.2 | |
| QT | 0.6 | 0.6 | 16.8 | 101.5 | |
| VEGAS | PR | 32.5 | 8.2 | 26.0 | 34.0 |
| QRS | 26.0 | 11.9 | 23.9 | 29.8 | |
| QT | 25.8 | 14.1 | 27.1 | 33.0 | |
| LASSO | PR | 0.1 | 0.1 | 0.2 | 0.4 |
| QRS | 0.1 | 0.1 | 0.3 | 0.3 | |
| QT | 0.1 | 0.1 | 0.2 | 0.4 | |
The minimal memory requirement and the total CPU time to finish one genome-wide study are reported for both a null (shuffled) trait and the real trait. Benchmarks were obtained from AMD Operon 2.3GHz or similar processors. The memory and CPU requirements include the model selection and the calculation of the gene-based p-values (or selection index). Costs for the genome-wide permutations to establish genome-wide significance thresholds are not included in the estimates. LASSO consumes the least resources because it pre-filters the SNPs (only uses SNPs having p-values 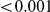 ) and does not require permutations to calculate the selection index. The real phenotypes require more CPU time because more permutations are required to calculate genome-wide significant p-values for true associations.
) and does not require permutations to calculate the selection index. The real phenotypes require more CPU time because more permutations are required to calculate genome-wide significant p-values for true associations.