

Abstract
Background:
We propose that a computerized, internet-based graphical description language for systems biology will be essential for describing, archiving and analyzing complex problems of biological function in health and disease.
Results:
We outline here a conceptual basis for designing such a language and describe BioD, a prototype language that we have used to explore the utility and feasibility of this approach to functional biology. Using example models, we demonstrate that a rather limited lexicon of icons and arrows suffices to describe complex cell-biological systems as discrete models that can be posted and linked on the internet.
Conclusions:
Given available computer and internet technology, BioD may be implemented as an extensible, multidisciplinary language that can be used to archive functional systems knowledge and be extended to support both qualitative and quantitative functional analysis.
Background
Standard graphical representations of complex systems have been developed for many disciplines in order to communicate, archive and analyze systems knowledge. Electronic circuit diagrams and architectural plans, for instance, can be created, read and analyzed by knowledgeable persons, yet there is no such common graphical language for describing functional systems in biology. A variety of representations are used in print or online to archive knowledge in particular domains such as metabolic pathways [1,2], gene networks [3], signaling networks [4,5], and molecular interactions [6,7], yet are not sufficiently standardized to represent cross-disciplinary systems - the interactions of gene expression and metabolism at the inter- and intra-molecular levels, for instance. To describe such multidisciplinary biological problems at multiple levels of abstraction (for example, from intramolecular to disease phenotype), biologists customarily resort to informal cartoon diagrams which, although expressive, are often ambiguous and must be annotated to be interpreted properly.
Here we suggest that a standardized visual biological description language would provide more readable and less ambiguous communication and, with computational implementation, provide a basis for distributed searchable archives of functional (as opposed to structural) knowledge, and serve as a 'computer-aided design' (CAD) language for simulating and analyzing biological systems. To explore these possibilities, we are developing a prototype biological description language, BioD, as a platform to test its conceptual basis, explore its utility and identify key issues surrounding its implementation.
Results and discussion
Functional properties: a basis for a biological description language
If the goal were simply to standardize the graphics of cartoon diagrams, one could collect and standardize sets of icons and arrows for use as 'clip art' in general-purpose graphics software or as templates in diagramming software such as Visio™ (Microsoft™; for Windows™) or TopDown™ (Kaetron™ Software; for MacOS™ and Windows™). To build a sophisticated software editor for a visual language and, especially, to design the language to support computer archiving and analysis, one must, however, establish deeper foundations for creating, organizing and using graphic elements.
As a starting point, consider the common metaphor of biological systems as 'circuits'. Indeed, viewing metabolic pathways as electrical circuits is particularly apt as, in each case, a limited set of entity classes (metabolites versus resistors, capacitors and inductors) are represented by icons that are linked by lines (or arrows) representing interactions between iconified entities. The benefit of such pathway and circuit diagrams is, of course, that they help us infer and track how a change in a 'state property' (for example, metabolite concentration versus electrical voltage) of one icon acts via an 'action property' (metabolic flux rate versus electrical current) on the state properties of linked icons. The problem for biologists is that metabolite concentrations and flux rates, while absolutely fundamental to many biological systems, are not the only functional properties that need to be considered.
Just as architectural plans must display and distinguish several interacting circuit systems with different properties (such as power, water, drainage, ventilation, telephone, security, and digital cable), diagrams of multidisciplinary biological systems must distinguish multiple functions (metabolic pathways, enzyme kinetics, ion fluxes, fluid flow) characterized by different properties (concentration, catalytic activity, voltage, pressure, respectively). Thus, the key challenge for describing, archiving and analyzing complex biological systems is not simply to collect expressive icons and arrows but to establish a system of expressive icons and arrows that consistently and unambiguously represents the functional entities and their specific functional properties. Does an arrow increase (or decrease) the concentration or the catalytic activity of an enzyme icon? Can we consistently represent the separate effects of transmembrane ion flow on both ion concentration and transmembrane voltage?
Whereas state and action properties are generally implicit in most diagrams, computer implementation of a visual language requires that language elements be formalized as computational objects in order to edit, archive and analyze models. To this end, we began designing BioD by creating building-block icons (Figure 1) that represent primitive functional entities such as atoms, molecules, compartments and so on, where each icon is understood implicitly (as in metabolic pathway and electrical circuit diagrams), but specifically, to possess one or more state properties. For instance, 'atom' and 'molecule' icons each have a 'concentration' property. A 'compartment' icon has 'volume' and (surface) 'area' properties. A 'membrane-bound compartment' icon, derived from a simple compartment, is used to represent cells and organelles and inherits the compartment's volume and area properties while including a (transmembrane) 'voltage' property. In this manner, an extensible hierarchical lexicon of building-block icons can be built by deriving new icons from existing icons. Furthermore, additional state properties (such as temperature, ionic charge, isotopy, pressure) can be created and appended to new or existing icons as needed to create new functional entities; an 'ion' icon is an atom icon with an ionic charge property, for instance.
Figure 1.
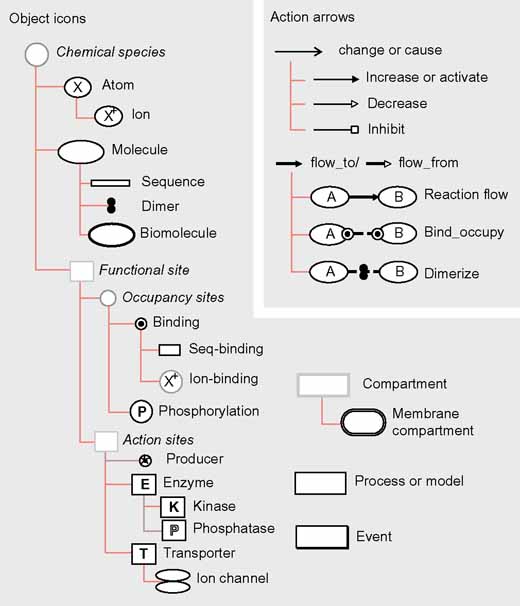
A basic lexicon of icons and arrows for describing the function of complex biological systems using BioD. Object icons are provided for common biological structures such as atoms, molecules, molecular sites and compartments, and for events and processes. 'Action' arrows represent the actions that the functional properties of one object can have on the functional properties of other objects and actions.
In addition to creating new icons by derivation, compound icons representing complex structures can be built by aggregating icons according to common-sense structural rules (for example, a 'molecular site', but not a cell, can be part of a molecule). In the examples below, a simple model of the cell-cycle kinase Cdk4 begins as a molecule icon to which is appended a 'kinase site' (a square with a K), a 'phosphorylation site' (a circle with a P) and a 'binding site' (a small circle with a dot) (Figure 2a). As another example, the membrane-bound compartment representing a cell could contain other compartments representing internal organelles or even compartments defined solely by diffusion-limitations (for example a submembrane compartment).
Figure 2.
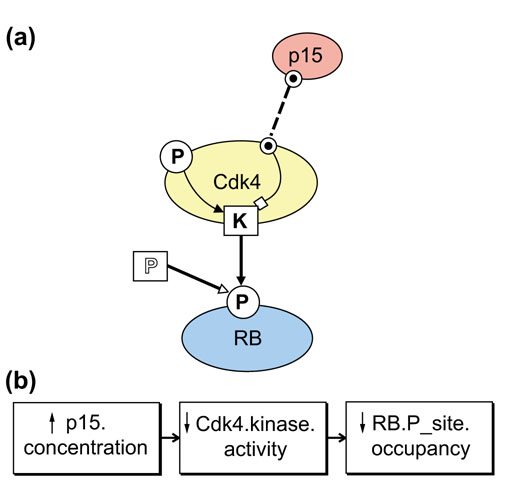
(a) A simplified model of a Cdk4 kinase molecule illustrates how basic BioD icons and action arrows can concisely represent intra- and inter-molecular actions. The Cdk4 molecule includes a kinase site (K) that, when active, phosphorylates a phosphorylation site (P) on the RB protein. The kinase site on Cdk4 is activated (filled arrow) by occupancy of its phosphorylation site and inhibited (open-squared arrow) by occupancy of the binding site (dotted circle) that binds the Cdk4 inhibitor p15. (b) An 'event model' derived from the model above. Events are defined as changes of state of one or more functional properties of icons in a state model. Here, for instance, the event model displays a chain of events triggered by an increase of p15 concentration (see text).
Functional interactions between building-block icons, whether aggregated as compound icons or not, are represented by 'action' arrows (Figure 1, upper right panel) that are designed to minimize the functional ambiguity of informal cartoon diagrams. It is required, for instance, that arrows consistently distinguish: which (if there is more than one) of an icon's functional properties are affected either by context (as in cartoons) or by appearance (for example, double-weight arrows change concentration properties, by chemical reactions or transport, whereas single-weight arrows activate or inhibit activity properties); and how the property is affected (for example, filled heads mean 'increase', open heads mean 'decrease'). As for the lexicon of icons, the lexicon of action arrows in Figure 1 could be extended to include other forms to represent, say, thermal fluxes (which change temperature properties) and fluid flows (which change compartmental volume and pressure properties).
In the Cdk4 kinase model of Figure 2 we use single-weight arrows to represent how the activity of the kinase site is inhibited by occupancy of the p15 binding site and is activated by occupancy of the phosphorylation site. A double-weight, solid-headed 'phosphorylate' arrow represents the transfer of phosphate groups by the kinase site of Cdk4 to increase occupancy of the retinoblastoma protein (RB) phosphorylation site. A phosphatase molecular site (a square with open-style P) represents how an unspecified phosphatase activity reduces the occupancy of the RB phosphorylation site. A dashed, double-weight 'occupy' arrow represents the binding to and increased occupancy of p15 to its binding site on Cdk4. In this manner the diagram can represent both intra- and inter-molecular site-site interactions while distinguishing activation/inhibition (as by conformational changes) from chemical reactions between species.
Consistent coding of qualitative effects as 'increase' or 'decrease' arrows allows qualitative 'what if experiments where an initiating 'event' - an increase or decrease of a functional property of one icon, say - can be tracked through a model (or a network of models, see below) as a chain of events. Such events chains can be displayed in 'event diagrams' to represent some events (but not necessarily all events) as chains of 'event' icons (each representing a change of a specific functional property) linked by 'cause' arrows. For instance, the event diagram in Figure 2b displays the consequences (as events icons) of increasing the concentration of p15 in the model in Figure 2a. In complex systems, such consistent qualitative representations can reveal previously unappreciated causal links - redundant parallel pathways or feedback loops - in models built solely from pairwise interactions of many functional parts.
As we will discuss below, formalizing a functional descriptive language for biology can offer additional benefits for exploring and understanding the function of complex biological systems if the functional structures and processes can be adequately and unambiguously described.
Extending BioD to other domains
A major criterion for the utility of any technical language is whether it can fully express concepts within a domain of knowledge; that is, is the language 'complete'? For a domain such as biology which is almost limitless in terms of physical (not to mention psychic, social and evolutionary) phenomena, it is unlikely that any single language will suffice for all purposes - nor could such a language be proved complete by some inductive process. Rather, in our approach to designing and developing BioD, we have begun with basic cell-biological models as test beds for BioD's conceptual framework (that is, of functional properties, icons and arrows), its lexicon and its icon-arrow linking rules (tantamount to a 'graphical grammar'). We have iteratively tested and modified the language to describe an expanding array of biological functions that now includes online models of: the cell-cycle G1/S checkpoint machinery as it may be controlled by ion channel activation [8]; and the metabolic, signaling and neuronal pathways by which the ketogenic diet may affect seizure susceptibility [9]. In this early phase of design we expect modifications in basic as well as superficial features (as for any software design process), but we also expect that the basic precepts of the functional-property approach can be extended to other phenomenological domains such as fluid flow, heat exchange, neural circuitry, and so forth. To demonstrate the progress of language development within the cell-biological domain, we will describe and discuss the G1/S checkpoint control model.
Extending the lexicon to describe G1/S checkpoint machinery
With a few additional icons and arrows, the complex molecular signaling network that includes Cdk4 and controls the G1/S transition can be described compactly (Figure 3). First, a simple rectangle is used to represent a 'process' that may be, but need not be, specified by a separate model (see below). Second, we use an arrow with a starred circle as a tail to mean 'produces' to correspond to commonly used shorthand for saying that a commodity is produced where the specific mechanism of production is either unknown or not germane to the model. Figure 3 can then represent a very complex network of intramolecular and intermolecular actions controlling the G1/S cell-cycle transition [6,10,11,12,13,14], including Cdk2, cyclins A and E, the inhibitor proteins p21 and p27 and inputs from transforming growth factor-β (TGF-β) stimulation and DNA damage processes.
Figure 3.
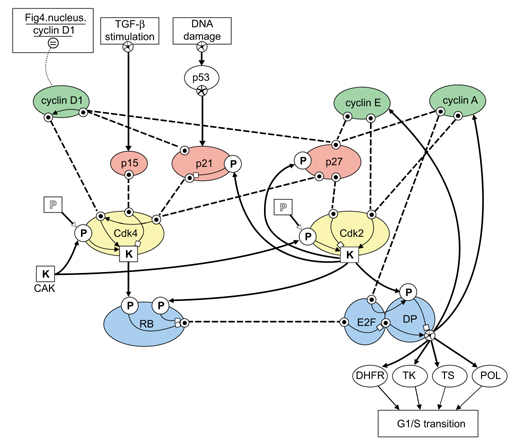
A BioD model of structures and functional elements controlling the G1/S transition of the cell cycle [6,10,11,12,13,14]. Key molecular players are represented by binding sites, phosphorylation sites and kinase sites whose occupancies and activities interact to control the cell cycle. The critical synthesis and degradation kinetics of cyclin D1 are represented by an 'identity' link to the nuclear cyclin D1 icon in the 'process' modeled in Figure 4. Additional inputs to the model (TGF-β stimulation and DNA damage) and outputs (G1/S transition) are included as unmodeled processes. CAK, cyclin-activating kinase; DP, DRTF1-polypeptide; DRTF, differentiation-regulated transcription factor; E2F, E2F transcription factor; TK, thymidine kinase; TS,thymidylate synthetase; POL, DNA polymerase.
Extensions for compartmental transport, gene expression and membrane biophysics
BioD's iconography can be extended to accommodate additional cell-biological phenomena. For instance, the model of Figure 4 introduces 'membrane-bound compartments' in order to model the dependence of the nuclear concentration of cyclin D1 on the complex kinetics of its synthesis, degradation and cytoplasmic-nuclear transport. The figure also introduces two derivatives of the 'molecule' icon: a 'dimer' icon (adapted from Kohn [6]) to represent dimers such as cyclin D1-Cdk4 and a 'biomolecule' icon as a shorthand to represent catalytically active molecules with both 'concentration' and 'activity' properties (for example the kinases in the mitogen-to-ERK and -Akt kinase cascades). The 'dimerization' action arrow (dashed, double-weight) is derived from the 'bind occupy' arrow to link to species participating in the dimerization reaction.
Figure 4.
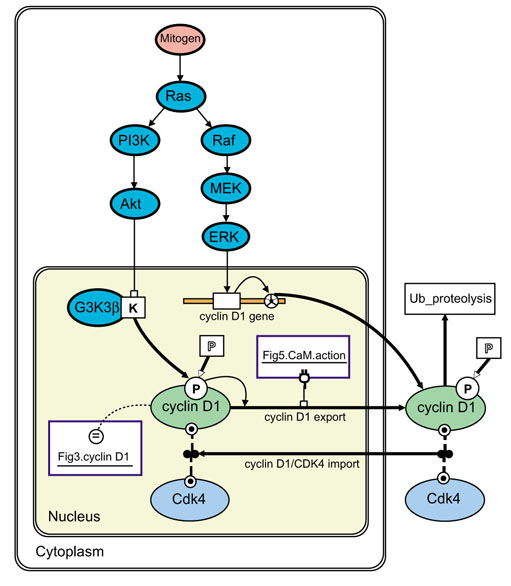
The activation of cyclin D1 gene expression by parallel Ras kinase cascades [44] modeled with BioD. Nuclear cyclin D1 levels depend on cytoplasmic-nuclear transport [45] and cytoplasmic degradation of cyclin D1 via an unmodeled ubiquitin proteolysis ('Ub proteolysis') process. The cyclin D1 icon in this model is identified with the cyclin D1 icon in Figure 3 using an identity link (Fig 3.cyclin D1; which is reciprocal to the Fig 4.nucleus.cyclin D1 link in Figure 3). The inhibition of nuclear export of cyclin D1 is inhibited by an 'action' link (Fig 5.CaM.action) that allows activated calmodulin (CaM) in the model of Figure 5 to reach into and affect this model. ERK, extracellular-regulated kinase; G3K3β, glycogen-3-synthase kinase-3β; MEK, MAP/ERK kinase; PI3K, phosphatidylinositol trisphosphate kinase.
To describe the possible effects of membrane potential and ion fluxes on cellular activity in Figure 5, we have introduced an electrical 'voltage' property (represented by the 'depol' (depolarization) process icon) of membrane-bound compartments and an ionic 'charge' property used to derive 'ion' icons from atom icons. Ion channel function is modeled as a 'transporter'-derivative 'ion channel' that is used to represent the K+ and Ca2+ ion transport pathways as parts of ATP-sensitive potassium channels (KATP) and calcium channels, respectively. A 'depol' process icon is appended to the cytoplasmic compartment's membrane to represent how depolarization is decreased by K+ efflux, increased by Ca2+ influx, and further activates voltage-dependent Ca2+ channels. With these additions, we can represent the hypothetical action of 'K-channel blockers' that bind to and inhibit ATP-sensitive potassium channels so that increased intracellular Ca2+ activates intranuclear calmodulin.
Figure 5.
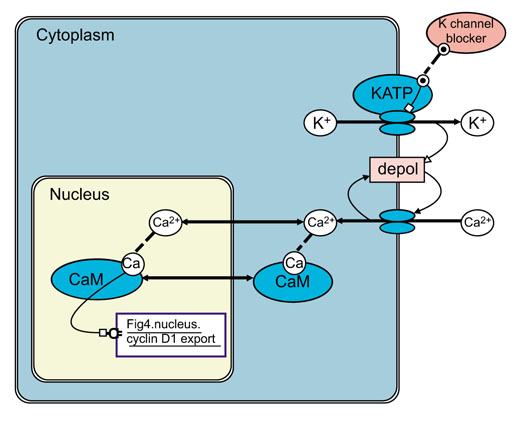
A model of the activation of calmodulin (CaM) by calcium influx in which K-channel blockers [46,47,48] inhibit ATP-sensitive potassium channels (KATP) and thus cause membrane depolarization (depol). Membrane depolarization activates voltage-dependent calcium 'ion channel' transporters and Ca2+ influx which have the dual effects of increasing intracellular Ca2+ concentration and further increasing depolarization. The action of Ca2+-activated CaM is linked via an action link (Fig 4.nucleus.cyclinD1.export) to the inhibition of cyclin D1 export in Figure 4.
The lexicon and example models presented so far have focused primarily on cell signaling and transduction pathways where intramolecular site-site interactions (for example conformational regulation of kinase activity) are prominent. However, intermolecular steric interactions figure just as prominently in gene-regulatory systems, as exemplified in the classic problem of the lysis-lysogeny 'genetic switch' of lambda phage [15]. This system is modeled in Figure 6 to illustrate how activation and inhibition arrows can be adapted to represent both steric hindrance and mutual cooperativity of binding. For instance, steric hindrance between the binding of the transcription factor Cro to its operator (Cro-OR1) and RNA polymerase (RNAP) to the promoter PR (RNAP-PR) is represented by a double-headed 'inhibition' arrow linking the respective binding-reaction arrows. The positive cooperativity of the binding reactions CI-OR2, RNAP-PRM and CI-RNAP is represented by three pairs of double-tailed 'activation' arrows that each link two binding reactions to a third reaction that is enhanced by the two previous binding events. The cooperativity being mutual, the three sets of double-headed arrows are superimposed to form a triangle of arrows linking the three binding arrows. Similar cooperativity in the formation of CI-CI and CI-OR1 bonds with the CI-OR2 bond further stabilizes the association of RNAP with PRM (see Figure 6 legend for further discussion).
Figure 6.
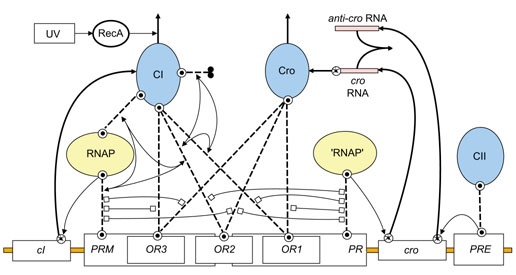
The use of 'inhibit' and 'activate' arrows to represent steric hindrance and cooperative binding reactions in a model of the lysis-lysogeny decision network of phage lambda [15]. A single-molecule icon is used to represent transcription factors that are actually homodimers of CI, Cro and CII. Key pathways for the synthesis and degradation of CI and Cro are as follows. Binding of RNA polymerase (RNAP) to the PRM promoter activates production of CI via the cI gene. The binding of 'RNAP' (the quotation marks signify that the icon is a duplicate of the RNAP icon and is not a separate entity) to promoter PR activates production of cro RNA via the cro gene. Binding of CII to promoter PRE at the opposite end of the cro gene activates production of antisense cro RNA ('anti-cro'). cro RNA required to produce Cro protein is degraded and removed from the system by a quenching reaction with anti-cro. The degradation of CI (the vertical reaction arrow at the top of the CI icon) depends on the proteolytic enzyme RecA, which is activated by ultraviolet light (UV). Cro degradation is unregulated, and CII degradation is not represented. The diagram is laid out to emphasize the symmetry between the CI and Cro synthesis and degradation pathways. The symmetry is broken, however, when one considers how competitive binding of the transcription factors (CI and Cro) to the operators (OR1, OR2 and OR3) controls access of RNA polymerase to the back-to-back promoters PRM and PR. Transcription repression by CI and Cro is nearly symmetric as they each limit access (by steric hindrance; shown as double-headed 'inhibit' arrows) of RNA polymerase to the promoters. RNA polymerase access to PR is limited in four instances by CI or Cro binding to either OR1 or OR2, whereas access to PRM is limited in three instances by Cro binding to either OR2 or OR3, or by CI binding to OR3. The break in symmetry occurs because, rather than being repressive, CI binding to OR2 actually enhances PRM transcription, which is stabilized in two ways. First, CI bound to OR3 directly binds RNA polymerase to stabilize its binding at its promoter site. Second, CI stabilizes its own binding to OR2 by establishing a homodimerization bond (the dimerization arrow extending to the right of the CI icon) with a CI molecule bound to OR1. The result is two trimers (CI-gene-RNAP and CI-gene-CI) within which the ability of pairs of bonds to stabilize a third bond is indicated by a double-tailed activation arrow. As such bond stabilization is mutually cooperative, the three double-tailed arrows are superimposed to form the triadic arrows shown in the figure.
The example models in Figures 3,4,5,6 demonstrate that a functional description language can be extended by defining new functional properties, associating the properties with existing or new icons, and using the new icons to represent a wide variety of biological phenomena. In creating the BioD lexicon and drawing conventions we have opted for simple icons and arrows (more elaborate and expressive graphics can readily be imagined) so that diagrams can be easily drawn by hand or with a variety of general purpose computer graphics applications. Creating and editing models is greatly facilitated, however, by applications such as TopDown™ (as used here) or Visio™ that provide 'templates' of reuseable icons and arrows and that also maintain icon-icon arrow links as icons are moved about in the field of the diagram. Even at this stage of development, BioD diagrams created by these general graphic and diagramming tools can be readily imported to available web page design tools to create, edit, link and post models on the internet (see sample BioD model sites [8,9]).
Design considerations for a BioD editor
To fully realize the advantages of a standardized visual language, however, custom editors will be required for creating new icon and arrow objects, for maintaining icon and arrow lexicons, and for creating and editing models. The key first step in developing editors (as well as archiving and analysis software, see below) is to develop standardized, platform-independent computer representations of core BioD language elements (functional properties, icons, arrows and models) in object-oriented programming (OOP) terms so that editors and browsers can be developed on a number of platforms.
In OOP parlance, for instance, BioD icons and arrow objects would 'know' how to draw themselves, 'inherit' functional and graphical properties from 'parent' icons and arrows, and know how to link to each other. The tasks of a BioD editor, as we envisage, would be to create icons and arrows, organize them into lexicons and palettes, and allow users to draw and link them into models. A particularly attractive, but considerably more sophisticated, feature would allow models to be built and formatted automatically according to pairwise functional interactions as available in metabolic pathway, gene network and protein interaction databases.
Model-model links for archiving and searching system models
A standardized biological description language that is formalized in terms of computational and not just graphical objects offers two very attractive benefits. First, it would be possible to build models of very large systems by linking individual submodels into distributed model networks on the internet. Second, it would be possible to treat such model networks as searchable archives of functional knowledge that can be queried for specific functional relationships.
The first possibility can be illustrated by linking models (see Figures 3,4,5) into networks using only two kinds of model-model links: 'action' and 'identity' links. Action links allow a functional property of an icon in one model to act on a functional property of an icon or action in another model, as if in a single model. For instance, an action link (iconified as a wall plug) allows the calcium-dependent activation of calmodulin in Figure 4 to inhibit the export of cyclin D1 from the nucleus in Figure 3. Identity links, on the other hand, allow models to share a common icon and its properties. For instance, an identity link (iconified as an = sign) establishes that the cyclin D1 concentration in Figure 2 represents the same cyclin D1 concentration as in the nucleus in Figure 3. Additional examples of model-model links can be seen in a network of models created to explain how the ketogenic diet might affect seizure activity [9].
The second possibility is to establish online models as searchable archives of functional, as opposed to structural, relationships. Just as modern bioinformatics provides immense databases and search tools for molecular structures and polymer sequences [16], we envisage that with suitable computational implementation, distributed online models may be queried not only for local pairwise interactions (such as site-site interactions) but also for long-distance chains of events (for example 'Find models where a potassium channel blocker affects cell-cycle progression'), a capability not available in any other approach to biological systems. Implementing this capability would depend, first, on encoding and storing BioD language elements and models as objects in XML (Extensible Markup Language [17]). This is a standard language for describing and exchanging information on the internet and is rapidly beginning to fulfill its promise [18] as a means of representing biological knowledge such as polymer sequences [19], cellular and anatomical structures [20] and mathematical models of biological systems [21]. Second, to link model elements into distributed networks on the internet, XML-encoded BioD objects and models can be linked using XLink (XML Linking Language [22]) and queried for patterns of functional interactions using query languages such as Lore (Lightweight Object Repository [23]) that have been developed for distributed, semistructured data.
Beyond description: model simulation and analysis
Visual languages in other fields [24,25] have been implemented as formal computer languages so that the diagrams are not only comprehensible to humans but can be analyzed by computers. This includes CAD systems for electronic circuit design and visual languages for computer programming (for example ProLog) as well as mathematical modeling software that translates diagrams into mathematical equations for quantitative analysis. Indeed, visual languages suitable for mathematical modeling in biology (for example Stella™ [26], SAAM II™ [27,28] and KineCyte™ [29,30]) have contributed to the establishment of quantitative modeling as a 'gold standard' and critical element of biological analysis.
Despite the successes and value of quantitative analytical methods, it must be recognized that progress in the knowledge of biological systems proceeds largely on qualitative information and reasoning ('A binds to B.' 'If C increases then D decreases.') rather than quantitative analysis. This is due in large part to the huge range of experimental conditions and phenomena that are encountered and must be considered, and the great time, effort and expense required to generate consistent sets of quantitative system properties (concentrations, reaction rates and binding constants, for instance) many of which are experimentally inaccessible. In the face of such difficulties, biology, perhaps more than other disciplines, consists of a vast amount of qualitative information that forms the basis for speculation, reasoning and decision-making at all levels from the basic science laboratory to the clinic. To exploit this reservoir of qualitative biological knowledge, a variety of qualitative and hybrid qualitative/quantitative simulation and analysis tools that have been developed in the artificial intelligence community [31,32,33,34] have been adapted to the analysis of biological problems [35,36,37,38].
The advantage of hybrid qualitative/quantitative methods with BioD is that they allow simulation and analysis of the behavior of complex systems at the prevailing level of systems knowledge; BioD models may be brainstormed in descriptive terms early in the discovery process and then upgraded in stages as qualitative information and then quantitative data become available. Model development and testing can, therefore, become an integral part of early discovery and hypothesis testing all within the context of a single graphical description, rather than developing models simply as validations of well understood phenomena. In effect, BioD models would fill the descriptive and analytical gap between informal cartoons and fully quantitative mathematical models.
To accomplish this, BioD object data structures would have to be extended to first represent functional properties and then provide action arrows with logical rules or mathematical functions that determine how the states of linked icons affect each other. Within a BioD editor, such rules and functions would be selectable in preprogrammed form from context-sensitive menus of appropriate and specific action arrows (a feature in KineCyte™ simulation software; RainTown [39]). Encoding BioD data structures in XML according to the Systems Biology Markup Language (SBML), a recently proposed 'open standard' for exchanging mathematical models of biological systems [21], would allow BioD models to be exported and quantitatively simulated in a variety of analytical software platforms.
Dissociating the specification of functional properties and values from the graphical description of the model means that models can be created on a purely speculative basis, and then, as system knowledge matures, qualitative values and action rules can be selected (or specified) to support qualitative reasoning and hypothesis testing. As quantitative values and functions are warranted, they can then used to constrain the qualitative predictions of the model.
Discussion
We propose a conceptual basis for an extensible biological description language that is designed to be implemented as visual computer language for describing, archiving and analyzing complex, multidisciplinary biological systems. We view this suggestion not as radical but as a natural, perhaps inevitable, extension to biomedicine of the visual computer language paradigm [24,25,40] that has been successfully applied in other technical disciplines. The major challenge is that biology offers perhaps some of the most complex and functionally diverse systems known.
With example models here and online, we have shown that a limited, but extensible, lexicon of icons and arrows is capable of describing complex systems that involve a range of cell-biological disciplines from gene regulation to the biophysics of ion transport. The value of such standardization is increased conciseness, readability and clarity for communication of knowledge about complex systems. Beyond simple graphic standardization, computer implementation of a graphic language for biology is not only feasible with currently available computer and internet technology, but could yield substantial benefits to biomedical education, investigation and industry. For instance, development of a common language to build accessible common models are critical elements in the 'blackboard' approach [41,42] for multidisciplinary collaborations on complex problems. We suggest that a standard language for building and posting cross-disciplinary functional models on the internet will be a necessary stage in the evolution of bioinformatics, as there is a growing need to express and understand whole-systems behavior and the complex systems that underlie disease. We further suggest that additional extensions to a standard graphical language can serve as a user interface for a variety of qualitative and quantitative simulation and analysis methods used in other disciplines.
The prototype language we present here is designed primarily for describing cell-biological systems as discrete networks of interacting functional objects at multiple conceptual levels. As such, it may prove cumbersome for describing massively parallel systems (such as the coupled expression of thousands of genes) or continuum systems (such as the wave propagation of cytoplasmic calcium). Even in these modeling domains, however, a standardized visual computational interface would allow modelers to describe discrete parts ('finite elements') of systems and/or to embed the systems in larger BioD models that both control and depend on the massive or continuum systems models.
How might BioD evolve and be extended from a core set of elements as described above to more general biological description language? In our approach, we have followed the model of recent developments of internet protocols, operating systems and computer languages by envisioning that if a core computational framework can be established, it can be extended and implemented for a variety of methods, languages and operating systems. Thus we envision that, given the development of one (or more) editor applications, users could invent BioD language elements (functional properties, icons, arrows and models) as needed to describe specific domains. As domain lexicons and model-model networks expand, with inevitable conflicts and duplications, we would suggest more global standardization be negotiated according to 'open-source' mechanisms of distributed, cooperative development [43] used for developing the Linux operating system and now being applied to SBML development [21].
By whatever mechanism, language development raises other key issues. How can one maintain the delicate balance of expressiveness and readability - can the language concisely describe all systems of interest while still being understandable without extensive training? When does the ability to derive more expressive icons begin to diminish readability as viewers are required to recognize and interpret subtle differences between similar icons? Even if successfully developed for selected domains and purposes, inevitably there will be edges to any language for describing all domains and for all analytical purposes - hence the continued appearance of new computer languages such as Java.
Conclusions
We have proposed that a generalized biological descriptive language akin to schematic diagramming languages in other disciplines is a necessary step in the evolution of functional bioinformatics. Toward this, we have developed a prototype language, BioD, that we have used to test the value of this approach and to explore the feasibility of computational implementation using object-oriented programming methods in the context of Internet-based communications. We suggest that a sufficiently formalized descriptive language built on the BioD concept of 'functional properties' can anchor a computational framework capable of supporting the archiving of extended, web-linked model networks and model analysis using hybrid qualitative reasoning and quantitative simulations.
Acknowledgments
Acknowledgements
We thank Jim Roberts, Dan Gottschling and Barbara Trask for helpful discussion during the preparation of this manuscript.
References
- Karp PD. Metabolic databases. Trends Biochem Sci. 1998;23:114–116. doi: 10.1016/s0968-0004(98)01184-0. [DOI] [PubMed] [Google Scholar]
- From sequence to function. An introduction to the KEGG project http://kegg.genome.ad.jp/kegg/kegg2.html
- McAdams HH, Shapiro L. Circuit simulation of genetic networks. Science. 1995;269:650–656. doi: 10.1126/science.7624793. [DOI] [PubMed] [Google Scholar]
- Takai-Igarashi T, Nadaoka Y, Kaminuma T. A database for cell signaling networks. J Comput Biol. 1998;5:747–754. doi: 10.1089/cmb.1998.5.747. [DOI] [PubMed] [Google Scholar]
- Transpath home page http://193.175.244.148/index.html
- Kohn KW. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Mol Biol Cell. 1999;10:2703–2734. doi: 10.1091/mbc.10.8.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MODULES in extracellular proteins http://www.bork.embl-heidelberg.de/Modules/extra.html
- BioD model of G1/S checkpoint machinery http://www.rainbio.com/BioD_G1-S.html
- BioD model of the ketogenic diet and epilepsy http://depts.washington.edu/perc/infoscientists/ketogenicdiet/KD_Events.html
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev. 1996;6:56–64. doi: 10.1016/s0959-437x(96)90011-8. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Kaldis P. The cdk-activating kinase (CAK): from yeast to mammals. Cell Mol Life Sci. 1999;55:284–296. doi: 10.1007/s000180050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Ptashne M. A Genetic Switch Cambridge, MA: Cell Press; 1992. pp. 20–28.
- Gelbart WM. Databases in genomic research. Science. 1998;282:659–661. doi: 10.1126/science.282.5389.659. [DOI] [PubMed] [Google Scholar]
- Extensible Markup Language (XML) 1.0 (Second Edition) http://www.w3.org/TR/REC-xml
- Mackenzie D. New language could meld the Web into a seamless database. Science. 1998;280:1840–1841. [Google Scholar]
- Fenyo D. The Biopolymer Markup Language. Bioinformatics. 1999;15:339–340. doi: 10.1093/bioinformatics/15.4.339. [DOI] [PubMed] [Google Scholar]
- The Physiome Markup Languages http://www.physiome.org.nz/
- Systems Biology Markup Language [SBML] http://www.cds.caltech.edu/erato/index.html
- XML Linking Language (XLink) Version 1.0 http://www.w3.org/TR/xlink/
- McHugh J, Abiteboul S, Goldman R, Quass D, Widom J. Lore: A database management system for semistructured data. SIGMOD Record. 1997;26:54–66. [Google Scholar]
- Backlund B, Hagsand O, Pehrson B. Generation of visual language-oriented design environments. J Vis Lang Comput. 1990;1:333–354. [Google Scholar]
- Meyers BA. Taxonomies of visual programming and program visualization. J Vis Lang Comput. 1990;1:97–123. [Google Scholar]
- Hargrove JL, Hulsey MG, Summers AO. From genotype to phenotype: computer-based modeling of gene expression with STELLA II. Biotechniques. 1993;15:1096–1101. [PubMed] [Google Scholar]
- Barrett PH, Bell BM, Cobelli C, Golde H, Schumitzky A, Vicini P, Foster DM. SAAM II: Simulation, Analysis, and Modeling Software for tracer and pharmacokinetic studies. Metabolism. 1998;47:484–492. doi: 10.1016/s0026-0495(98)90064-6. [DOI] [PubMed] [Google Scholar]
- Cobelli C, Foster DM. Compartmental models: theory and practice using the SAAM II software system. Adv Exp Med Biol. 1998;445:79–101. doi: 10.1007/978-1-4899-1959-5_5. [DOI] [PubMed] [Google Scholar]
- Cook DL, Atkins WM. Enhanced detoxification due to distributive catalysis and toxic thresholds: A kinetic analysis. Biochemistry. 1997;36:10801–10806. doi: 10.1021/bi971284b. [DOI] [PubMed] [Google Scholar]
- Cook DL, Gerber AN, Tapscott SJ. Modeling stochastic gene expression: implications for haploinsufficiency. Proc Natl Acad Sci USA. 1998;95:15641–15646. doi: 10.1073/pnas.95.26.15641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonarini A, Maniezzo V. Integrating qualitative and quantitative modeling. Int J Expert Systems Res Appl. 1991;4:51–70. [Google Scholar]
- Fishwick PA, Zeigler BP. Creating Qualitative and Combined Models with Discrete Events IEEE/Computer Society Press: Los Alamitos, CA, 1991.
- Kuipers BJ. Reasoning with qualitative models. Artificial Intelligence. 1993;59:1–2. [Google Scholar]
- Kuipers BJ, Shults B. Reasoning in Logic About Continuous Systems San Francisco, CA: Morgan Kaufman Publ; 1994.
- Eisenberg M. The kineticist's workbench: qualitative/quantitative simulation of chemical reaction mechanisms. Expert Systems Appl. 1991;3:367–377. [Google Scholar]
- Ursino M, Avanzolini G, Barbini P. Qualitative simulation of dynamic physiological models using the KEE environment. Artificial Intelligence Med. 1992;4:53–73. [Google Scholar]
- Tyson JJ, Novak B, Odell GM, Chen K, Thron CD. Chemical kinetic theory: understanding cell-cycle regulation. Trends Biochem Sci. 1996;21:89–96. [PubMed] [Google Scholar]
- Thieffry D, Thomas R. Qualitative analysis of gene networks. Pac Symp Biocomput. 1998:77–88. [PubMed] [Google Scholar]
- KineCyte™ biological systems simulator http://www.rainbio.com/KineCyte.html
- Glasgow J, Narayanan NH, Chandrasekaran B. Diagrammatic Reasoning: Cognitive and Computational Perspectives Cambridge, MA, The AAAI Press; 1995.
- Corkill DD. Blackboard systems. AI Expert. 1991;6:40–47. [Google Scholar]
- Buschmann F, Meunier R, Rohnert H, Sommerlad P, Stal M. Pattern-Oriented Software Architecture; A System of Patterns New York: John Wiley and Sons; 1996.
- DiBona C, Ockman S, Stone M. Open Sources: Voices from the Open Source Revolution Sebastopol, CA: O'Reilly & Associates, Inc; 1999)
- Gille H, Downward J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J Biol Chem. 1999;274:22033–22040. doi: 10.1074/jbc.274.31.22033. [DOI] [PubMed] [Google Scholar]
- Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfork KA, Wonderlin WF, Peterson VA, Strobl JS. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J Cell Physiol. 1995;162:163–171. doi: 10.1002/jcp.1041620202. [DOI] [PubMed] [Google Scholar]
- Heist EK, Schulman H. The role of Ca2+/calmodulin-dependent protein kinases within the nucleus. Cell Calcium. 1998;23:103–114. doi: 10.1016/s0143-4160(98)90108-7. [DOI] [PubMed] [Google Scholar]
- Wang S, Melkoumian Z, Woodfork KA, Cather C, Davidson AG, Wonderlin WF, Strobl JS. Evidence for an early G1 ionic event necessary for cell cycle progression and survival in the MCF-7 human breast carcinoma cell line. J Cell Physiol. 1998;176:456–464. doi: 10.1002/(SICI)1097-4652(199809)176:3<456::AID-JCP2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]