

Abstract
The endothelium is a highly dynamic structure lining the inside of blood vessels that exhibits physical and chemical properties that are critical determinants of overall vascular function. Physically, the endothelium constitutes a semipermeable barrier. Chemically, the endothelium synthesizes numerous factors such as reactive oxygen species (ROS) that can act as autocrine and paracrine signaling molecules. Oxidative stress results when ROS levels increase to levels that cause cellular injury, and, in the endothelium oxidative stress leads to barrier disruption. Endothelial barrier disruption also results from increased cytosolic calcium through store-operated calcium (SOC) entry channels. Although it is known that ROS can interact with and regulate some ion channels, relatively little is known about the interaction of these species with components of endothelial SOC entry channels, the canonical transient receptor potential (TRPC) proteins. Here we review our current understanding of ROS-mediated TRPC channel function and how it affects SOC entry and endothelial barrier disruption. Antioxid. Redox Signal. 15, 1567–1582.
Introduction
Endothelial cells line the inside of blood vessels and form a semipermeable barrier across which macromolecules and solutes are selectively transported. The physical barrier is formed by tight cell–cell and cell–matrix adhesions (32), and its integrity is tightly coupled to cytosolic calcium ([Ca2+]i) levels (105). Increases in [Ca2+]i lead to formation of interendothelial cell gaps that allow for the exudation in fluid and proteinaceous material from the blood vessel into the interstitium. In endothelial cells a very specific calcium pathway has been identified that is important for triggering gap formation. This pathway is calcium entry from the extracellular space through the so-called store-operated calcium (SOC) entry channels. SOC entry occurs downstream of Gq or receptor tyrosine kinase activation and subsequent hydrolysis of phosphoinositol-4,5-bisphosphate (PIP2) to inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). DAG, which is membrane localized, can directly or indirectly activate cation entry channels called receptor operated channels (ROC). InsP3, on the other hand, must diffuse through the cytosol to bind to its receptor on the endoplasmic reticulum, a calcium store. Once activated, the InsP3 receptor releases calcium from the endoplasmic reticulum into the cytosol. Upon store depletion, an unidentified signal is relayed to the plasma membrane to open channels that allow for calcium influx into the cell (Fig. 1). These channels are known as SOC entry channels [reviewed in (17)]. While store depletion activates multiple SOC entry channels, it is activation of a specific channel mediating the calcium-selective current known as ISOC (store-operated calcium), which is an important step leading to the formation of interendothelial cell gaps (133).
FIG. 1.

Activation of store-operated calcium (SOC) entry channels. Inflammatory mediators act on Gq receptors (R) or receptor tyrosine kinases to activate phospholipase C (PLC). PLC hydrolyzes phosphoinositol-4,5-bisphosphate (PIP2) forming inositol-1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). InsP3 diffuses through the cytosol to bind to and activate its receptor on the endoplasmic reticulum (ER). Upon receptor activation, calcium is released from the ER into the cytosol increasing [Ca2+]i. Once the ER is depleted of calcium, an unidentified signal is relayed to the plasma membrane to open up SOC entry channels that allow for the influx of calcium and further increase of [Ca2+]i. Thapsigargin (Tg) and 2,5-di-t-butylhydroquinone (BHQ) are inhibitors of the Ca2+/ATPase pump on the ER and are often used to directly activate SOC entry. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
In addition to the physical barrier it provides, the endothelium is also a highly dynamic structure capable of producing numerous factors that play fundamental roles in blood vessel function. Reactive oxygen species (ROS), for example, are synthesized by the endothelium and act in a paracrine and autocrine manner as signaling molecules. Increased levels of ROS, however, can lead to oxidative stress and endothelial barrier disruption. ROS-mediated endothelial barrier disruption [reviewed in (10)] has been studied in both the systemic and pulmonary vasculatures. Molecular players important in the mechanisms underlying barrier disruption include the ROS themselves as well as cytoskeletal proteins and proteins involved in cell–cell and cell–matrix adhesions, which are important in determining cell shape and barrier integrity. In endothelium of the blood–brain barrier, for example, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase- or xanthine oxidase-mediated oxidative stress resulted in activation of contractile proteins, downregulation of the tight junction molecule occludin, and endothelial barrier disruption (42). In human lung endothelium, particulate matter derived from airborne pollutants induced ROS generation, which resulted in endothelial barrier disruption via p38 mitogen-activated protein (MAP) kinase-dependent and heat-shock protein-27 (HSP27)-dependent pathways (127). In bovine lung microvascular cells, 4-hydroxy-2-nonenal (HNE), a bioactive aldehyde product formed upon lipid peroxidation [reviewed in (120)], induced mitochondrial-dependent ROS production and depleted cellular glutathione (GSH) (122). Further, HNE caused remodeling of the actin cytoskeleton and endothelial barrier disruption via a mechanism involving extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK). and p38 MAP kinases (121).
Disruption of the endothelial barrier is a contributing factor to the development and/or progression of numerous pathological states, including atherosclerosis [reviewed in (98)], ischemia/reperfusion [reviewed in (112)], acute respiratory distress syndrome [reviewed in (128)], high altitude pulmonary edema [reviewed in (90)], and hyperoxia (97). To date mechanisms underlying endothelial barrier disruption are not clearly understood. Indeed increased [Ca2+]i levels, in general, and increased [Ca2+]i levels due to calcium entry through SOC entry channels, more specifically, promote endothelial barrier disruption. Further, ROS can be important mediators of endothelial barrier disruption. However, to date relatively little is known about the relationship between ROS and SOC entry in endothelial cells and further how this relationship affects endothelial barrier integrity. It is therefore the focus of this review to discuss what is currently known about endothelial ROS in the context of SOC entry.
Molecular Makeup of Endothelial SOC Entry Channels
The SOC entry pathway was first described just over 20 years ago by Putney (101). Initially, Putney coined the term “capacitative calcium entry” because he believed that the calcium that entered the cell was directly funneled into the endoplasmic reticulum to refill the calcium store. It was later determined that the calcium that entered the cell first traversed the cytosol before entering the endoplasmic reticulum [reviewed in (102)]. Since then, it has come to be appreciated that in addition to serving the purpose of refilling the calcium store, calcium entry through SOC entry channels contributes to multiple cellular functions that require calcium influx (87). It turns out that not all SOC entry channels and currents are the same. One particular SOC entry current, first described in mast cells (41) and subsequently in Jurkat human leukemic T (147) and rat basophilic leukemia cells (5), is calcium release-activated calcium current (ICRAC). The ICRAC is a highly calcium-selective current that is the prototype SOC entry current and is mediated by the CRAC channel. Endothelial cells also possess a calcium-selective SOC entry current, the ISOC (26, 79, 123). This current is similar, but not identical, to the ICRAC and is likely mediated by a channel different than the CRAC channel. Still, there are other SOC entry currents that are not at all calcium-selective. Since Putney's initial description, SOC entry has been widely and intensely studied in the pursuit to answer two major questions: First, what is the nature of the signal(s) that links store depletion to channel activation? Second, what is the molecular makeup of these channels? To date, the identity of the signal(s) remains unclear. As to the molecular makeup of the channels, only recently has the identity of the CRAC channel been described, whereas still the identity of other SOC entry channels remains unclear.
The identity of proteins that make up SOC entry channels has been and still is hotly debated. Currently, they are thought to be made up of mammalian homologs of the transient receptor potential (TRP) protein family and/or orai1. The TRP proteins were the first candidates described for SOC entry channel subunits. Orai1, on the other hand, has just been recently identified and implicated in the molecular makeup of the CRAC channel and possibly other SOC entry channels. TRP proteins were originally identified in the Drosophila melanogaster phototransduction signaling cascade, when it was observed that mutant flies lacking TRP protein expression are blinded by bright, sustained light (19). It was subsequently determined that the TRP protein constitutes a calcium-selective ion channel that, when activated, allows for calcium entry into the cell (36). When expressed in vitro, the Drosophila TRP protein can be activated by store depletion; that is, it acts as a SOC entry channel (124). Homologs of Drosophila TRP proteins have been identified in mammals, Caenorhabditis elegans, and zebrafish, and now TRP proteins constitute a superfamily in which there are seven subfamilies [reviewed in (74, 76)] (Fig. 2a). The mammalian isoforms most closely resembling Drosophila TRP proteins are referred to as the TRPC (canonical) subfamily [reviewed in (91)]. The TRP proteins all exhibit six transmembrane spanning domains (S1–S6), with the pore region residing between S5 and S6, and have cytosolic amino and carboxy termini. Similar to voltage-gated potassium, calcium, and sodium channels, the functional channel is believed to be formed from the coalescence of four subunits (Fig. 2b). The TRPC subfamily members 1, 3, 4, and 5 can exhibit SOC entry channel characteristics, as demonstrated mainly in studies utilizing overexpression systems (33, 65, 66, 95, 96, 126, 129, 134–136, 142).
FIG. 2.

Transient receptor potential (TRP) protein family. (a) The TRP proteins constitute a superfamily of which there are seven subfamilies. The TRPC (canonical) subfamily most closely resembles the TRP protein (red) originally identified in Drosophila melanogaster. Adapted from Minke (74). (b) Functional cation channels are believed to be formed by oligomerization of TRP proteins into hetero- or homotetramers. Reprinted with permission from (61) and (83). (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
In endothelial cells, several studies have looked at the molecular makeup of the endothelial ISOC channel. Two in particular focused on endogenous TRPC proteins and their role ISOC channel function. In the TRPC4 knockout mouse, ISOC in aortic endothelial cells was nearly abolished (29). A second study used small interfering RNA (siRNA) to knockdown TRPC1 and demonstrated a reduced ISOC (11). From these observations, it was determined that the endothelial ISOC channel is comprised of at least TRPC1 and TRPC4 subunits. More specifically, from these data we know that there is at least one TRPC1 subunit and one TRPC4 subunit. However, since the functional channel is likely a tetramer, the question remains as to the identity of the other two subunits. To date, the identity of these other subunits remains to be determined. While there may be more than one TRPC1 or TRPC4 in the functional channel, other TRPC proteins, for example, TRPC3, also represent promising candidates. Another candidate may be orai1, yet the contribution of orai1 to the endothelial ISOC channel structure and function is unknown.
Unlike the TRP proteins, orai1 is a four-transmembrane-spanning domain protein (28). Similar to the TRPs it has cytosolic amino and carboxy termini, and the functional channel is also proposed to be a tetramer. Orai1 was first identified by Feske et al. (28) in 2006 in a study of two patients with severe combined immunodeficiency whose T cells exhibited impaired SOC entry, and orai1 was identified as the mutated protein responsible for the defects in SOC entry. By 2006 another protein involved in SOC entry, stromal interaction molecule (STIM), had also been identified (104) and characterized (138) as a calcium sensor that links store depletion to CRAC channel activation. In their study, Zhang et al. (138) eloquently demonstrated that the endoplasmic reticulum-localized STIM1 translocates to the plasma membrane upon store depletion. They went on to show that when STIM1 translocation can first be detected, ICRAC has also begun to develop, and that the maximum current is observed when the maximal translocation has been achieved. Thus, at this point, both orai1 and STIM1 had been linked to crucial roles in CRAC events. It was not long thereafter that Soboloff et al. (113) drew a connection between orai1 and STIM1. In their study, coexpression of orai1 and STIM1 in human embryonic kidney 293 (HEK293) cells and rat basophilic leukemia cells resulted in greatly enhanced SOC entry and ICRAC, respectively. They went on to propose that orai1 is the actual channel component of SOC entry. Almost at the same time, Prakriya et al. (100) concluded from their study that orai1 is a pore-forming subunit of the CRAC channel. Subsequently, it was determined that orai1 subunits coalesce in a tetramer (69, 92, 141) and further that a tetramer configuration is sufficient to recapitulate the ICRAC in cells stably expressing STIM1 (72).
To date, little is known about the role(s) of orai1 in endothelial SOC entry. Abdullaev et al. (1) examined ICRAC and SOC entry in human umbilical vein endothelial cells (HUVECs). Here they demonstrated that HUVECs exhibit very low amplitude ICRAC, which was abolished by silencing STIM1 or orai1, and which was rescued by ectopic expression of the respective proteins. However, when they inhibited expression of TRPC1 or TRPC4, there was no effect on the ICRAC, collectively suggesting that it is orai1, and not the TRPC proteins, that mediates the endothelial ICRAC. Contrary to these findings, Sundivakkam et al. (115) recently reported that knockdown of orai1 in mouse lung endothelial cells did not prevent calcium entry after thrombin stimulation. However, in mouse lung endothelial cells from TRPC4 knockout mice as well as wild-type cells treated with siRNA to TRPC4, the calcium entry response to thrombin was abolished. Further, in agreement with an earlier study by Brough et al. (11), knockdown of TRPC1 only partially inhibited thrombin-induced calcium entry. Collectively, these results suggest that TRPC4 and TRPC1 play a role in endothelial SOC entry, whereas orai1 does not. Clearly then, a discord exists. On one hand, orai1 appears to play a critical role in endothelial SOC entry; on the other hand, TRPC4 and TRPC1 are the key players. This then leads to the question of why such controversy exists.
Controversy in the SOC entry field is nothing new. Indeed, in the 20 plus years since Putney's initial observation, this field has been hotly debated not only the identity of the channels but also the identity of the signal(s) linking store depletion to channel activation. In addressing the question of which proteins make up SOC entry channels, one must consider that there are a multitude of factors involved in SOC entry as well as in studies thereof. To begin, the proteins that play a role in SOC entry are not just the proteins that make up the pore-forming channel. Indeed, there are likely multiple, very specific protein–protein interactions between channel proteins, regulatory proteins, and even proteins in the endoplasmic reticulum that form an intricate complex that is responsible for overall channel behavior and current properties. However, to date, the molecular players involved in these complexes are only partially resolved. A second factor contributing to the controversy is that there is very likely cell-type specificity in which the molecular makeup of channel complexes varies from one cell type to another. Yet, disparate results have been observed even when comparing studies using the same cell type. This leads to a third confounding factor, the use of overexpression systems. To date, the majority of studies focused on identifying contribution of either TRPC proteins or orai1 to SOC entry have relied on protein expression systems, in which only one type of subunit is overexpressed. However, at least in the case of the TRPC proteins, as they can form either homo- or heteromultimers, one may expect that upon overexpression of a single subunit, the formation of the homomultimer, or a heteromultimer with a different subunit stoichiometry, will be favored. Thus, the channel formed in the overexpression system may or may not faithfully recapitulate the native state. Taking into account these factors, and likely other factors as well, it is not surprising that controversy exists.
To date, the roles of TRPC and orai1 proteins in SOC entry are still being debated. However, should the contributions of these proteins to SOC entry be mutually exclusive? Perhaps not. Indeed, several investigators have already demonstrated a functional synergy between TRPC proteins and orai1. Liao et al. (62–64) showed that orai1 structurally and functionally interacts with TRPC proteins to mediate SOC entry in HEK cells stably expressing TRPC proteins. Ong et al. (86) examined the interaction of orai1 and TRPC1 in human salivary gland cells and dispersed mouse submandibular gland cells. They observed a dynamic interaction between TRPC1, orai1, and STIM1 that was increased after store depletion, and at least in the human salivary gland cells was important for the activation of SOC entry. Cheng et al. (15) also demonstrated functional interaction of orai1 with TRPC1 and STIM1. In human platelets, Jardin et al. (46) observed increased coimmunoprecipitation between TRPC1 and orai1 upon store depletion. In the presence of an orai1 antibody, SOC entry was decreased and interaction between TRPC1 and STIM1 was interrupted leading the authors to conclude that orai1 functions to link STIM1 to TRPC1. In endothelial cells, interaction of TRPC1 and/or TRPC4 with orai1 has not yet been examined. Indeed, it may be that functional interaction between TRPC proteins and orai1 is a critical determinant of endothelial SOC entry activation and barrier disruption.
SOC Entry Channels and Endothelial Barrier Disruption
The observation that calcium entry through SOC entry channels plays a key role in endothelial barrier disruption was first made in the pulmonary circulation (16, 49). In these and other studies, the plant alkaloid thapsigargin was, and still is, used to activate SOC entry. Thapsigargin is a Ca2+/ATPase inhibitor that causes the endoplasmic reticulum calcium store to become depleted (118, 124). After store depletion, the signal will be initiated to activate SOC entry channels. In pulmonary artery endothelial cells (PAECs), thapsigargin application activates multiple SOC entry channels, including the calcium-selective ISOC channel. PAEC monolayers treated with thapsigargin exhibit disruption of cell–cell and cell–matrix adhesions, an increase in myosin light chain phosphorylation and formation of actin-based stress fibers, leading to formation of interendothelial cell gaps and increased permeability (49, 78, 79) (Fig. 3). In situ, lung permeability increased in a dose-dependent manner after thapsigargin application, with the principal site of fluid leak from large, extra-alveolar vessels (16).
FIG. 3.

Activation of SOC entry leads to endothelial barrier disruption. (a) Monolayer of cultured rat pulmonary artery endothelial cells in 2 mM extracellular calcium (left panel) exhibits tight cell–cell adhesion and no intercellular gaps. Addition of thapsigargin leads to activation of SOC entry and the formation of large interendothelial cell gaps (right panel). Reprinted with permission from Moore et al. (79). (b) Endothelial barrier function was assessed by determining the permeability of fluorescein isothiocyanate-labeled dextran across confluent monolayers in the absence (open symbols) and presence (closed symbols) of thapsigargin (1 μM) at 30, 60, and 120 min. With both 12 and 72 kDa dextrans, the permeability of the monolayer was increased after addition of thapsigargin. Reprinted with permission from Kelly et al. (49).
As described above, we know that TRPC1 and TRPC4 contribute subunits to the functional ISOC channel. Importantly, in the TRPC4 knockout mouse thrombin-induced increases in lung permeability were attenuated (119), whereas permeability was increased in TRPC1-overexpressing human dermal microvascular endothelium (88). These observations reflect the critical roles of TRPC1 and TRPC4 and the ISOC channel in endothelial barrier disruption.
Endothelial ROS
Chemical and biochemical reactions that lead to generation of ROS are complex and numerous, and have been reviewed in detail elsewhere (68, 85). ROS and reactive nitrogen species (RNS) can act in the cell via direct protein oxidation and nitration, respectively, or can be short-lived signaling molecules in specific pathways [reviewed in (73)]. Reactive molecular species include superoxide (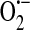 ), nitric oxide (NO), hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and the extremely reactive hydroxyl radical (OH•). Importantly, ROS and RNS exist in all cells, and cells strive to maintain a certain redox homeostasis through a balance of oxidants and antioxidants.
), nitric oxide (NO), hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and the extremely reactive hydroxyl radical (OH•). Importantly, ROS and RNS exist in all cells, and cells strive to maintain a certain redox homeostasis through a balance of oxidants and antioxidants.
Endothelium synthesizes and responds to ROS. In the 1980s to early and mid-1990s, it was of keen interest to identify specific ROS generated and the generating source. In general, endothelium generates 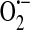 and H2O2 in both unstimulated (50, 51, 116) and stimulated (40, 51, 70, 103, 116, 117) conditions. In addition to the mitochondrial electron transport chain, the enzymes NADPH oxidase, xanthine oxidoreductase, cytochrome P450, and uncoupled endothelial NO synthase (eNOS) play roles in the generation of endothelial ROS [reviewed in (13, 59)] (Fig. 4). Xanthine oxidase and NAD(P)H oxidase, in particular, are principally responsible for
and H2O2 in both unstimulated (50, 51, 116) and stimulated (40, 51, 70, 103, 116, 117) conditions. In addition to the mitochondrial electron transport chain, the enzymes NADPH oxidase, xanthine oxidoreductase, cytochrome P450, and uncoupled endothelial NO synthase (eNOS) play roles in the generation of endothelial ROS [reviewed in (13, 59)] (Fig. 4). Xanthine oxidase and NAD(P)H oxidase, in particular, are principally responsible for 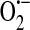 generation in endothelial cells.
generation in endothelial cells.
FIG. 4.

Sources of reactive oxygen species (ROS) in endothelial cells. Xanthine dehydrogenase (XDH), xanthine oxidase (XO), glutathione (GSH), endothelial nitric oxide synthase (eNOS), and oxidized glutathione (GSSG). Adapted from Li and Shah (59).
Xanthine oxidase
Xanthine oxidase is one form of the enzyme xanthine oxidoreductase. The mammalian xanthine oxidoreductase exists in two interconvertable forms, xanthine dehydrogenase and xanthine oxidase [reviewed in (37)]. While either form can reduce molecular oxygen to form 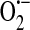 and H2O2, only the dehydrogenase form can reduce oxidized nicotinamide adenine dinucleotide (NAD+). Xanthine oxidase was shown to be expressed in endothelium by Jarasch et al. (45) in 1981 using immunolocalization with light and electron microscopy. Interestingly, tissues of the mammary gland, liver, heart, lung, and intestine stained positive for xanthine oxidase in the capillary endothelium but not the endothelium of larger blood vessels. Further, the endothelial cells exhibited a cytoplasmic distribution of xanthine oxidase. Indeed xanthine oxidoreductase is generally described as cytosolic (106); however, the exact subcellular localization is somewhat less clear (43). In 1992 Partridge et al. (89) observed a constitutive release of xanthine oxidase from endothelial cells. It was subsequently determined that xanthine oxidase localizes to the outer surface of endothelial cells (2, 106). Rouquette et al. (106) further demonstrated that this extracellular enzyme is often localized on “surfaces apposed to those of closely neighboring cells” leading to the idea that it plays a role in cell–cell interactions. Along a similar line, Adachi et al. (2) proposed that the extracellular xanthine oxidase generated
and H2O2, only the dehydrogenase form can reduce oxidized nicotinamide adenine dinucleotide (NAD+). Xanthine oxidase was shown to be expressed in endothelium by Jarasch et al. (45) in 1981 using immunolocalization with light and electron microscopy. Interestingly, tissues of the mammary gland, liver, heart, lung, and intestine stained positive for xanthine oxidase in the capillary endothelium but not the endothelium of larger blood vessels. Further, the endothelial cells exhibited a cytoplasmic distribution of xanthine oxidase. Indeed xanthine oxidoreductase is generally described as cytosolic (106); however, the exact subcellular localization is somewhat less clear (43). In 1992 Partridge et al. (89) observed a constitutive release of xanthine oxidase from endothelial cells. It was subsequently determined that xanthine oxidase localizes to the outer surface of endothelial cells (2, 106). Rouquette et al. (106) further demonstrated that this extracellular enzyme is often localized on “surfaces apposed to those of closely neighboring cells” leading to the idea that it plays a role in cell–cell interactions. Along a similar line, Adachi et al. (2) proposed that the extracellular xanthine oxidase generated 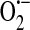 on the outside of the cell that could cause oxidative damage to transmembrane proteins and unsaturated fatty acids that comprise the membrane. Alternatively, or in addition, the
on the outside of the cell that could cause oxidative damage to transmembrane proteins and unsaturated fatty acids that comprise the membrane. Alternatively, or in addition, the 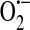 generated extracellularly could attract and injure neutrophils leading to further tissue injury. As xanthine oxidoreductase is activated by anoxia and reoxygenation (145), its function in endothelium is of particular interest in ischemia/reperfusion injury (71).
generated extracellularly could attract and injure neutrophils leading to further tissue injury. As xanthine oxidoreductase is activated by anoxia and reoxygenation (145), its function in endothelium is of particular interest in ischemia/reperfusion injury (71).
Zweier et al. (145) demonstrated the generation of 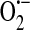 and OH• in bovine aortic endothelial cells exposed to anoxia and reoxygenation using electron paramagnetic resonance measurements. In an effort to determine whether xanthine oxidase activity contributed to the generation of these reactive species, the inhibitors allopurinol and oxypurinol were employed. Radical generation was reduced and/or scavenged with these inhibitors, leading the authors to conclude that xanthine oxidase activity is a major source of radical production in endothelial cells exposed to anoxia and reoxygenation. As it was not necessarily clear that what was observed in bovine endothelial cells also occurred in human cells, Zweier et al. (146) in a later study, demonstrated that human endothelial cells, in this case HUVECs, exposed to anoxia followed by reoxygenation do indeed generate oxygen-free radicals. The mechanisms underlying generation of these radicals were concluded to include xanthine oxidase in addition to redox cycling of chelatable iron. In yet another study, Zweier et al. (144), using human aortic endothelial cells exposed to anoxia then reoxygenation, determined that indeed xanthine oxidase is present in these cells and is the principal free radical generator. The principal substrate for xanthine oxidase is xanthine, yet it can also convert hypoxanthine to xanthine, and it is hypoxanthine, which accumulates as adenosine triphosphate (ATP) is depleted in conditions of low oxygen [reviewed in (108)]. Finally, in this study the authors determined that
and OH• in bovine aortic endothelial cells exposed to anoxia and reoxygenation using electron paramagnetic resonance measurements. In an effort to determine whether xanthine oxidase activity contributed to the generation of these reactive species, the inhibitors allopurinol and oxypurinol were employed. Radical generation was reduced and/or scavenged with these inhibitors, leading the authors to conclude that xanthine oxidase activity is a major source of radical production in endothelial cells exposed to anoxia and reoxygenation. As it was not necessarily clear that what was observed in bovine endothelial cells also occurred in human cells, Zweier et al. (146) in a later study, demonstrated that human endothelial cells, in this case HUVECs, exposed to anoxia followed by reoxygenation do indeed generate oxygen-free radicals. The mechanisms underlying generation of these radicals were concluded to include xanthine oxidase in addition to redox cycling of chelatable iron. In yet another study, Zweier et al. (144), using human aortic endothelial cells exposed to anoxia then reoxygenation, determined that indeed xanthine oxidase is present in these cells and is the principal free radical generator. The principal substrate for xanthine oxidase is xanthine, yet it can also convert hypoxanthine to xanthine, and it is hypoxanthine, which accumulates as adenosine triphosphate (ATP) is depleted in conditions of low oxygen [reviewed in (108)]. Finally, in this study the authors determined that 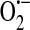 generated reacts with iron to form the highly reactive OH•.
generated reacts with iron to form the highly reactive OH•.
NAD(P)H oxidase
Whereas xanthine oxidoreductase function appears to require activation, such as by exposure to anoxia and reoxygenation, nicotinamide adenine dinucleotide (NADH) oxidoreductase does not require exogenous stimulation to produce ROS (75). In bovine coronary artery endothelium, NADH oxidoreductase was shown to be the major synthesizer of 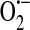 (75). Jones et al. (48) later showed that HUVECs do indeed express components of the NADPH oxidase system, but concluded that it is most likely not a significant source of endothelial
(75). Jones et al. (48) later showed that HUVECs do indeed express components of the NADPH oxidase system, but concluded that it is most likely not a significant source of endothelial 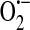 . In the next year, however, Zulueta et al. (143) demonstrated that a “plasma membrane-bound NADPH oxidase-like enzyme” is active in bovine PAECs after exposure to anoxia and reoxygenation and is the source of H2O2 that is released extracellularly. In the same work, these authors also showed that xanthine oxidase was responsible for intracellular
. In the next year, however, Zulueta et al. (143) demonstrated that a “plasma membrane-bound NADPH oxidase-like enzyme” is active in bovine PAECs after exposure to anoxia and reoxygenation and is the source of H2O2 that is released extracellularly. In the same work, these authors also showed that xanthine oxidase was responsible for intracellular 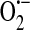 generation, which then could react with NO resulting in the formation of ONOO−.
generation, which then could react with NO resulting in the formation of ONOO−.
The NADPH oxidase system expressed in endothelial cells is very similar to that of neutrophils. Bayraktutan et al. (8) revealed that in rat coronary microvascular endothelial cells, the heterodimer cytochrome b558, with subunits p22-phox and gp91-phox, which is the principal component of the phagocyte NADH/NADPH oxidase complex, is expressed at both the message and protein levels and is active. While the cDNA isolated in this study exhibited high homology to the phagocyte, and vascular smooth muscle cell, sequences, the authors noted that there are some critical biochemical differences between the endothelial and phagocyte NADPH oxidase systems, including substrate specificity and activation properties. The NADPH oxidase inhibitor diphenyleneiodonium (DPI) was “more effective in inhibiting NADPH- than NADH- evoked activity.” Since DPI can also inhibit other oxidant producing flavoproteins such as xanthine oxidase and NO synthase, specific inhibitors of these compounds were tested and found to have no effect. Thus overall, it was concluded that in coronary microvascular endothelial cells NAD(P)H oxidase is a functional ROS-generating species. Further, whereas the phagocyte system is only functional after specific triggers in initiation of nonspecific host defense pathways, the endothelial system is always active, although to a lesser degree, such that there is continuous production of 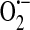 and other ROS. This constitutive production might therefore suggest that endothelial ROS are involved in cellular signaling mechanisms other than those involved in cell injury.
and other ROS. This constitutive production might therefore suggest that endothelial ROS are involved in cellular signaling mechanisms other than those involved in cell injury.
Following up on these early studies, Bayraktutan et al. (7) proceeded to determine sequence differences and hence likely structural differences between the neutrophil NADPH oxidase system components p22-phox and gp91-phox and the analogous endothelial NAD(P)H proteins. The p22-phox sequence was observed to exhibit >90% homology between the predicted sequence of rat coronary microvascular endothelial cell protein with neutrophil sequences from pig, mouse, human, and cow. Likewise the endothelial gp91-phox also exhibited a high degree of homology (>90%) with neutrophils of other species. However, two differences were noted. First, in the NADPH binding domain C, a serine to alanine mutation exists at residue 416, which the authors suggested might lead to changes in enzyme activity and substrate specificity, as compared to the neutrophil system. The second difference noted was in potential glycosylation sites. The consensus motif N-X-(S/T), with X being any amino acid residue, occurs four or five times in the phagocyte sequences of murine, human, and bovine, but only three times in the predicted rat endothelial sequence. The authors speculated that the cellular localization of the p22-phox/gp91-phox complex in endothelial cells is influenced by the glycosylation patterns. Indeed, immunofluorescence revealed that endothelial p22-phox and gp91-phox were mainly localized to the perinuclear region with a “more diffuse reticular staining extending toward the cell membrane.” This is in contrast to the phagocyte NADPH oxidase, where in the active neutrophil system, p22-phox and gp91-phox are associated with the plasma membrane [(47, 132) and reviewed in (58)]. Li et al. (60) pursued the observation that the cytochrome b558 subunits, p22-phox and gp91-phox, appear to be localized intracellularly in unstimulated endothelial cells. In their study, the authors used a combination of techniques, including coimmunoprecipitation, subcellular fractionation, and confocal microscopy, to determine the localization of p22-phox and gp91-phox as well as the cytosolic subunits p47-phox, p67-phox, and p40-phox. From their observations they determined that in the resting state these subunits interact in a preformed cytosolic complex that is associated with the cytoskeletal microtubule system. Importantly, this preformed complex is functional and thus capable of generating ROS.
Collectively, from these studies of endothelial xanthine oxidase and NAD(P)H oxidase, it became clear that endothelium does indeed generate 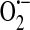 , and it does so both constitutively and after activation. It was further determined that
, and it does so both constitutively and after activation. It was further determined that 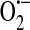 generation occurs both intracellularly and extracellularly.
generation occurs both intracellularly and extracellularly.
ROS Regulation of TRPC Proteins in Endothelium
It is well established that ROS can lead to increased [Ca2+]i due to calcium release from intracellular stores as well as calcium entry across the plasma membrane [reviewed in (21)]. Endothelial cells are generally considered to be nonexcitable cells [reviewed in (84)], although pulmonary microvascular endothelial cells express a functional voltage-gated T-type calcium channel (140). Without voltage-gated channels, the principal calcium entry pathways in endothelial cells are through SOC and ROC entry. As TRPC proteins are believed to underlie the molecular makeup of many ROC entry channels and at least some SOC entry channels, it is logical to question whether TRPC proteins are sensitive to regulation by ROS. To date, TRPC7 (currently known as transient receptor potential melastatin 2 [TRPM2]) and TRPC3/TRPC4 have been shown to be regulated by ROS in endothelium.
Transient receptor potential melastatin 2
TRPM2, of the melastatin subfamily, was originally named TRPC7 or long transient receptor potential canonical 2 (LTRPC2) (77). Although no longer considered a member of the TRPC subfamily, as much more is known about the mechanism underlying oxidant-induced channel activation for this channel as compared to other TRPC channels, we felt it appropriate to include it in this section. The TRPM subfamily only exhibits limited homology to other TRP proteins, and functional channels comprised of members of this subfamily are believed to be homotetramers (81).
Early work by two groups, Sano et al. (107) and Perraud et al. (93), examined activation of LTRPC2 by intracellular pyrimidine nucleotides based on the fact that LTRPC2 possesses a Nudix motif. The term “Nudix” was originally coined by Bessman et al. (9) to describe a motif found in proteins that is important in the hydrolysis of a nucleoside diphosphate linked to some other moiety, X (i.e., Nudix). The Nudix motif was originally described in the MutT protein of Escherichia coli, and as such was also referred to as a MutT motif, and later was also found in the MutX protein of Streptococcus pneumonia; proteins bearing the Nudix motif contain a conserved signature sequence that imparts nucleoside-triphosphate pyrophosphohydrolase activity [reviewed in (9)]. Perraud et al. (93) observed that in LTRPC2-expressing HEK293 cells, 100 μM adenosine 5′-diphosphoribose (ADPR) activated LTRPC2, but the same concentration of other closely related compounds, including NAD, NAD+, cyclic ADPR (cADPR), ATP, or ADP, did not (Fig. 5). Similarly, Sano et al. (107) showed that LTRPC2 expressed in HEK293 cells was activated in a dose-dependent manner by ADPR, as well as by 1 mM β-NAD. Using an inside-out excised patch configuration, both groups further determined that these intracellular pyrimidine nucleotides act directly, without the aid of cytoplasmic or membrane components, to activate LTRPC2 yielding a nonselective cation current permeable to calcium.
FIG. 5.

Biochemical pathways and inter-relationships between different oxidative species. Superoxide (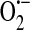 ), GSH, GSSG, glutathione peroxidase (GSH-Px), glutathione reductase (GSH-Red.), total nitric oxide synthase (tNOS), nitric oxide (NO), peroxynitrite (ONOO−), hydrogen peroxide (H2O2), hydroxide anion (−OH), hydroxyl radical (OH•), polyunsaturated fatty acid (PUFA), oxidized nicotinamide adenine dinucleotide phosphate (NADP+), adenosine monophosphate (AMP), and adenosine 5′-diphosphoribose (ADPR). Modified from Naziroğlu (80). (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
), GSH, GSSG, glutathione peroxidase (GSH-Px), glutathione reductase (GSH-Red.), total nitric oxide synthase (tNOS), nitric oxide (NO), peroxynitrite (ONOO−), hydrogen peroxide (H2O2), hydroxide anion (−OH), hydroxyl radical (OH•), polyunsaturated fatty acid (PUFA), oxidized nicotinamide adenine dinucleotide phosphate (NADP+), adenosine monophosphate (AMP), and adenosine 5′-diphosphoribose (ADPR). Modified from Naziroğlu (80). (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Subsequent studies used H2O2 to induce oxidant stress. Wehage et al. (130) reasoned that under oxidant stress NAD is elevated, and NAD is metabolized to ADPR, a known activator of LTRPC2. In their patch-clamp studies, 0.3 mM ADPR in the patch pipette or 5 mM H2O2 in the bath were able to activate a cation current in LTRPC2 expressing HEK293 cells. Unlike the results of Sano et al. (107), however, 1 mM NAD was not able to activate a current, which the authors attribute to potential experimental differences. An experiment in nature then led to more mechanistic insight into channel activation. Neutrophil granulocytes express a LTRPC2 deletion mutant (Δ1292-1325). As this mutant was activated by H2O2 but not by ADPR, the authors concluded that the mechanism of ADPR-induced LTRPC2 activation is independent of H2O2. In the same year, Hara et al. (35) performed an in depth study in which calcium entry was measured, using the Fura/2 approach, in LTRPC2-expressing HEK293 cells treated with H2O2. In support of a role for the MutT or Nudix motif, upon deletion of this sequence no calcium entry was detected. Further, the idea that channel activation is mediated by direct interaction between β-NAD+ and LTRPC2 was confirmed again using inside-out patches. In a whole-cell configuration, H2O2 application induced a unitary LTRPC2 current; however, when a patch was isolated in the inside-out configuration, the current disappeared and only was restored by β-NAD+ (not H2O2). Further, 32P-labeled β-NAD+ strongly bound to a glutathione S-transferase (GST)—LTRPC2 carboxy terminus fusion protein.
If H2O2-mediated activation of LTRPC2 requires conversion of H2O2 to NAD+ or ADPR, then OH• that is upstream of ADPR formation (Fig. 5) should also be able to activate the current. Along this train of thought, Ishii et al. (44) demonstrated the ability of intracellularly generated OH• to activate LTRPC2 (by now more commonly referred to as TRPM2) in both TRPM2-expressing HEK293 cells and in the rat β-cell line RIN-5F. Kolisek et al. (53) studied the cADPR part of the pathway and demonstrated that at concentrations >100 μM, cADPR was sufficient to gate TRPM2, but at lower concentrations of 10 μM, it was only able to act in an ancillary way to potentiate ADPR channel gating. In a study by Buelow et al. (12) it was demonstrated that the enzyme responsible for converting NAD+ to pADPR, poly(ADP-ribose) polymerase (PARP) isoform 1 was critical for TRPM2 currents and Ca2+ entry in DT40 B cells. From these studies it is clear that H2O2 and many of its metabolites are capable of activating TRPM2. However, the question of which species directly interact with and gate the channel and which species act indirectly as second messengers persists. Indeed, in a recent study, Naziroğlu and Luckhoff (81) measured single-channel conductances in TRPM2-expressing Chinese hamster ovary cells. Here they added H2O2 to the bath solution, or ADPR or NAD+ in the patch pipette. Using any of the three agonists in the whole-cell patch mode they observed a nonselective current. H2O2 in the bath led to current activation typically after a 2–5 min delay and wash-out did not terminate channel activity, suggesting that the H2O2-induced current did not rely on generation of a soluble factor that could be washed away in the inside-out patch configuration. However, the authors recognized that since the channels were first activated by H2O2 in the whole-cell configuration, it could be that H2O2 is not directly acting on the channel but is initiating a cascade of events that culminate in channel activation. To determine whether this was the case, they began in the cell-attached mode and treated cells with H2O2 for several minutes after which the inside-out patch configuration was established, and they observed that H2O2 activated the channels. However, in inside-out patches from cells that had never been exposed to H2O2, no current was detected until addition of ADPR. They thus concluded that both H2O2 and ADPR are sufficient to activate TRPM2, but that they act independently in their gating mechanisms. This is in support of observations of Wehage et al. (130) and Kraft et al. (56) where a carboxy terminal-truncated TRPM2 maintained H2O2 gating but lost ADPR gating. Finally, Perraud et al. (94) proposed that oxidative/nitrosative stress activates a biochemical pathway within the mitochondria, which results in the production and release of ADPR from the mitochondria, after which ADPR acts as diffusible second messenger to activate TRPM2.
Even though there currently are still some uncertainties as to which compounds act directly and which act indirectly to gate the TRPM2 channel, it is clear that TRPM2 does indeed act as a cellular oxidant sensor. What then are the downstream physiological effects of channel activation? Hara et al. (35) determined that activation of LTRPC2 leads to increased susceptibility to cell death and reduced cell survival. More recently, Hecquet et al. (38) described a role for TRPM2 in endothelial permeability. In this study TRPM2 was activated in human PAECs by exposure to H2O2 or an ADPR analog (3-deaza-cADP-ribose). Studies were performed to demonstrate activation of calcium entry by H2O2 and to determine whether this agonist causes release of calcium from intracellular stores. They observed that at H2O2 concentrations <300 μM, calcium was not released from intracellular stores, indicating that the calcium entry that occurred was not through SOC entry channels. Endothelial barrier integrity was monitored by measuring transendothelial electrical resistance. With H2O2 they observed a dose-dependent decrease in resistance with the maximal effect achieved at 300 μM H2O2. Both TRPM2 siRNA and TRPM2 blocking antibody inhibited H2O2-induced calcium entry and also reduced the decrease in transendothelial resistance.
TRPC3/4 channel
Early observations by Balzer et al. (6) revealed that in the presence of oxidants, channels comprised, at least in part, of TRPC3 were activated and contributed to endothelial cell depolarization. Groschner et al. (34) pursued the idea that TRPC3 activation was somehow sensitive to, or regulated by, oxidant stress. They proceeded to describe a mechanism whereby TRPC3 channel function, likely in combination with TRPC4, is activated following “oxidant-induced disruption of cholesterol-rich lipid rafts.” The proposed mechanism provided insight into how TRPC channel function might be intimately coupled to cellular redox status. To test their hypothesis, oxidative stress was induced in TRPC3-expressing HEK293 cells by treatment with tert-butylhydroperoxide or cholesterol oxidase. Caveolin1, which is a marker of caveolae, moved from the membrane into the cytosol, which was interpreted as disruption of caveolae. TRPC3, on the other hand, remained localized to the membrane and importantly was activated upon oxidative stress. As TRPC3 activation is regulated by phospholipase C (PLC), and activated PLC hydrolyzes membrane-bound PIP2, the authors next looked at membrane localization of PIP2. Using a PIP2-binding GFP fusion protein (GFP-PH-PLCδ) they observed that after tert-butylhydroperoxide application, GFP-PH-PLCδ lost its membrane association; however, they did not see the same effect after cholesterol oxidase treatment, leading them to conclude that the mechanism underlying oxidative stress-mediated TRPC3 activation does not involve PIP2 hydrolysis. The authors proposed a potential mechanism of cross-talk between TRPC channels and signaling molecules that reside in caveolae (or lipid rafts) and further speculated that membrane cholesterol oxidation might actually be the signaling molecule that activates TRPC3.
The observation that oxidative stress leads to TRPC3 activation leads to two questions. First, as TRPC channels are most likely tetramers, the question arises as to whether this particular channel is a TRPC3 homomer or heteromer, and if it is a heteromer, what are the identities of the other subunits? Second is the question of the mechanism underlying redox regulation of the TRPC3 containing channel. Poteser et al. (99) resolved, in part, the first question. As in their earlier study they demonstrated that TRPC3-expressing HEK293 cells possessed a redox-sensitive current activated by treatment with tert-butylhydroperoxide or cholesterol oxidase. The current–voltage plot revealed a linear relation with reversal potentials <0 mV, suggesting activation of a nonselective cation current. Additionally, native porcine aortic endothelial cells exhibited a similar current. While the aortic endothelial cells were shown to express several TRPC homologs, including TRPC1, TRPC3, TRPC4, and TRPC6, only TRPC3 and TRPC4 were resolved at the plasma membrane; further through immunoprecipitation and Förster Resonance Energy Transfer experiments, it was shown that TRPC4 interacts with TRPC3. The authors thus concluded that TRPC3 and TRPC4 contribute subunits to the redox-sensitive channel. It is still unclear as to the identity of the other two subunits. It could be any combination of TRPC3 and TRPC4, or it may be that another TRPC homolog is present. In the current study, even though TRPC1 was resolved in aortic endothelial cells and it appeared to interact, though weakly, with TRPC3, it was not detected at the plasma membrane. However, the authors did not rule out a contribution from TRPC1, as they recognized that Strübing et al. (114) had previously identified a channel complex comprised of TRPC1 plus TRPC4 or TPRC5 plus TRPC3 or TRPC6 in mammalian embryonic brain. Nonetheless, to date, the subunit stoichiometry of the endothelial TRPC3/TRPC4 redox-sensitive channel remains unresolved. Further the mechanism by which oxidants lead to channel activation and the specific downstream function(s) of this channel have not yet been explored.
Redox Regulation of SOC Entry in Endothelial Cells
Activation of SOC entry, in general, and ISOC in particular, is an important step leading to interendothelial cell gap formation and barrier disruption. ROS can also contribute to mechanisms underlying endothelial barrier disruption [reviewed in (10)]. In this section we review studies that specifically examined redox regulation of SOC entry in endothelial cells.
Elliott et al. (23) performed early experiments looking at the effect of oxidant stress on agonist-induced calcium entry in vascular endothelial cells. In this study, bovine aortic endothelial cells were subjected to oxidative stress using tert-butylhydroperoxide and then were stimulated with the agonist bradykinin. Earlier studies (18, 111) had already established that bradykinin stimulates calcium release from the endoplasmic reticulum followed by entry across the plasma membrane, that is, SOC entry. Using 0.4 mM tert-butylhydroperoxide, the authors observed, using the fluorescent calcium indicator Fura/2, that after 30 min incubation the bradykinin-induced calcium entry was inhibited by over 50%. Importantly, they also tested for effects of tert-butylhydroperoxide on calcium release from the endoplasmic reticulum and observed no change in release after 30 min incubation, overall indicating that the tert-butylhydroperoxide-induced oxidant stress inhibits calcium entry in bovine aortic endothelial cells. These observations were supported in a follow-up study (25) whereby uptake of 45Ca2+ was measured in calf PAECs treated with 0.4 mM tert-butylhydroperoxide followed by exposure to bradykinin. In line with results from the previous study, after tert-butylhydroperoxide treatment, uptake of 45Ca2+ into the cells was decreased. It was not until 1993 that Elliott and Doan (22) specifically focused on the effect of oxidant stress on SOC entry in endothelial cells. Here they used the Ca2+/ATPase inhibitors thapsigargin or 2,5-di-t-butylhydroquinone (BHQ) to directly activate SOC entry. Application of either thapsigargin or BHQ after 30-min and 1-h incubation with 0.4 mM tert-butylhydroperoxide resulted in reduced calcium entry with no changes in the basal calcium levels or calcium release. As these results mirror those obtained using the Gq agonist bradykinin, the authors concluded that the mechanism underlying oxidant-mediated inhibition of calcium entry does not involve decreased synthesis or increased metabolism of InsP3. Instead they proposed that it is likely that the oxidants directly inhibit the calcium entry channels located on the plasma membrane or interfere with the link between the calcium entry channels and the calcium store fill state.
Wesson and Elliott (131) expanded these early studies by exogenously adding xanthine oxidase in the presence of its substrate hypoxanthine to cultured calf PAECs. Xanthine oxidase caused increased basal calcium levels and inhibited bradykinin-induced calcium release from the endoplasmic reticulum as well as subsequent entry. However, when the substrate concentration was decreased, they observed that xanthine oxidase specifically inhibited calcium entry without decreasing calcium release. To determine the molecular species underlying this inhibitory effect on calcium signaling, they used superoxide dismutase (SOD) and catalase as inhibitors of 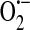 and H2O2, respectively. When SOD was added during exposure to hypoxanthine/xanthine oxidase, the calcium response to bradykinin was still inhibited, whereas catalase addition protected the calcium response, thus suggesting that H2O2 is the species important for inhibition of calcium signaling. This was further demonstrated when glucose–glucose oxidase was used to generate H2O2 and the results closely mirrored those of the hypoxanthine/xanthine oxidase experiments. Henschke and Elliott (39) reasoned that within the cell peroxide metabolism is coupled with GSH oxidation, and as such, perhaps oxidized glutathione (GSSG) is the molecular species important for inhibition of calcium signaling. 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU), an inhibitor of GSSG-reductase, was used to achieved increased cytosolic levels of GSSG after treatment with tert-butylhydroperoxide in calf PAECs. Endothelial cells were then treated with BHQ to inhibit the Ca2+/ATPase leading to release of calcium from the endoplasmic reticulum. The amount of calcium released from the store was less in the treatment group (tert-butylhydroperoxide plus BCNU) as compared to controls (tert-butylhydroperoxide alone or no tert-butylhydroperoxide), leading to the conclusion that increased GSSG after oxidative stress causes depletion of releasable calcium from the InsP3-sensitive store. They then went on to determine the effect of elevated GSSG on calcium entry through the SOC entry mechanism. Here they first depleted the stores, again using BHQ, in low extracellular calcium, after which calcium was then added back to the extracellular media. In this calcium add-back experiment it was observed that the calcium increase due to entry was attenuated in the BCNU/tert-butylhydroperoxide treatment group as compared to controls, and even potentiated the effect of tert-butylhydroperoxide alone. This observation led the authors to postulate that oxidation of thiol residues is occurring directly on the SOC entry channel protein or on a protein involved in the signal that links store depletion to channel activation.
and H2O2, respectively. When SOD was added during exposure to hypoxanthine/xanthine oxidase, the calcium response to bradykinin was still inhibited, whereas catalase addition protected the calcium response, thus suggesting that H2O2 is the species important for inhibition of calcium signaling. This was further demonstrated when glucose–glucose oxidase was used to generate H2O2 and the results closely mirrored those of the hypoxanthine/xanthine oxidase experiments. Henschke and Elliott (39) reasoned that within the cell peroxide metabolism is coupled with GSH oxidation, and as such, perhaps oxidized glutathione (GSSG) is the molecular species important for inhibition of calcium signaling. 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU), an inhibitor of GSSG-reductase, was used to achieved increased cytosolic levels of GSSG after treatment with tert-butylhydroperoxide in calf PAECs. Endothelial cells were then treated with BHQ to inhibit the Ca2+/ATPase leading to release of calcium from the endoplasmic reticulum. The amount of calcium released from the store was less in the treatment group (tert-butylhydroperoxide plus BCNU) as compared to controls (tert-butylhydroperoxide alone or no tert-butylhydroperoxide), leading to the conclusion that increased GSSG after oxidative stress causes depletion of releasable calcium from the InsP3-sensitive store. They then went on to determine the effect of elevated GSSG on calcium entry through the SOC entry mechanism. Here they first depleted the stores, again using BHQ, in low extracellular calcium, after which calcium was then added back to the extracellular media. In this calcium add-back experiment it was observed that the calcium increase due to entry was attenuated in the BCNU/tert-butylhydroperoxide treatment group as compared to controls, and even potentiated the effect of tert-butylhydroperoxide alone. This observation led the authors to postulate that oxidation of thiol residues is occurring directly on the SOC entry channel protein or on a protein involved in the signal that links store depletion to channel activation.
In two follow-up studies, Koliwad et al. (54, 55) identified a potential oxidation target. It was known from an earlier study (24) that cytosolic sodium increased after tert-butylhydroperoxide treatment of endothelial cells. In the first study (55) it was determined that treatment of calf PAECs with tert-butylhydroperoxide led to activation of a current that was equally selective for sodium and potassium. Interestingly, the channel underlying this current also permits calcium entry, which the authors suggested could explain the increased basal calcium after tert-butylhydroperoxide exposure. Second, they observed membrane depolarization, likely due to activation of this nonselective cation channel. As SOC entry is inhibited by membrane depolarization (20, 110), the authors proposed that the mechanism underlying tert-butylhydroperoxide-induced inhibition of SOC entry is due to opening of the nonselective cation channel leading to membrane depolarization. In the second study patch-clamp electrophysiology was performed to identify the molecular species important for activating this nonselective channel. Based on the earlier study (39) that identified GSSG as a critical mediator of SOC entry inhibition, Koliwad et al. (54) examined whether GSSG could activate the nonselective current. Using excised membrane patches, internal but not external GSSG did indeed activate this current. Additionally, internal tert-butylhydroperoxide and reduced GSH were unable to activate the current leading the authors to conclude that under oxidant stress endogenous GSSG mediates opening of a nonselective cation channel activation and membrane depolarization. While GSSG washout did not reverse channel activity, the addition of GSH, to increase the [GSH]:[GSSG] ratio, or dithiothreitol did result in reversal. These observations further suggested that GSSG acts via covalent sulfhydryl oxidation or at least binds with high affinity to some internal receptor.
While these early studies revealed that oxidative stress, when induced by 0.4 mM tert-butylhydroperoxide, leads to inhibition of SOC entry, it appears that not all oxidative species act in the same manner. Favre et al. (27) were interested in the effects of NO and S-nitrosylation on SOC entry. Using membrane-permeant NO donors they demonstrated that calcium entry was activated by NO and that calcium store depletion augments the entry response. Calcium entry was also triggered by alkylating agents that react with thiol groups, to mimic S-nitrosylation, and this entry was also enhanced by store depletion. From these observations, the authors concluded that channel S-nitrosylation activates calcium entry that is also activated by store depletion. In a follow-up study, Ma et al. (67) probed the relationship between S-nitrosylation and SOC entry. Here they examined differences between SOC entry activated by store depletion and calcium entry activated by an NO donor. In an ion selectivity experiment, they observed that with barium as a charge carrier, there were differences in cation entry between the two groups. Further, there were also small differences in sensitivity to lanthanum when used to block entry. Collectively, these results suggested that the NO-mediated calcium entry mechanism was distinct for store-mediated calcium entry. In the next study, van Rossum et al. (125) compared calcium entry mediated by store depletion, that is, SOC entry, with that induced by S-nitrosylation and observed that calcium entry induced by an NO donor was insensitive to the InP3 receptor blocker 2-aminoethoxydiphenyl borate (2-APB), thus reinforcing the earlier results indicating that NO-mediated entry is via a mechanism distinct from SOC entry. More recently, Yoshida et al. (137) described activation of TRP proteins, including TRPC1, TRPC4, and TRPC5, by NO. They went on to demonstrate that in TRPC5, two cysteine residues, Cys553 and Cys558, are S-nitrosylated by NO. Interestingly, and perhaps importantly, the other TRP proteins that are activated by NO also have conserved cysteine residues, leading to the idea that the other TRP homologs are also S-nitrosylated. While still unknown at this time, it is interesting to consider whether S-nitrosylation of TRPC1 and TRPC4, when configured in the ISOC channel arrangement, leads to channel activation.
It is clear that endothelial SOC entry is sensitive to redox regulation, at least in part through an indirect mechanism in which a nonselective cation channel is activated by GSSG oxidation leading to membrane depolarization and inhibition of SOC entry. What is less clear is whether there is also direct redox regulation of SOC entry channels and especially whether the endothelial ISOC channel is directly or indirectly redox regulated. As activation of the endothelial ISOC is an important step leading to endothelial barrier disruption (133), and conditions of oxidative stress have been shown to lead to endothelial barrier disruption [reviewed in (10)], it is important for future studies to determine whether ISOC function is regulated by cellular redox status as well. Based on our current understanding of the field, we propose a model describing how ISOC-induced endothelial barrier disruption can be influenced by cellular redox status. To begin, in an unstimulated state, NAD(P)H oxidase is localized within the cytosol in a perinuclear manner (7). As it is constitutively active (75), 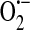 and H2O2 are generated.
and H2O2 are generated. 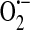 is reduced to H2O2 through the action of SOD. H2O2, in turn, is rapidly metabolized to H2O in a reaction coupled with the oxidation of GSH and dependent upon GSH peroxidase. GSSG is then reduced through the action of GSH reductase. Thus, the GSH:GSSG ratio remains high and redox homeostasis is preserved. In this unstimulated state, when a Gq -linked agonist, for example, the inflammatory mediator thrombin, is introduced to endothelial cells, SOC entry and ISOC will be activated leading to endothelial barrier disruption. Redox homeostasis can be perturbed in different physiological/pathophysiological settings, resulting in a stimulated redox state. In sepsis for example, ROS are synthesized by endothelial cells as well as by activated neutrophils [reviewed in (14)]. In the endothelial cell, NAD(P)H oxidase translocates to the membrane and along with xanthine oxidase produces ROS both intracellularly and extracellularly. Elevated levels of
is reduced to H2O2 through the action of SOD. H2O2, in turn, is rapidly metabolized to H2O in a reaction coupled with the oxidation of GSH and dependent upon GSH peroxidase. GSSG is then reduced through the action of GSH reductase. Thus, the GSH:GSSG ratio remains high and redox homeostasis is preserved. In this unstimulated state, when a Gq -linked agonist, for example, the inflammatory mediator thrombin, is introduced to endothelial cells, SOC entry and ISOC will be activated leading to endothelial barrier disruption. Redox homeostasis can be perturbed in different physiological/pathophysiological settings, resulting in a stimulated redox state. In sepsis for example, ROS are synthesized by endothelial cells as well as by activated neutrophils [reviewed in (14)]. In the endothelial cell, NAD(P)H oxidase translocates to the membrane and along with xanthine oxidase produces ROS both intracellularly and extracellularly. Elevated levels of 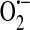 and H2O2 lead to a decreased GSH:GSSG ratio, resulting in increased levels of GSSG. Calcium loading in the endoplasmic reticulum is sensitive to GSSG and is decreased in the presence of GSSG (39). Additionally, GSSG can react with available sulfhydryl groups on proteins resulting in glutathionylation [reviewed in (31)]. Of particular interest here is the glutathionylation of a nonselective cation channel that leads to channel activation, influx of sodium, and membrane depolarization (54, 55), which ultimately reduces the driving force for calcium entry. In this stimulated state, in the presence of Gq-linked agonists, the decreased calcium levels in the endoplasmic reticulum plus membrane depolarization will lead to attenuated SOC entry and ISOC activation (Fig. 6a). Thus, in this setting of oxidative stress, ISOC activation is not a contributing factor to endothelial barrier disruption. However, mechanisms underlying endothelial barrier disruption in sepsis likely involve other redox-sensitive reactions. For example, nitration of cytoskeletal proteins such as actin and β-catenin by the RNS ONOO− has been implicated in endothelial barrier disruption (52, 82). While ISOC activation may indeed be inhibited in the presence of elevated
and H2O2 lead to a decreased GSH:GSSG ratio, resulting in increased levels of GSSG. Calcium loading in the endoplasmic reticulum is sensitive to GSSG and is decreased in the presence of GSSG (39). Additionally, GSSG can react with available sulfhydryl groups on proteins resulting in glutathionylation [reviewed in (31)]. Of particular interest here is the glutathionylation of a nonselective cation channel that leads to channel activation, influx of sodium, and membrane depolarization (54, 55), which ultimately reduces the driving force for calcium entry. In this stimulated state, in the presence of Gq-linked agonists, the decreased calcium levels in the endoplasmic reticulum plus membrane depolarization will lead to attenuated SOC entry and ISOC activation (Fig. 6a). Thus, in this setting of oxidative stress, ISOC activation is not a contributing factor to endothelial barrier disruption. However, mechanisms underlying endothelial barrier disruption in sepsis likely involve other redox-sensitive reactions. For example, nitration of cytoskeletal proteins such as actin and β-catenin by the RNS ONOO− has been implicated in endothelial barrier disruption (52, 82). While ISOC activation may indeed be inhibited in the presence of elevated 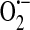 and H2O2, it is important to recognize that not all oxidative stress produces the same ROS. For instance, in ischemia/reperfusion settings the highly reactive OH• is formed downstream of
and H2O2, it is important to recognize that not all oxidative stress produces the same ROS. For instance, in ischemia/reperfusion settings the highly reactive OH• is formed downstream of 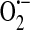 production (4, 57). OH• is synthesized from H2O2 via the Fenton reaction [reviewed in (30)]. A critical factor in the Fenton reaction is that H2O2 must come into contact with reduced metal cations such as iron(II) in order for the reaction to proceed. Importantly, ischemia promotes release of free iron from cellular storage sites (3, 139), thus setting the stage for the Fenton reaction to occur. The OH• is a powerful oxidant that, in the presence of membrane lipids, can cause lipid peroxidation and the formation of HNE [reviewed in (120)]. HNE reacts with available sulfhydryl groups, such as those of GSH [reviewed in (109)] and will ultimately deplete the cellular GSH pool. As the overall GSH pool disappears, levels of GSSG will also decrease. In the absence of GSSG, the nonselective cation channel will no longer be activated. As such, the membrane potential will become more hyperpolarized, restoring the driving force for calcium entry. Additionally, calcium levels in the endoplasmic reticulum will return to a normal fill state. In this setting, as Gq-linked agonists promote calcium store depletion, the signal will be sent to the membrane, and ISOC will be activated (Fig. 6b) leading to intercellular gap formation and endothelial barrier disruption. Thus, our model proposes that ISOC channel function is indeed regulated by cellular redox status, and importantly, is exquisitely sensitive to the particular ROS species formed.
production (4, 57). OH• is synthesized from H2O2 via the Fenton reaction [reviewed in (30)]. A critical factor in the Fenton reaction is that H2O2 must come into contact with reduced metal cations such as iron(II) in order for the reaction to proceed. Importantly, ischemia promotes release of free iron from cellular storage sites (3, 139), thus setting the stage for the Fenton reaction to occur. The OH• is a powerful oxidant that, in the presence of membrane lipids, can cause lipid peroxidation and the formation of HNE [reviewed in (120)]. HNE reacts with available sulfhydryl groups, such as those of GSH [reviewed in (109)] and will ultimately deplete the cellular GSH pool. As the overall GSH pool disappears, levels of GSSG will also decrease. In the absence of GSSG, the nonselective cation channel will no longer be activated. As such, the membrane potential will become more hyperpolarized, restoring the driving force for calcium entry. Additionally, calcium levels in the endoplasmic reticulum will return to a normal fill state. In this setting, as Gq-linked agonists promote calcium store depletion, the signal will be sent to the membrane, and ISOC will be activated (Fig. 6b) leading to intercellular gap formation and endothelial barrier disruption. Thus, our model proposes that ISOC channel function is indeed regulated by cellular redox status, and importantly, is exquisitely sensitive to the particular ROS species formed.
FIG. 6.

Model of ROS-dependent store-operated calcium current (ISOC) regulation. (a) In the setting of oxidative stress, such as sepsis, NAD(P)H oxidase (multicolor) translocates from the cytosol along microtubules (blue) to the plasma membrane where it produces intra- and extracellular 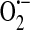 . Additionally, XO, which is found both in the cytosol as well as extracellularly near sites of cell–cell contact (106), also produces
. Additionally, XO, which is found both in the cytosol as well as extracellularly near sites of cell–cell contact (106), also produces 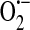 . In the presence of superoxide dismutase (SOD),
. In the presence of superoxide dismutase (SOD), 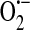 is converted to hydrogen peroxide (H2O2), which in turn oxidizes GSH to GSSG. Elevated levels of GSSG lead to decreased levels of calcium in the ER (39). Additionally, GSSG reacts with available thiol bearing amino acid residues on the nonselective cation channel to form and activate a GSH channel adduct. Channel activation allows for influx of cations (principally sodium) and promotes membrane depolarization. The combination of decreased calcium store levels with membrane depolarization inhibits agonist-induced SOC entry and ISOC activation. (b) In the setting of oxidative stress such as in ischemia/reperfusion, in addition to
is converted to hydrogen peroxide (H2O2), which in turn oxidizes GSH to GSSG. Elevated levels of GSSG lead to decreased levels of calcium in the ER (39). Additionally, GSSG reacts with available thiol bearing amino acid residues on the nonselective cation channel to form and activate a GSH channel adduct. Channel activation allows for influx of cations (principally sodium) and promotes membrane depolarization. The combination of decreased calcium store levels with membrane depolarization inhibits agonist-induced SOC entry and ISOC activation. (b) In the setting of oxidative stress such as in ischemia/reperfusion, in addition to 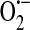 production, Fe(II) is released from cellular storage sites (3, 139). This sets the stage for formation of the extremely reactive OH• via Fenton reaction chemistry. OH• will react with PUFAs of membranes forming 4-hydroxy-2-nonenal (HNE). HNE readily reacts with GSH resulting in GSSG depletion. In the absence of GSSG, the nonselective cation channel is no longer activated and membrane depolarization is reversed. Additionally, the calcium store will return to its normal fill state. Now in the presence of Gq-linked agonists, the signal will be sent from the ER to the plasma membrane leading to activation of the ISOC channel. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
production, Fe(II) is released from cellular storage sites (3, 139). This sets the stage for formation of the extremely reactive OH• via Fenton reaction chemistry. OH• will react with PUFAs of membranes forming 4-hydroxy-2-nonenal (HNE). HNE readily reacts with GSH resulting in GSSG depletion. In the absence of GSSG, the nonselective cation channel is no longer activated and membrane depolarization is reversed. Additionally, the calcium store will return to its normal fill state. Now in the presence of Gq-linked agonists, the signal will be sent from the ER to the plasma membrane leading to activation of the ISOC channel. (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
Summary
Activation of the endothelial ISOC channel is an important step leading to intercellular gap formation and endothelial barrier disruption. Likewise, oxidative stress can trigger endothelial barrier disruption. To date it is unclear whether there is a link between ISOC channel function and oxidative stress. Indeed, it is conceivable that the ISOC channel is indirectly regulated by cellular redox status through a nonselective cation channel that is covalently modified by GSSG. Alternatively, the ISOC channel may itself be a direct target of GSSG or some other ROS species. Along this line, we searched for the Nudix motif in TRPC1 and TRPC4, two subunits that contribute to the molecular makeup of the endothelial ISOC channel, but did not find one (unpublished data). However, this does not rule out direct oxidative modification by other species such as RNS, in a manner similar to what has been observed for NO modification of TRPC5. Overall, we have much to learn about redox regulation of SOC entry channels, in general, and the endothelial ISOC channel, in particular. Indeed, we need to better understand the relationship between cellular redox status and ISOC channel function to more fully appreciate the downstream physiological and pathophysiological effects.
Abbreviations Used
- 2-APB
2-aminoethoxydiphenyl borate
- ADPR
adenosine 5′-diphosphoribose
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- BCNU
1,3-bis-(2-chloroethyl)-1-nitrosourea
- BHQ
2,5-di-t-butylhydroquinone
- [Ca2+]i
cytosolic calcium
- cADPR
cyclic adenosine 5′-diphosphoribose
- cDNA
complementary DNA
- CRAC
calcium release activated calcium
- DAG
diacylglycerol
- DPI
diphenyleneiodonium
- eNOS
endothelial nitric oxide synthase
- EOH
hydroxide anion
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinase
- GFP-PH-PLCδ
PIP2-binding GFP fusion protein
- GSH
glutathione
- GSH-Px
glutathione peroxidase
- GSH-Red.
glutathione reductase
- GSSG
oxidized glutathione
- GST
glutathione S-transferase
- HEK293
human embryonic kidney 293
- HNE
4-hydroxy-2-nonenal
- H2O
water
- HSP27
heat-shock protein-27
- HUVECs
human umbilical vein endothelial cell
- ICRAC
calcium release activated calcium current
- InsP3
inositol-1,4,5-trisphosphate
- ISOC
store-operated calcium current
- JNK
c-Jun N-terminal kinase
- kDa
kilodalton
- LTRPC
long transient receptor potential canonical
- MAP
mitogen-activated protein
- mM
millimolar
- mV
millivolt
- NAD+
oxidized nicotinamide adenine dinucleotide
- NADH
nicotinamide adenine dinucleotide
- NADP+
oxidized nicotinamide adenine dinucleotide phosphate
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- Nudix
nucleoside diphosphate linked to some other moiety, X
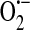
superoxide
- OH•
hydroxyl radical
- ONOOE
peroxynitrite
- pADPR
poly(ADP-ribose)
- PAEC
pulmonary artery endothelial cell
- PARP
poly(ADP-ribose) polymerase
- PIP2
phosphoinositol-4,5-bisphosphate
- PLC
phospholipase C
- PUFA
polyunsaturated fatty acid
- RNS
reactive nitrogen species
- ROC
receptor operated channels
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- SOC
store-operated calcium
- SOD
superoxide dismutase
- STIM
stromal interaction molecule
- Tg
thapsigargin
- tNOS
total nitric oxide synthase
- TRP
transient receptor potential
- TRPC
transient receptor potential canonical
- TRPM
transient receptor potential melastatin
- XDH
xanthine dehydrogenase
- XO
xanthine oxidase
- μM
micromolar
Acknowledgments
The author would like to thank Ms. Sidra Rasool for her help in preparing figures for this article. Funding is provided by NIH 5R00HL089361-03.
References
- 1.Abdullaev IF. Bisaillon JM. Potier M. Gonzalez JC. Motiani RK. Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adachi T. Fukushima T. Usami Y. Hirano K. Binding of human xanthine oxidase to sulphated glycosaminoglycans on the endothelial-cell surface. Biochem J. 1993;289(Pt 2):523–527. doi: 10.1042/bj2890523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Al-Mehdi AB. Zhao G. Tozawa K. Fisher AB. Depolarization-associated iron release with abrupt reduction in pulmonary endothelial shear stress in situ. Antioxid Redox Signal. 2000;2:335–345. doi: 10.1089/ars.2000.2.2-335. [DOI] [PubMed] [Google Scholar]
- 4.Arroyo CM. Kramer JH. Dickens BF. Weglicki WB. Identification of free radicals in myocardial ischemia/reperfusion by spin trapping with nitrone DMPO. FEBS Lett. 1987;221:101–104. doi: 10.1016/0014-5793(87)80360-5. [DOI] [PubMed] [Google Scholar]
- 5.Bakowski D. Parekh AB. Voltage-dependent conductance changes in the store-operated Ca2+ current ICRAC in rat basophilic leukaemia cells. J Physiol. 2000;529(Pt 2):295–306. doi: 10.1111/j.1469-7793.2000.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balzer M. Lintschinger B. Groschner K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc Res. 1999;42:543–549. doi: 10.1016/s0008-6363(99)00025-5. [DOI] [PubMed] [Google Scholar]
- 7.Bayraktutan U. Blayney L. Shah AM. Molecular characterization and localization of the NAD(P)H oxidase components gp91-phox and p22-phox in endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:1903–1911. doi: 10.1161/01.atv.20.8.1903. [DOI] [PubMed] [Google Scholar]
- 8.Bayraktutan U. Draper N. Lang D. Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res. 1998;38:256–262. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 9.Bessman MJ. Frick DN. O'Handley SF. The MutT proteins or “Nudix” hydrolases, a family of versatile, widely distributed, “housecleaning” enzymes. J Biol Chem. 1996;271:25059–25062. doi: 10.1074/jbc.271.41.25059. [DOI] [PubMed] [Google Scholar]
- 10.Boueiz A. Hassoun PM. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc Res. 2009;77:26–34. doi: 10.1016/j.mvr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 11.Brough GH. Wu S. Cioffi D. Moore TM. Li M. Dean N. Stevens T. Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 2001;15:1727–1738. [PubMed] [Google Scholar]
- 12.Buelow B. Song Y. Scharenberg AM. The poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cai H. Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 14.Cepinskas G. Wilson JX. Inflammatory response in microvascular endothelium in sepsis: role of oxidants. J Clin Biochem Nutr. 2008;42:175–184. doi: 10.3164/jcbn.2008026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng KT. Liu X. Ong HL. Ambudkar IS. Functional requirement for Orai1 in store-operated TRPC1-STIM1 channels. J Biol Chem. 2008;283:12935–12940. doi: 10.1074/jbc.C800008200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chetham PM. Babal P. Bridges JP. Moore TM. Stevens T. Segmental regulation of pulmonary vascular permeability by store-operated Ca2+ entry. Am J Physiol Lung Cell Mol Physiol. 1999;276:L41–L50. doi: 10.1152/ajplung.1999.276.1.L41. [DOI] [PubMed] [Google Scholar]
- 17.Cioffi DL. Wu S. Stevens T. Transient receptor potential cation channels and store-operated Ca2+ channels. In: Yuan JX-J, editor. Ion Channels in the Pulmonary Vasculature. Boca Raton: Taylor & Francis Group; 2005. pp. 125–146. [Google Scholar]
- 18.Colden-Stanfield M. Schilling WP. Ritchie AK. Eskin SG. Navarro LT. Kunze DL. Bradykinin-induced increases in cytosolic calcium and ionic currents in cultured bovine aortic endothelial cells. Circ Res. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- 19.Cosens DJ. Manning A. Abnormal electroretinogram from a Drosophila mutant. Nature. 1969;224:285–287. doi: 10.1038/224285a0. [DOI] [PubMed] [Google Scholar]
- 20.Di Virgilio F. Lew PD. Andersson T. Pozzan T. Plasma membrane potential modulates chemotactic peptide-stimulated cytosolic free Ca2+ changes in human neutrophils. J Biol Chem. 1987;262:4574–4579. [PubMed] [Google Scholar]
- 21.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 22.Elliott SJ. Doan TN. Oxidant stress inhibits the store-dependent Ca(2+)-influx pathway of vascular endothelial cells. Biochem J. 1993;292(Pt 2):385–393. doi: 10.1042/bj2920385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elliott SJ. Eskin SG. Schilling WP. Effect of t-butyl-hydroperoxide on bradykinin-stimulated changes in cytosolic calcium in vascular endothelial cells. J Biol Chem. 1989;264:3806–3810. [PubMed] [Google Scholar]
- 24.Elliott SJ. Schilling WP. Oxidant stress alters Na+ pump and Na(+)-K(+)-Cl- cotransporter activities in vascular endothelial cells. Am J Physiol. 1992;263:H96–H102. doi: 10.1152/ajpheart.1992.263.1.H96. [DOI] [PubMed] [Google Scholar]
- 25.Elliott SJ. Schilling WP. Oxidative stress inhibits bradykinin-stimulated 45Ca2+ flux in pulmonary vascular endothelial cells. Am J Physiol. 1991;260:H549–H556. doi: 10.1152/ajpheart.1991.260.2.H549. [DOI] [PubMed] [Google Scholar]
- 26.Fasolato C. Nilius B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflugers Arch Eur J Physiol. 1998;436:69–74. doi: 10.1007/s004240050605. [DOI] [PubMed] [Google Scholar]
- 27.Favre CJ. Ufret-Vincenty CA. Stone MR. Ma HT. Gill DL. Ca2+ pool emptying stimulates Ca2+ entry activated by S-nitrosylation. J Biol Chem. 1998;273:30855–30858. doi: 10.1074/jbc.273.47.30855. [DOI] [PubMed] [Google Scholar]
- 28.Feske S. Gwack Y. Prakriya M. Srikanth S. Puppel SH. Tanasa B. Hogan PG. Lewis RS. Daly M. Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 29.Freichel M. Suh SH. Pfeifer A. Schweig U. Trost C. Weissgerber P. Biel M. Philipp S. Freise D. Droogmans G. Hofmann F. Flockerzi V. Nilius B. Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat Cell Biol. 2001;3:121–127. doi: 10.1038/35055019. [DOI] [PubMed] [Google Scholar]
- 30.Fridovich I. Oxygen toxicity: a radical explanation. J Exp Biol. 1998;201:1203–1209. doi: 10.1242/jeb.201.8.1203. [DOI] [PubMed] [Google Scholar]
- 31.Gallogly MM. Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Garcia JG. Concepts in microvascular endothelial barrier regulation in health and disease. Microvasc Res. 2009;77:1–3. doi: 10.1016/j.mvr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Groschner K. Hingel S. Lintschinger B. Balzer M. Romanin C. Zhu X. Schreibmayer W. Trp proteins form store-operated cation channels in human vascular endothelial cells. FEBS Lett. 1998;437:101–106. doi: 10.1016/s0014-5793(98)01212-5. [DOI] [PubMed] [Google Scholar]
- 34.Groschner K. Rosker C. Lukas M. Role of TRP channels in oxidative stress. Novartis Found Symp. 2004;258:222–230. discussion 231–225, 263–226, [PubMed] [Google Scholar]
- 35.Hara Y. Wakamori M. Ishii M. Maeno E. Nishida M. Yoshida T. Yamada H. Shimizu S. Mori E. Kudoh J. Shimizu N. Kurose H. Okada Y. Imoto K. Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 36.Hardie RC. Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 37.Harrison R. Structure and function of xanthine oxidoreductase: where are we now? Free Radic Biol Med. 2002;33:774–797. doi: 10.1016/s0891-5849(02)00956-5. [DOI] [PubMed] [Google Scholar]
- 38.Hecquet CM. Ahmmed GU. Vogel SM. Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- 39.Henschke PN. Elliott SJ. Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell ins(1,4,5)P3-sensitive Ca2+ store. Biochem J. 1995;312(Pt 2):485–489. doi: 10.1042/bj3120485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holland JA. Pritchard KA. Pappolla MA. Wolin MS. Rogers NJ. Stemerman MB. Bradykinin induces superoxide anion release from human endothelial cells. J Cell Physiol. 1990;143:21–25. doi: 10.1002/jcp.1041430104. [DOI] [PubMed] [Google Scholar]
- 41.Hoth M. Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 42.Huppert J. Closhen D. Croxford A. White R. Kulig P. Pietrowski E. Bechmann I. Becher B. Luhmann HJ. Waisman A. Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- 43.Ichikawa M. Nishino T. Nishino T. Ichikawa A. Subcellular localization of xanthine oxidase in rat hepatocytes: high-resolution immunoelectron microscopic study combined with biochemical analysis. J Histochem Cytochem. 1992;40:1097–1103. doi: 10.1177/40.8.1619276. [DOI] [PubMed] [Google Scholar]
- 44.Ishii M. Shimizu S. Hara Y. Hagiwara T. Miyazaki A. Mori Y. Kiuchi Y. Intracellular-produced hydroxyl radical mediates H2O2-induced Ca2+ influx and cell death in rat beta-cell line RIN-5F. Cell Calcium. 2006;39:487–494. doi: 10.1016/j.ceca.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Jarasch ED. Grund C. Bruder G. Heid HW. Keenan TW. Franke WW. Localization of xanthine oxidase in mammary-gland epithelium and capillary endothelium. Cell. 1981;25:67–82. doi: 10.1016/0092-8674(81)90232-4. [DOI] [PubMed] [Google Scholar]
- 46.Jardin I. Lopez JJ. Salido GM. Rosado JA. Orai1 mediates the interaction between STIM1 and hTRPC1 and regulates the mode of activation of hTRPC1-forming Ca2+ channels. J Biol Chem. 2008;283:25296–25304. doi: 10.1074/jbc.M802904200. [DOI] [PubMed] [Google Scholar]
- 47.Jesaitis AJ. Buescher ES. Harrison D. Quinn MT. Parkos CA. Livesey S. Linner J. Ultrastructural localization of cytochrome b in the membranes of resting and phagocytosing human granulocytes. J Clin Invest. 1990;85:821–835. doi: 10.1172/JCI114509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones SA. O'Donnell VB. Wood JD. Broughton JP. Hughes EJ. Jones OT. Expression of phagocyte NADPH oxidase components in human endothelial cells. Am J Physiol. 1996;271:H1626–H1634. doi: 10.1152/ajpheart.1996.271.4.H1626. [DOI] [PubMed] [Google Scholar]
- 49.Kelly JJ. Moore TM. Babal P. Diwan AH. Stevens T. Thompson WJ. Pulmonary microvascular and macrovascular endothelial cells: differential regulation of Ca2+ and permeability. Am J Physiol Lung Cell Mol Physiol. 1998;274:L810–L819. doi: 10.1152/ajplung.1998.274.5.L810. [DOI] [PubMed] [Google Scholar]
- 50.Kinnula VL. Mirza Z. Crapo JD. Whorton AR. Modulation of hydrogen peroxide release from vascular endothelial cells by oxygen. Am J Respir Cell Mol Biol. 1993;9:603–609. doi: 10.1165/ajrcmb/9.6.603. [DOI] [PubMed] [Google Scholar]
- 51.Kinnula VL. Whorton AR. Chang LY. Crapo JD. Regulation of hydrogen peroxide generation in cultured endothelial cells. Am J Respir Cell Mol Biol. 1992;6:175–182. doi: 10.1165/ajrcmb/6.2.175. [DOI] [PubMed] [Google Scholar]
- 52.Knepler JL., Jr. Taher LN. Gupta MP. Patterson C. Pavalko F. Ober MD. Hart CM. Peroxynitrite causes endothelial cell monolayer barrier dysfunction. Am J Physiol Cell Physiol. 2001;281:C1064–C1075. doi: 10.1152/ajpcell.2001.281.3.C1064. [DOI] [PubMed] [Google Scholar]
- 53.Kolisek M. Beck A. Fleig A. Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. doi: 10.1016/j.molcel.2005.02.033. [DOI] [PubMed] [Google Scholar]
- 54.Koliwad SK. Elliott SJ. Kunze DL. Oxidized glutathione mediates cation channel activation in calf vascular endothelial cells during oxidant stress. J Physiol. 1996;495(Pt 1):37–49. doi: 10.1113/jphysiol.1996.sp021572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koliwad SK. Kunze DL. Elliott SJ. Oxidant stress activates a non-selective cation channel responsible for membrane depolarization in calf vascular endothelial cells. J Physiol. 1996;491(Pt 1):1–12. doi: 10.1113/jphysiol.1996.sp021191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kraft R. Grimm C. Grosse K. Hoffmann A. Sauerbruch S. Kettenmann H. Schultz G. Harteneck C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol. 2004;286:C129–C137. doi: 10.1152/ajpcell.00331.2003. [DOI] [PubMed] [Google Scholar]
- 57.Kramer JH. Arroyo CM. Dickens BF. Weglicki WB. Spin-trapping evidence that graded myocardial ischemia alters post-ischemic superoxide production. Free Radic Biol Med. 1987;3:153–159. doi: 10.1016/s0891-5849(87)80011-4. [DOI] [PubMed] [Google Scholar]
- 58.Leusen JH. Verhoeven AJ. Roos D. Interactions between the components of the human NADPH oxidase: intrigues in the phox family. J Lab Clin Med. 1996;128:461–476. doi: 10.1016/s0022-2143(96)90043-8. [DOI] [PubMed] [Google Scholar]
- 59.Li JM. Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 60.Li JM. Shah AM. Intracellular localization and preassembly of the NADPH oxidase complex in cultured endothelial cells. J Biol Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- 61.Li SW. Westwick J. Poll CT. Receptor-operated Ca2+ influx channels in leukocytes: a therapeutic target? Trends Pharmacol Sci. 2002;23:63–70. doi: 10.1016/s0165-6147(00)01897-6. [DOI] [PubMed] [Google Scholar]
- 62.Liao Y. Erxleben C. Abramowitz J. Flockerzi V. Zhu MX. Armstrong DL. Birnbaumer L. Functional interactions among Orai1, TRPCs, and STIM1 suggest a STIM-regulated heteromeric Orai/TRPC model for SOCE/Icrac channels. Proc Natl Acad Sci U S A. 2008;105:2895–2900. doi: 10.1073/pnas.0712288105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y. Erxleben C. Yildirim E. Abramowitz J. Armstrong DL. Birnbaumer L. Orai proteins interact with TRPC channels and confer responsiveness to store depletion. Proc Natl Acad Sci U S A. 2007;104:4682–4687. doi: 10.1073/pnas.0611692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y. Plummer NW. George MD. Abramowitz J. Zhu MX. Birnbaumer L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci U S A. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu X. Singh BB. Ambudkar IS. TRPC1 is required for functional store-operated Ca2+ channels. Role of acidic amino acid residues in the S5-S6 region. J Biol Chem. 2003;278:11337–11343. doi: 10.1074/jbc.M213271200. [DOI] [PubMed] [Google Scholar]
- 66.Liu X. Wang W. Singh BB. Lockwich T. Jadlowiec J. O'Connell B. Wellner R. Zhu MX. Ambudkar IS. Trp1, a candidate protein for the store-operated Ca(2+) influx mechanism in salivary gland cells. J Biol Chem. 2000;275:3403–3411. doi: 10.1074/jbc.275.5.3403. [DOI] [PubMed] [Google Scholar]
- 67.Ma HT. Favre CJ. Patterson RL. Stone MR. Gill DL. Ca(2+) entry activated by S-nitrosylation. Relationship to store-operated ca(2+) entry. J Biol Chem. 1999;274:35318–35324. doi: 10.1074/jbc.274.50.35318. [DOI] [PubMed] [Google Scholar]
- 68.Martinez MC. Andriantsitohaina R. Reactive nitrogen species: molecular mechanisms and potential significance in health and disease. Antioxid Redox signal. 2009;11:669–702. doi: 10.1089/ars.2007.1993. [DOI] [PubMed] [Google Scholar]
- 69.Maruyama Y. Ogura T. Mio K. Kato K. Kaneko T. Kiyonaka S. Mori Y. Sato C. Tetrameric Orai1 is a teardrop-shaped molecule with a long, tapered cytoplasmic domain. J Biol Chem. 2009;284:13676–13685. doi: 10.1074/jbc.M900812200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matsubara T. Ziff M. Superoxide anion release by human endothelial cells: synergism between a phorbol ester and a calcium ionophore. J Cell Physiol. 1986;127:207–210. doi: 10.1002/jcp.1041270203. [DOI] [PubMed] [Google Scholar]
- 71.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 72.Mignen O. Thompson JL. Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mikkelsen RB. Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene. 2003;22:5734–5754. doi: 10.1038/sj.onc.1206663. [DOI] [PubMed] [Google Scholar]
- 74.Minke B. TRP channels and Ca2+ signaling. Cell Calcium. 2006;40:261–275. doi: 10.1016/j.ceca.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohazzab KM. Kaminski PM. Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol. 1994;266:H2568–H2572. doi: 10.1152/ajpheart.1994.266.6.H2568. [DOI] [PubMed] [Google Scholar]
- 76.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 77.Montell C. Birnbaumer L. Flockerzi V. Bindels RJ. Bruford EA. Caterina MJ. Clapham DE. Harteneck C. Heller S. Julius D. Kojima I. Mori Y. Penner R. Prawitt D. Scharenberg AM. Schultz G. Shimizu N. Zhu MX. A unified nomenclature for the superfamily of TRP cation channels. Mol Cell. 2002;9:229–231. doi: 10.1016/s1097-2765(02)00448-3. [DOI] [PubMed] [Google Scholar]
- 78.Moore T. Brough G. Kelly J. Babal P. Li M. Stevens T. Regulation of pulmonary endothelial cell shape by Trp-mediated calcium entry. Chest. 1998;114:36S–38S. doi: 10.1378/chest.114.1_supplement.36s. [DOI] [PubMed] [Google Scholar]
- 79.Moore TM. Brough GH. Babal P. Kelly JJ. Li M. Stevens T. Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1. Am J Physiol Lung Cell Mol Physiol. 1998;275:L574–L582. doi: 10.1152/ajplung.1998.275.3.L574. [DOI] [PubMed] [Google Scholar]
- 80.Naziroğlu M. New molecular mechanisms on the activation of TRPM2 channels by oxidative stress and ADP-ribose. Neurochem Res. 2007;32:1990–2001. doi: 10.1007/s11064-007-9386-x. [DOI] [PubMed] [Google Scholar]
- 81.Naziroğlu M. Luckhoff A. A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: single channel events. Neurochem Res. 2008;33:1256–1262. doi: 10.1007/s11064-007-9577-5. [DOI] [PubMed] [Google Scholar]
- 82.Neumann P. Gertzberg N. Vaughan E. Weisbrot J. Woodburn R. Lambert W. Johnson A. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am J Physiol Lung Cell Mol Physiol. 2006;290:L674–L684. doi: 10.1152/ajplung.00391.2005. [DOI] [PubMed] [Google Scholar]
- 83.Nilius B. Mahieu F. A road map for TR(I)Ps. Mol Cell. 2006;22:297–307. doi: 10.1016/j.molcel.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 84.Nilius B. Viana F. Droogmans G. Ion channels in vascular endothelium. Annu Rev Physiol. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- 85.Nordberg J. Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 86.Ong HL. Cheng KT. Liu X. Bandyopadhyay BC. Paria BC. Soboloff J. Pani B. Gwack Y. Srikanth S. Singh BB. Gill DL. Ambudkar IS. Dynamic assembly of TRPC1-STIM1-Orai1 ternary complex is involved in store-operated calcium influx. Evidence for similarities in store-operated and calcium release-activated calcium channel components. J Biol Chem. 2007;282:9105–9116. doi: 10.1074/jbc.M608942200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parekh AB. Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 88.Paria BC. Vogel SM. Ahmmed GU. Alamgir S. Shroff J. Malik AB. Tiruppathi C. Tumor necrosis factor-alpha-induced TRPC1 expression amplifies store-operated Ca2+ influx and endothelial permeability. Am J Physiol Lung Cell Mol Physiol. 2004;287:L1303–L1313. doi: 10.1152/ajplung.00240.2004. [DOI] [PubMed] [Google Scholar]
- 89.Partridge CA. Blumenstock FA. Malik AB. Pulmonary microvascular endothelial cells constitutively release xanthine oxidase. Arch Biochem Biophys. 1992;294:184–187. doi: 10.1016/0003-9861(92)90155-p. [DOI] [PubMed] [Google Scholar]
- 90.Patterson CE. Lum H. Update on pulmonary edema: the role and regulation of endothelial barrier function. Endothelium. 2001;8:75–105. doi: 10.3109/10623320109165319. [DOI] [PubMed] [Google Scholar]
- 91.Pedersen SF. Owsianik G. Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 92.Penna A. Demuro A. Yeromin AV. Zhang SL. Safrina O. Parker I. Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Perraud AL. Fleig A. Dunn CA. Bagley LA. Launay P. Schmitz C. Stokes AJ. Zhu Q. Bessman MJ. Penner R. Kinet JP. Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- 94.Perraud AL. Takanishi CL. Shen B. Kang S. Smith MK. Schmitz C. Knowles HM. Ferraris D. Li W. Zhang J. Stoddard BL. Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 95.Philipp S. Cavalié A. Freichel M. Wissenbach U. Zimmer S. Trost C. Marquart A. Murakami M. Flockerzi V. A mammalian capacitative calcium entry channel homologous to Drosophila TRP and TRPL. EMBO J. 1996;15:6166–6171. [PMC free article] [PubMed] [Google Scholar]
- 96.Philipp S. Trost C. Warnat J. Rautmann J. Himmerkus N. Schroth G. Kretz O. Nastainczyk W. Cavalie A. Hoth M. Flockerzi V. TRP4 (CCE1) protein is part of native calcium release-activated Ca2+-like channels in adrenal cells. J Biol Chem. 2000;275:23965–23972. doi: 10.1074/jbc.M003408200. [DOI] [PubMed] [Google Scholar]
- 97.Phillips PG. Tsan MF. Hyperoxia causes increased albumin permeability of cultured endothelial monolayers. J Appl Physiol. 1988;64:1196–1202. doi: 10.1152/jappl.1988.64.3.1196. [DOI] [PubMed] [Google Scholar]
- 98.Poredos P. Endothelial dysfunction in the pathogenesis of atherosclerosis. Clin Appl Thromb Hemost. 2001;7:276–280. doi: 10.1177/107602960100700404. [DOI] [PubMed] [Google Scholar]
- 99.Poteser M. Graziani A. Rosker C. Eder P. Derler I. Kahr H. Zhu MX. Romanin C. Groschner K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- 100.Prakriya M. Feske S. Gwack Y. Srikanth S. Rao A. Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 101.Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 102.Putney JW., Jr. McKay RR. Capacitative calcium entry channels. Bioessays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 103.Ratych RE. Chuknyiska RS. Bulkley GB. The primary localization of free radical generation after anoxia/reoxygenation in isolated endothelial cells. Surgery. 1987;102:122–131. [PubMed] [Google Scholar]
- 104.Roos J. DiGregorio PJ. Yeromin AV. Ohlsen K. Lioudyno M. Zhang S. Safrina O. Kozak JA. Wagner SL. Cahalan MD. Velicelebi G. Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rotrosen D. Gallin JI. Histamine type I receptor occupancy increases endothelial cytosolic calcium, reduces F-actin, and promotes albumin diffusion across cultured endothelial monolayers. J Cell Biol. 1986;103:2379–2387. doi: 10.1083/jcb.103.6.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rouquette M. Page S. Bryant R. Benboubetra M. Stevens CR. Blake DR. Whish WD. Harrison R. Tosh D. Xanthine oxidoreductase is asymmetrically localised on the outer surface of human endothelial and epithelial cells in culture. FEBS Lett. 1998;426:397–401. doi: 10.1016/s0014-5793(98)00385-8. [DOI] [PubMed] [Google Scholar]
- 107.Sano Y. Inamura K. Miyake A. Mochizuki S. Yokoi H. Matsushime H. Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. doi: 10.1126/science.1062473. [DOI] [PubMed] [Google Scholar]
- 108.Saugstad OD. Role of xanthine oxidase and its inhibitor in hypoxia: reoxygenation injury. Pediatrics. 1996;98:103–107. [PubMed] [Google Scholar]
- 109.Schaur RJ. Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol Aspects Med. 2003;24:149–159. doi: 10.1016/s0098-2997(03)00009-8. [DOI] [PubMed] [Google Scholar]
- 110.Schilling WP. Rajan L. Strobl-Jager E. Characterization of the bradykinin-stimulated calcium influx pathway of cultured vascular endothelial cells. Saturability, selectivity, and kinetics. J Biol Chem. 1989;264:12838–12848. [PubMed] [Google Scholar]
- 111.Schilling WP. Ritchie AK. Navarro LT. Eskin SG. Bradykinin-stimulated calcium influx in cultured bovine aortic endothelial cells. Am J Physiol. 1988;255:H219–H227. doi: 10.1152/ajpheart.1988.255.2.H219. [DOI] [PubMed] [Google Scholar]
- 112.Seal JB. Gewertz BL. Vascular dysfunction in ischemia-reperfusion injury. Ann Vasc Surg. 2005;19:572–584. doi: 10.1007/s10016-005-4616-7. [DOI] [PubMed] [Google Scholar]
- 113.Soboloff J. Spassova MA. Tang XD. Hewavitharana T. Xu W. Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 114.Strübing C. Krapivinsky G. Krapivinsky L. Clapham DE. Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem. 2003;278:39014–39019. doi: 10.1074/jbc.M306705200. [DOI] [PubMed] [Google Scholar]
- 115.Sundivakkam P. Freichel M. Levitan I. Vogel SM. Flockerzi V. Malik AB. Tiruppathi C. Binding of STIM1, but not Orai1 to TRPC4 activates store-operated calcium entry in mouse lung endothelial cells. FASEB J. 2010;24:869–863. [Google Scholar]
- 116.Sundqvist T. Bovine aortic endothelial cells release hydrogen peroxide. J Cell Physiol. 1991;148:152–156. doi: 10.1002/jcp.1041480118. [DOI] [PubMed] [Google Scholar]
- 117.Tesfamariam B. Cohen RA. Role of superoxide anion and endothelium in vasoconstrictor action of prostaglandin endoperoxide. Am J Physiol. 1992;262:H1915–H1919. doi: 10.1152/ajpheart.1992.262.6.H1915. [DOI] [PubMed] [Google Scholar]
- 118.Thastrup O. Cullen PJ. Drobak BK. Hanley MR. Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tiruppathi C. Freichel M. Vogel SM. Paria BC. Mehta D. Flockerzi V. Malik AB. Impairment of store-operated Ca2+ entry in TRPC4−/− mice interferes with increase in lung microvascular permeability. Circ Res. 2002;91:70–76. doi: 10.1161/01.res.0000023391.40106.a8. [DOI] [PubMed] [Google Scholar]
- 120.Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- 121.Usatyuk PV. Natarajan V. Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J Biol Chem. 2004;279:11789–11797. doi: 10.1074/jbc.M311184200. [DOI] [PubMed] [Google Scholar]
- 122.Usatyuk PV. Parinandi NL. Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- 123.Vaca L. Kunze DL. Depletion of intracellular Ca2+ stores activates a Ca2+-selective channel in vascular endothelium. Am J Physiol. 1994;267:C920–C925. doi: 10.1152/ajpcell.1994.267.4.C920. [DOI] [PubMed] [Google Scholar]
- 124.Vaca L. Sinkins WG. Hu Y. Kunze DL. Schilling WP. Activation of recombinant trp by thapsigargin in Sf9 insect cells. Am J Physiol. 1994;267:C1501–C1505. doi: 10.1152/ajpcell.1994.267.5.C1501. [DOI] [PubMed] [Google Scholar]
- 125.van Rossum DB. Patterson RL. Ma HT. Gill DL. Ca2+ entry mediated by store depletion, S-nitrosylation, and TRP3 channels. Comparison of coupling and function. J Biol Chem. 2000;275:28562–28568. doi: 10.1074/jbc.M003147200. [DOI] [PubMed] [Google Scholar]
- 126.Vazquez G. Lievremont JP. St. J Bird G. Putney JW., Jr Human Trp3 forms both inositol trisphosphate receptor-dependent and receptor-independent store-operated cation channels in DT40 avian B lymphocytes. Proc Natl Acad Sci U S A. 2001;98:11777–11782. doi: 10.1073/pnas.201238198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang T. Chiang ET. Moreno-Vinasco L. Lang GD. Pendyala S. Samet JM. Geyh AS. Breysse PN. Chillrud SN. Natarajan V. Garcia JG. Particulate matter disrupts human lung endothelial barrier integrity via ROS- and p38 MAPK-dependent pathways. Am J Respir Cell Mol Biol. 2010;42:442–449. doi: 10.1165/rcmb.2008-0402OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ware LB. Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 129.Warnat J. Philipp S. Zimmer S. Flockerzi V. Cavalié A. Phenotype of a recombinant store-operated channel: highly selective permeation of Ca2+ J Physiol. 1999;518:631–638. doi: 10.1111/j.1469-7793.1999.0631p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wehage E. Eisfeld J. Heiner I. Jungling E. Zitt C. Luckhoff A. Activation of the cation channel long transient receptor potential channel 2 (LTRPC2) by hydrogen peroxide. A splice variant reveals a mode of activation independent of ADP-ribose. J Biol Chem. 2002;277:23150–23156. doi: 10.1074/jbc.M112096200. [DOI] [PubMed] [Google Scholar]
- 131.Wesson DE. Elliott SJ. Xanthine oxidase inhibits transmembrane signal transduction in vascular endothelial cells. J Pharmacol Exp Ther. 1994;270:1197–1207. [PubMed] [Google Scholar]
- 132.Wientjes FB. Segal AW. Hartwig JH. Immunoelectron microscopy shows a clustered distribution of NADPH oxidase components in the human neutrophil plasma membrane. J Leukoc Biol. 1997;61:303–312. doi: 10.1002/jlb.61.3.303. [DOI] [PubMed] [Google Scholar]
- 133.Wu S. Cioffi EA. Alvarez D. Sayner SL. Chen H. Cioffi DL. King J. Creighton JR. Townsley M. Goodman SR. Stevens T. Essential role of a Ca2+-selective, store-operated current (ISOC) in endothelial cell permeability: determinants of the vascular leak site. Circ Res. 2005;96:856–863. doi: 10.1161/01.RES.0000163632.67282.1f. [DOI] [PubMed] [Google Scholar]
- 134.Wu X. Babnigg G. Villereal ML. Functional significance of human trp1 and trp3 in store-operated Ca(2+) entry in HEK-293 cells. Am J Physiol Cell Physiol. 2000;278:C526–C536. doi: 10.1152/ajpcell.2000.278.3.C526. [DOI] [PubMed] [Google Scholar]
- 135.Xu SZ. Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ Res. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- 136.Yoshida J. Ishibashi T. Imaizumi N. Takegami T. Nishio M. Capacitative Ca2+ entries and mRNA expression for TRPC1 and TRPC5 channels in human epidermoid carcinoma A431 cells. Eur J Pharmacol. 2005;510:217–222. doi: 10.1016/j.ejphar.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 137.Yoshida T. Inoue R. Morii T. Takahashi N. Yamamoto S. Hara Y. Tominaga M. Shimizu S. Sato Y. Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 138.Zhang SL. Yu Y. Roos J. Kozak JA. Deerinck TJ. Ellisman MH. Stauderman KA. Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao G. Ayene IS. Fisher AB. Role of iron in ischemia-reperfusion oxidative injury of rat lungs. Am J Respir Cell Mol Biol. 1997;16:293–299. doi: 10.1165/ajrcmb.16.3.9070614. [DOI] [PubMed] [Google Scholar]
- 140.Zhou C. Wu S. T-type calcium channels in pulmonary vascular endothelium. Microcirculation. 2006;13:645–656. doi: 10.1080/10739680600930289. [DOI] [PubMed] [Google Scholar]
- 141.Zhou Y. Ramachandran S. Oh-Hora M. Rao A. Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci U S A. 2010;107:4896–4901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zitt C. Zobel A. Obukhov AG. Harteneck C. Kalkbrenner F. Luckhoff A. Schultz G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]
- 143.Zulueta JJ. Sawhney R. Yu FS. Cote CC. Hassoun PM. Intracellular generation of reactive oxygen species in endothelial cells exposed to anoxia-reoxygenation. Am J Physiol. 1997;272:L897–L902. doi: 10.1152/ajplung.1997.272.5.L897. [DOI] [PubMed] [Google Scholar]
- 144.Zweier JL. Broderick R. Kuppusamy P. Thompson-Gorman S. Lutty GA. Determination of the mechanism of free radical generation in human aortic endothelial cells exposed to anoxia and reoxygenation. J Biol Chem. 1994;269:24156–24162. [PubMed] [Google Scholar]
- 145.Zweier JL. Kuppusamy P. Lutty GA. Measurement of endothelial cell free radical generation: evidence for a central mechanism of free radical injury in postischemic tissues. Proc Natl Acad Sci U S A. 1988;85:4046–4050. doi: 10.1073/pnas.85.11.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zweier JL. Kuppusamy P. Thompson-Gorman S. Klunk D. Lutty GA. Measurement and characterization of free radical generation in reoxygenated human endothelial cells. Am J Physiol. 1994;266:C700–C708. doi: 10.1152/ajpcell.1994.266.3.C700. [DOI] [PubMed] [Google Scholar]
- 147.Zweifach A. Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]