

Abstract
Cardiac hypertrophy develops most commonly in response to hypertension and is an independent risk factor for the development of heart failure. The mechanisms by which cardiac hypertrophy may be reversed to reduce this risk have not been fully determined to the point where mechanism-specific therapies have been developed. Recently, proteases in the calpain family have been implicated in regulating the development of cardiac hypertrophy in preclinical animal models. In this review, we summarize the molecular mechanisms by which calpain inhibition has been shown to modulate the development of cardiac (specifically ventricular) hypertrophy. The context within which calpain inhibition might be developed for therapeutic intervention of cardiac hypertrophy is then discussed.
Keywords: cardiac hypertrophy, calpain, calpastatin, NF-κB inhibition, HSP90, β3 integrins
Introduction
Cardiac hypertrophy develops most commonly in response to hypertension and is an independent risk factor for the development of heart failure and more generally an increased morbidity and mortality1. Although the mechanisms by which cardiac hypertrophy may be reversed to reduce the increased risk have not been fully determined to the point where mechanism-specific therapies have been developed, epidemiologic studies suggest that regression of hypertrophy is a salutary clinical goal 2, 3. The increase in cardiomyocyte mass involves the increase in protein synthesis stimulated by a variety of intracellular signaling pathways 4. In parallel, changes in the rate of protein degradation occur, both increasing and decreasing depending on the hypertrophic stimuli 5, 6,7-10. Therefore, the reversal of cardiac hypertrophy therapeutically would likely involve either decreasing protein synthesis and/or increasing the rate of protein degradation. In this review, we discuss the newly discovered role that the calpain proteolytic system plays in mediating signal transduction pathways involved in cardiac ventricular hypertrophy.
Degradation of proteins in the cardiomyocyte, as in other cells, involves 3 parallel systems that function both separately and cooperatively: 1) the ubiquitin proteasome system; 2) lysosomes and the process of autophagy; and 3) the calpain proteases. The ubiquitin proteasome system (UPS) includes a series of enzymes that target specific substrate proteins for degradation by the 26S proteasome. The UPS-mediated regulation of cardiac mass has been shown to be mediated by multiple ubiquitin ligases, the components of the UPS that give it its specificity, as well as the proteasome. The ubiquitin ligases muscle ring finger-1 (MuRF1) and MAFbx (aka atrogin-1) play a role in regulating cardiac mass 11-14. There is some evidence suggesting that inhibition of the proteasome may play a role in regulating cardiac hypertrophy in vivo, at least experimentally 15. However, there is also evidence that proteasomal inhibition actually causes cardiac hypertrophy under baseline conditions and enhances the development of hypertrophy in aortic-banded animals 16, leaving the issue unclear as to whether inhibition of the proteasome in the setting of cardiac hypertrophy is protective or detrimental.
The second system involved in cardiac protein degradation involves lysosomal proteolysis. Inhibiting lysosome function in the heart results in an approximately 25-30% reduction in the overall rate of protein degradation 17. While lysosome activity does not appear to affect myosin degradation, it does play a role in the degradation of organellar proteins, including mitochondrial cytochromes and microtubules 17, 18. Autophagy, which is involved with targeted lysosomal degradation of proteins and organelles, occurs constitutively at a low level during normal cardiac function 19. However, during times of cardiac stress, autophagic activity increases, presumably as an adaptive response to the significant amount of structural remodeling that accompanies the cardiac stress response 20-22.
The third proteolytic system active in the heart is the calpain system, which includes a family of calcium-dependent, non-lysosomal cysteine proteases that are expressed ubiquitously within all cells and whose function in muscle appears to involve both atrophic and hypertrophic pathways 23, 24. Several recent publications have reported the role of calpain proteases in regulating the development of cardiac hypertrophy. These studies add numerous novel details to our understanding of how calpains, and their interactions with specific cell signaling pathways, might be involved in the complex regulation of cardiac hypertrophy. With few therapies available to regulate or reverse cardiac hypertrophy, the identification of cardiac calpains as a potential therapeutic target is exciting since a large body of work already exists describing the regulatory pathways involved in this form of proteolysis. This review gives a brief background on the role of calpains in the heart and then focuses on their role in regulating cell signaling in cardiac hypertrophy.
The calcium ion-dependent papain-like protease (Calpain) family of proteases
Members of the calpain family of intracellular Ca2+-activated proteases are critical mediators of the action of calcium. At least 16 calpains have been described, most found ubiquitously, although some being tissue specific (see recent review by Bukowska et al., 2010 25). Calpains are generally localized to the cytosol as inactive pro-enzymes that may be activated by increases in intracellular calcium. Calpains operate by processing proteins, through interactions with a limited number of motifs, to transform their activities and structure. Calpain activity is specific and does not induce widespread degradation of proteins (see Table 1). The conventional calpains, calpain 1 and 2 (also known as μ and m-calpain), are tightly regulated by an endogenous inhibitor, called calpastatin23. The four inhibitory domains of calpastatin bind reversibly to the active calpain domains to inhibit their activity. The activity of calpain is also inhibited by post-translational modification by phosphate groups23. For example, phosphorylation of Ser369 by protein kinase A (PKA) prevents the formation of the active site necessary for calpain activity 26. Calpains have been implicated in degrading a diverse array of substrates, involved in various areas of biology (see Table 1).
Table 1. Examples of calpain substrates relevant to cardiac (patho)physiology.
Many calpain substrates have been described in non-muscle systems, with the exception of sarcomere proteins. Investigation of the role of calpain in muscle initially started with the realization of their role in meat “tenderization” (sarcomere breakdown), which is an active area of food research (see recent reviews95-97).
| Substrate | References |
|---|---|
| Actin | 98, 99 |
| Amyloid precursor protein | 100-102 |
| Bax | 103-105 |
| Calcineurin | 84 |
| Caspases | 31, 106 |
| Ca2+ ATPase | 107-109 |
| Ca2+/Calmodulin-protein kinase | 110 |
| c-Fos/c-Jun | 111-113 |
| Dystrophin, Utrophin, and Spectrin | 70 |
| Estrogen receptor | 114, 115 |
| Focal adhesion kinase | 85, 86 |
| G protein (α subunit) | 116 |
| IκBα | 117, 118 |
| Integrin β3 | 119-121 |
| L-type Ca2+ calcium channel | 48-50, 122, 123 |
| p53 | 124, 125 |
| Phospholipase C (PLC) | 126, 127 |
| Protein kinase A (PKA) | 128 |
| Protein kinase C (PKC) | 129-131 |
| Ryanodine receptors | 132 |
| Tau protein | 133, 134 |
Calpains in the heart in health and disease
Both calpain 1 and calpain 2 are present in moderate amounts within the muscle where they are localized to the Z disk of muscle fibers 27, 28and have been associated with the in vitro degradation of sarcomeric proteins such as α-tropomyosin 29, 30. The majority of the studies looking at calpain activity in the heart have focused on the role of this proteolytic system in response to pathological cardiac conditions, such as post-ischemic cardiac injury 31-33. However, at least one study has examined the role of the calpains in baseline cardiac function. In cultured cardiomyocytes, calpain 1, but not calpain 2, is found to be active at physiological levels of calcium, resulting in the proteolysis of specific substrates (e.g. desmin and protein kinase Cα) as well as increased protein ubiquitination and protein turnover by the 26S proteasome 34. Mice in which the calpain inhibitor calpastatin is ectopically expressed at increased levels in the heart exhibit a decrease in ubiquitination of some specific cardiac proteins, but no overall change in cardiac protein ubiquitination, suggesting that the effect of calpain 1 (the only calpain moiety affected by the overexpression of calpastatin) is on the actual ubiquitination step and not on 26S proteasome activity34. Most interestingly, however, is the finding that inhibition of calpain 1 activity by forced expression of calpastatin results in a progressive, dilated cardiomyopathy that is accompanied by an accumulation of aggregated protein complexes, formation of autophagasomes, and destruction of sarcomere integrity 34. Together, these findings suggest that calpain 1 activity is essential for normal cardiac function and is integral to the regulation of protein turnover of specific cardiac proteins (the identity of which have not yet been confirmed) whose accumulation leads to disruption of normal myofibril activity and subsequent cardiomyopathy. More broadly, calpains have been implicated in cell cycle (calpain 2), contraction, apoptosis (calpain 2), cell migration (calpain 1), cell differentiation (calpain 2) and cellular signal transduction in muscle (calpain 3) 35-40.
The involvement of calpain activity in the progression of cardiac pathologies is well known 41. Calpain activity mediates alterations in sarcomere structure and affects contractile dysfunction in ischemia reperfusion injury (calpains 1 and 2), myocardial stunning (calpain 1) and atrial fibrillation (calpain 1) 25, 32, 42-47. One mechanism by which calpain 1 activity may be linked to atrial fibrillation is through its cleavage of specific L-type Ca2+ channel proteins, leading to the disruption of the excitation-contraction coupling mediated by Ca2+ channels 48-50. Likewise, calpain activation in reperfusion following ischemic insult has been proposed as one possible mechanism mediating myocyte cell death, either by activation of apoptosis via Bid and/or necrosis by increasing fragility due to degradation of sarcomeric proteins (recently reviewed by Inserte et al., 2009 51). In isolated rabbit hearts exposed to global ischemic injury, calpain activity cleaves Bid, resulting in the enhanced release of cytochrome c from mitochondria leading to apoptosis 52. There is also evidence for calpain involvement in congestive heart failure as well as in the atrophic remodeling that accompanies cardiac unloading. Ventricular tissue isolated from congestive heart failure patients exhibits a marked increase in calpain expression 53. In milder cases of congestive heart failure (rated as class II on the New York Heart Association (NYHA) scale) an increase in the protein level of calpain 1, but not calpain 2 is observed. However, when heart failure progresses to a more severe level (NYHA III and IV) a significant increase in the protein levels of both calpains is seen. Mechanical unloading of the failing human heart results in a slight increase in calpain 1 expression and a significant increase in expression of calpain 2 54. Likewise, in the unloaded rat heart, both calpain 1 and 2 expression and activity levels are increased 54, providing further evidence of calpain’s involvement in the tissue remodeling associated with various cardiac pathologies.
Calpain regulation of signaling in cardiac hypertrophy
The development of pathologic cardiac hypertrophy, such as that induced by pressure overload, occurs in response to the stimulation of multiple signaling pathways that in turn activate a handful of transcription factors to activate pro-hypertrophic gene expression programs (see recent reviews4, 55, 56). Despite the complexity of these signaling pathways, only a relatively few number of transcription factors have been shown to drive this process, including NF-κB, GATA4, NFAT, SRF, and MEF2 55-59. The signaling pathways driven by these transcription factors facilitate hypertrophic growth of cardiomyocytes and activate so-called “fetal genes”. The concept of re-expressing genes normally expressed only during the fetal period of heart development is well established during the development of cardiac hypertrophy 4, 55. Briefly, the activation of transcription factors such as SRF and GATA4 induce specific gene expression and protein synthesis globally. In this respect, a number of influential signaling pathways have been identified as important mediators of cardiomyocyte hypertrophy. These pathways include the angiotensin II-induced NF-κB /NFAT pathway, the Akt signaling pathway, and the stretch-induced (β3 integrin-mediated) signaling pathways 4. Interestingly, recent studies have implicated calpain in regulating cardiac hypertrophy by its specific interaction through each of these signaling pathways.
Blocking calpain activity disrupts cardiac hypertrophy by inhibiting NF-κB activation
Angiotensin II (Ang II), a key component of the rennin-angiotensin-aldosterone system, induces cardiomyocyte hypertrophy by interacting with the angiotensin II type I receptor, a G protein coupled receptor. Chronic infusion of Ang II in mice results in the development of hypertension and cardiac hypertrophy 60. In parallel, increases in calpain activity and decreases in calpastatin (the endogenous inhibitor of calpain), expression are induced. In transgenic mice that constitutively express calpastatin, the chronic infusion of Ang II fails to induce cardiac hypertrophy, although these mice do still develop hypertension. Both Ang II and calpain 1 signaling activate the NF-κB 61-63 and calcineurin/NFAT signaling pathways 64, 65. Infusion of Ang II leads to a robust increase in expression of the p65 subunit of NF-κB in the nuclei of cardiomyocytes (indicating enhanced activity), an effect that is considerably blunted in calpastatin transgenic mice 60. Surprisingly, Ang II infusion induces equal amounts of NFAT activation in calpastatin transgenic mice and wild-type mice. Together, these results suggest that calpain 1 activity mediates Ang II-induced cardiac hypertrophy via a NFAT-independent, NF-κB-dependent pathway 60.
The mechanism by which Ang II activates NF-κB has been elucidated in recent studies published by Heidrich, et al., 200866. They have identified that Ang II induces calcium release after binding to the Ang II receptor via the inositol 1,4,5-triphosphate receptor (InsP3R) pathway in cardiomyocytes 66. They found that the InsP3R-dependent release of calcium, which turns on chromogranin B (CGB), leads to NF-κB activation and expression of brain natriuretic peptide, a protein whose expression is increased in cardiac hypertrophy and heart failure. It has been postulated that calpains may mediate chromogranin activation in response to increased calcium in this system67. NF-κB activation may be related to chromogranin B activation as well. The evidence for this comes from studies which demonstrate an attenuated NF-κB activity in cardiomyocytes with reduced chromogranin B expression 66. The proposed relationship of these signaling pathways is summarized in Figure 1 (highlighted in red). This figure describes the relationship between the increased Ang II, a hormone increased in most patients developing cardiac hypertrophy 68, which then activates calpain activity, resulting in enhanced downstream NF-κB activity to induce the “pro-hypertrophic” genes in cardiomyocytes.
Figure 1. Compiled schema of calpain-related signaling during the development of cardiac hypertrophy.
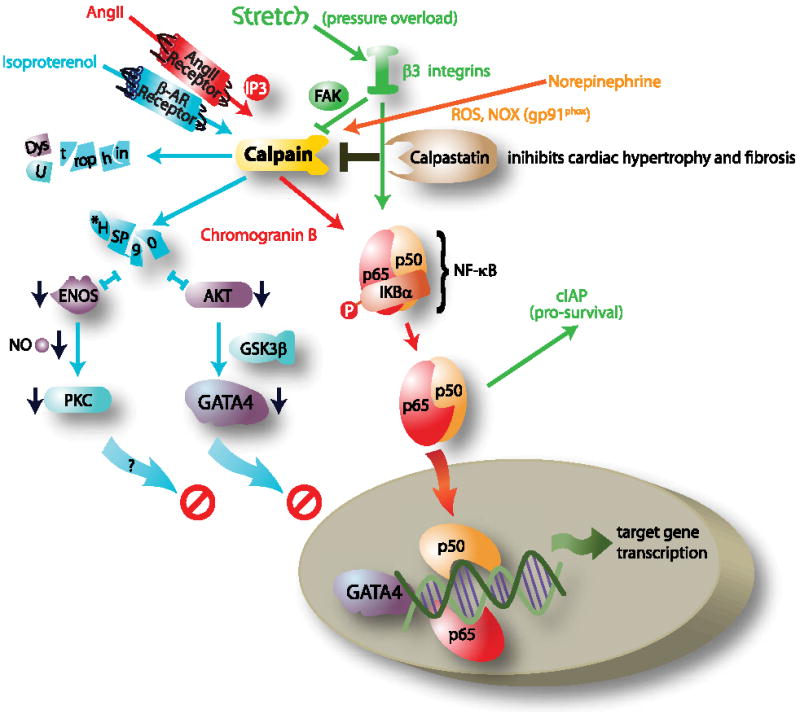
?=possible intermediates/connections not yet determined; FAK, focal adhesion kinase; HSP90, heat shock protein 90; GSK3β, glycogen synthase kinase 3 beta; GATA4, GATA binding protein 4; p65/p50, NF-κB heterodimer; NOX, NADPH oxidase; Ang II, angiotensin II. *indicates mechanisms identified in non-cardiomyocyte systems.
β-adrenergic stimulation of calpain activity blocks eNOS and Akt signaling
In addition to calpain 1’s role in activating cardiac hypertrophy by activating NF-κB, calpain activity can also lead to the inhibition of pro-hypertrophic signaling pathways. During the development of cardiac hypertrophy in humans, β-adrenergic stimulation occurs in parallel to stimulation by other G protein coupled receptors, like the Ang II receptor 69. β-adrenergic stimulation has recently been shown to activate calpains and block eNOS activity and Akt signaling, both of which have been implicated as “pro-hypertrophic” signaling pathways 70. Experimentally, cardiomyocyte stimulation with the β-adrenergic agonist isoproterenol increases calpain activity while decreasing the activity of calpastatin 71. Cardiac hypertrophy induced by chronic isoproterenol administration in ovariectomized female rats leads to calpain-mediated breakdown of the sarcomere, as evidenced by a decrease in the calpain substrate sarcomeric proteins dystrophin, utrophin, and spectrin protein expression (see Figure 1, pathways in blue) 70. In addition, a marked reduction in eNOS activity, a parallel decrease in HSP90 protein levels, and increase in caveolin 3 proteins levels were seen. Decreased Akt phosphorylation and increased glycogen synthase kinase 3β phosphorylation are also seen with chronic β-adrenergic stimulation. Although this study did not go so far as identifying the link between increased calpain and decreased eNOS and Akt activity, it is possible that HSP90 may be the commonality between these 2 effects, as described below.
The interaction of HSP90 with eNOS and Akt enhances their activity72, 73. Experimentally, calpain 2 degrades HSP90 in culture 74, 75. Evidence for the link between calpain and decreased Akt and eNOS activity comes from experiments performed in endothelial cells. Calpain inhibition in pulmonary artery endothelial cells leads to increased eNOS activity and nitric oxide production72, likely through the HSP90-mediated enhancement of eNOS activity 76, 77. Similar to the effects on eNOS, the HSP90/Akt complex formation is critical to Akt activity/phosphorylation 76. Calpain also inhibits Akt activity in diaphragmatic muscle by reducing HSP90 expression and decreasing Akt activity, an effect that coincides with reduced HSP90/Akt complex formation 73. Finally, isoproterenol administration in rats decreases HSP90 and eNOS activity in the heart at the same time Akt signaling is inhibited 70. Since both Akt-GATA4 and PKC activation are crucial to the development of cardiac hypertrophy experimentally 55, calpain’s destabilization of HSP90 may be one mechanism which inhibits pro-hypertrophic signaling pathways in cardiomyocytes. The proposed mechanisms by which isoproterenol activates calpain to inhibit downstream eNOS and Akt signaling through its disruption of HSP90 is proposed in Figure 1 (highlighted in blue).
β3 integrins induce calpain activity to enhance cell survival and induce cardiac hypertrophy
In the previous sections, we’ve discussed how Ang II stimulates calpain activity to enhance pro-hypertrophic signaling pathways via NF-κB and how β-adrenergic stimulation induces calpain activity to decrease eNOS/Akt stimulation, possibly inhibiting hypertrophic signaling. In addition to these roles for calpain activity in the development of cardiac hypertrophy, recent studies have reported that β3 integrin-dependent calpain 1 activation inhibits apoptosis in cardiomyocytes which in turn leads to the development of cardiac hypertrophy. A major mechanism by which mechanical forces activate cardiac hypertrophy is through integrins 78. Integrins are a class of receptors that extend through the plasma membrane and connect the intracellular sarcomere to the extracellular matrix 79. These receptors are located at specific sites in the plasma membrane: the intercalated discs and costameres. These receptors detect mechanical stress and act as initiators of downstream signaling through a number of signaling pathways including focal adhesion kinase 79. Recent studies have implicated integrin signaling in calpain activation and the development of cardiac hypertrophy. β3 integrin -/- mice subjected to trans-aortic constriction for four weeks to induce pressure overload cardiac hypertrophy exhibit both an increase in cardiomyocyte cell death (by TUNEL assay) and a decrease in ventricular mass compared to wild type control mice 78. Pressure overload in β3 integrin -/- mice also leads to an enrichment of calpain 1 in cardiac muscle 80, whereas pretreatment with calpeptin, a specific inhibitor of calpain, before pressure overload induction in β3 integrin -/- mice attenuates the enhanced cell death as determined by TUNEL staining 80. Although the role of calpain1 in β3 integrin-mediated cardiac hypertrophy has not been definitively determined, it is possible that it serves a regulatory function to balance the processes of cell survival and cell death80. In cultured cardiomyocytes, β3 integrin stimulation induces both calpain activity and NF-κB (independent of calpain activation NF-κB). This in turn leads to NF-κB-mediated enhancement of expression of the pro-survival factor cIAP 78. The absence of these pro-survival signals (and therefore the unabated pro-apoptotic influence of calpain activation) in the β3 integrin -/- mice may account for the enhanced cardiomyocyte apoptosis seen during pressure overload hypertrophy in these mice. Although much of this pathway still needs to be elucidated, these studies demonstrate a link between the mechanically-induced β3 integrins, calpain 1 activity, and the maintenance of cell survival, possibly involving the pro-hypertrophic cIAP and NF-κB signaling as summarized in Figure 1 (highlighted in green).
Calpains broadly consolidate stress signaling to induce cardiac hypertrophy
During the development of cardiac hypertrophy, calpain activities are enhanced by numerous stimuli, suggesting that calpain activation may represent a general mechanism by which the cell responds to external stress, including stress hormones (norepinephrine, AngII) and stretch (via β-integrins), as discussed above. Another way calpain activity influences cardiac hypertrophy by responding to external stress is by activation via reactive oxygen species. NADPH oxidases (NOXs) are membrane-bound enzymes found in the plasma membrane that function to generate superoxide by transferring electrons from NADPH to molecular oxygen to produce superoxide, a reactive free radical. Recent studies have shown that stimulating adult rat ventricular cardiomyocytes via norepinephrine increases NADPH oxidase (NOX) activity and reactive oxygen species (ROS) generation, leading to enhanced calpain 1 activation and apoptosis 81. Inhibiting the predominant NOX in cardiomyocytes, gp91phox-NADPH oxidase, using apocynin or diphenyleneiodonium, or inhibiting ROS using the antioxidant N-acetyl-cysteine protects cardiomyocytes from apoptosis at the same time as preventing the activation of calpain 1 81. Similarly, direct inhibition of calpain prevents cardiomyocyte apoptosis, presumably by blocking the norepinephrine-induced calpain activation that is mediated by NADPH-oxidase 81. These studies indicate a central role of calpains that intersect with numerous diverse stress signaling pathways to activate cardiac hypertrophy (see Figure 1, orange).
The role of calpains in protein degradation in cardiac hypertrophy
Many of the calpain substrate proteins listed in Table 1 play an important role in cardiac function, raising the obvious question of what would calpain degradation of these proteins mean for cardiac health? For example, the ability of calpains to degrade focal adhesion kinase, calcineurin, and caspases is striking given the prominent role these proteins play in cardiac hypertrophy. Focal adhesion kinase is a broadly expressed tyrosine kinase that detects biomechanical stress and then signals to induce cardiac hypertrophy79, 82 Subsequent calpain activation caused by this cardiac hypertrophy, could result in the degradation of focal adhesion kinase (or calcineurin, another purported calpain substrate) thereby explaining, in part, the inhibitory effect that calpain activation has on cardiac hypertrophy development 82, 83. Alternatively, if increased calpain activity enhances the degradation of caspases in cardiac hypertrophy, protection against cell death and development of cardiac hypertrophy might occur, confounding our understanding of how degradation of these reported calpain substrates might effect cardiac hypertrophy. In addition, many of the proteins listed in Table 1 (for example calcineurin, caspases, and G protein α subunit) were identified as calpain degradative targets in the brain 84,85, 86. Therefore, with the notable exception of calpain-mediated degradation of caspases, dystrophin, utrophin, spectrin and the L-type Ca2+ channels48-50 discussed in previous sections, the role of calpain degradation of known structural and signal transduction pathways in the heart has yet to be determined.
Calpastatin in cardiac health and disease
Although this review focuses mainly on the role that calpains play in the regulation of cardiac ventricular hypertrophy, a brief discussion on the role that the endogenous inhibitor of calpain, calpastatin, plays in physiological and pathological cardiac function is warranted. The regulation of calpastatin has been reported in experimental myocardial infarction and cardiac ischemia reperfusion injury (see Table 2)87, 88. In the left ventricular free wall, calpastatin protein levels are not affected days 1, 3, 7, and 14 after myocardial infarction in Wistar rats 87. Other studies have identified that ischemia reperfusion injury causes a down-regulation of calpastatin activity. When hearts from Wistar rats are perfused ex vivo and challenged with a 20 minute global ischemia, followed by reperfusion for up to 30 minutes, calpastatin activity was reduced 40-60% when assayed for their ability to inhibit calpain 1 and calpain 2 88. Parallel decreases in protein levels of calpastatin were also identified after reperfusion (summarized in Table 2) 88.
Table 2. Regulation of calpain and calpastatin activity and expression in cardiac disease.
| Cardiac Disease | Calpain Response | Calpastatin Response |
|---|---|---|
| Myocardial Infarction | N.D. | LV Free wall: Protein levels unaffected 1, 3, 7, 14 days after MI (Wistar Rats) 87 |
|
| ||
| Ischemia Ischemia/Reperfusion Injury |
I: m-calpain translocates to the membrane m-calpain not activated with ischemia alone 42 I/R: m-calpain translocates to the membrane m-calpain activates in reperfusion 42 |
Global I/R (20 min I/30 Min R): Calpastatin activity reduced ~40-60% 88 Protein levels reduced after reperfusion 88 Calpastatin protein levels decrease after I/R, but not after ischemia alone 42 |
|
| ||
| Congestive heart failure | MI induced heart failure: Calpain 1 and calpain 2 increased in viable LV muscle and RV muscle at 2 and 8 weeks. Calpain activities also increased 135 NYHA Class II: Increased calpain 1 protein levels. Calpain 2 levels not affected 54 NYHA Class III and IV: Increased Calpain 1 and Calpain 2 protein levels 54 Increased calpain 1 and calpain 2 protein expression 53 |
MI induced heart failure: calpastatin protein levels and activity not changed at 2 and 8 weeks after MI 135 |
|
| ||
| Atrophy associated with mechanical unloading | Unloaded (transplanted) heart: Calpain 1 and 2 protein expression and activity levels increased 54 | N.D. N.D. |
N.D. not determined.
A number of studies have been published detailing the effect of calpastatin overexpression, both systemically and specifically within the heart 34, 60, 89, 90. Since the methods used and the parameters evaluated differed between the various studies, it is difficult to get a clear idea of the effect of overexpression of calpastatin on cardiac function. For example, when calpastatin is overexpressed in all tissues, the baseline cardiac functions (as determined by heart rate, heart work, and rate of contraction and relaxation) do not differ from wild type mice 89. Likewise, no difference was seen between wild type and transgenic animals in relation to cardiac calpain activity (measured by the accumulation of 145/150-kDa spectrin BDP) or calpain 1 and calpain 2 expression 60. In unloaded hearts of mice overexpressing calpastatin cardiomyocyte size also decreases, suggesting that other proteolytic systems may compensate for calpain activity 54. However, when calpastatin is overexpressed specifically in the heart, a much different picture is seen. Mice in which cardiac calpastatin is increased such that myocardial calpain 1 activity is inhibited by 58% exhibit a slowly progressive dilated cardiomyopathy, illustrated by decreased ventricular ejection performance and responsiveness to ß-adrenergic stimulation 34. In addition, approximately half of the transgenic mice evaluated display atrial arrhythmias. Despite the difference in baseline phenotype of the systemic and cardiac-specific calpastatin mice, there is a common finding of decreased cardiac pathology in both types of transgenic mice when the mice are challenged with pathological stimuli. Mice in which calpastatin is systemically overexpressed, exhibit a decrease in the development of Ang II-induced cardiac hypertrophy and subsequent cardiac dysfunction when compared to wild type mice 60. Similarly, isolated rat hearts in which calpastatin is overexpressed (via adenoviral transfection) exhibit a significant decrease pathology associated with I/R injury, as evidenced by greater left ventricular functional recovery and a decrease in degraded cardiac troponin I levels (a target of calpain degradation)90.
Calpain inhibition as a therapeutic tool to treat cardiac hypertrophy
To date, only a handful of studies have been published examining the potential of calpain inhibition as a therapeutic approach for treatment of ventricular hypertrophy. In a feline model of right ventricular pressure overload, the calpain inhibitor calpeptin was administered intravenously both before and during the development of pressure overload 91. Control animals exhibited numerous physiological and pathological changes following 24 hours of pressure overload, including an increase in calpain protein expression and activity, a decrease in calpastatin levels, an increase in caspase-3 activation and an increase in cellular markers of programmed cell death in cardiomyocytes. In contrast, the animals that had been treated with calpeptin did not develop any of these changes, strongly suggesting the involvement of the calpain system in these cellular responses to the pressure overload as well as demonstrating a promising effect of calpain inhibition in the whole animal. Likewise, anesthetized, open-chested pigs, treated with the calpain inhibitor MDL-28170 before the induction of right ventricular pressure overload, exhibited a significant degree of protection from the development of right ventricular wall dysfunction compared to animals that were not treated with the calpain inhibitor 92. Lastly, rats treated with isoproterenol to induce ventricular hypertrophy exhibited a mild protection from hypertrophic changes when dosed with the cysteine protease inhibitor E-64c I hour prior to the treatment with isoproterenol, suggesting that calpain inhibition is effective in decreasing the effects of β-adrenergic-mediated cardiac hypertrophy 93. Although these studies hint at the possible effectiveness of calpain inhibition in the development of ventricular hypertrophy, the safety and long-term effects of calpain inhibition remains to be determined.
Summary
The studies reviewed here largely demonstrate that inhibiting calpain activity during the induction of cardiac hypertrophy attenuates or prevents the development of hypertrophy, suggesting that calpains may be a novel target for treating cardiac hypertrophy. A number of issues remain to be answered, however, if calpain is to be developed as a therapeutic target. Most importantly, it needs to be determined if inhibiting calpain has any long-term side effects in the heart. The pre-clinical studies reported so far do not look at long term outcomes of animals in which calpain inhibition prevents cardiac hypertrophy. Secondly, it needs to be determined if inhibiting calpain activity in established pressure overload-induced cardiac hypertrophy can reverse it enough to reduce the associated progression to heart failure and/or reduce the associated morbidity and mortality. Lastly, how does calpain inhibition affect other organ systems in both animals and humans that would undoubtedly be affected by a systemic anti-calpain approach. These questions are of primary importance given the array of calpain substrates found in the heart that have obvious relevance to cardiac health and disease (see Table 1). If calpain inhibition proves to be a viable target for cardiac therapies, studies have shown that calpains have a number of chemical qualities which make theoretically good targets for which synthetic inhibitors can be developed from a medicinal chemistry point of view94.
The recent studies described in this review demonstrate that calpain enzymes are emerging as unique entities within the protease systems active in the heart in that they appear to be able to respond to global stresses. As described above, calpains are capable of both activating and inhibiting signal transduction pathways involved in common hypertrophic responses to diverse external stimuli, including reactive oxygen species, stretch stimuli through β-integrins, and broadly through activation by G-protein coupled receptors such as Ang II and the β-adrenergic receptor (summarized in Figure 1). In addition, calpain activation mediates both pro- and anti-hypertrophic effects through NF-κB and eNOS/Akt signaling, respectively, although the contribution of each of these mechanisms in cardiac hypertrophy is not entirely worked out. Given the complexity of the multiple signal transduction pathways activated during cardiac hypertrophy, there are likely other pathways affected by calpain activation that have not been determined.
Acknowledgments
Sources of funding The authors are supported by the National Heart, Lung, and Blood Institute (R01HL065619 to Dr Patterson, R01HL104129 to Dr. Willis).
Non-standard abbreviations
- Akt
serine/threonine protein kinase
- Ang II
angiotensin II
- cAMP
cyclic adenosine monophosphate
- cGMP
cyclic guanosine monophosphate
- eNOS
endothelial nitric oxide synthase
- GSK3β
glycogen synthase kinase 3 beta
- InsP3R
inositol 1,4,5-triphosphate receptor
- NOX
NADPH-oxidases
- PKA
protein kinase A
- PKC
protein kinase C
- NFAT
Nuclear factor of activated T cells
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- ROS
reactive oxygen species
- TAC
trans-aortic constriction
Footnotes
Disclosures None.
References
- 1.Katz AM. Cardiomyopathy of overload. A major determinant of prognosis in congestive heart failure. N Engl J Med. 1990;322:100–110. doi: 10.1056/NEJM199001113220206. [DOI] [PubMed] [Google Scholar]
- 2.Verdecchia P, Angeli F, Borgioni C, Gattobigio R, de Simone G, Devereux RB, Porcellati C. Changes in cardiovascular risk by reduction of left ventricular mass in hypertension: a meta-analysis. Am J Hypertens. 2003;16:895–899. doi: 10.1016/s0895-7061(03)01018-5. [DOI] [PubMed] [Google Scholar]
- 3.Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, Reboldi G, Porcellati C. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation. 1998;97:48–54. doi: 10.1161/01.cir.97.1.48. [DOI] [PubMed] [Google Scholar]
- 4.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 5.Magid NM, Borer JS, Young MS, Wallerson DC, DeMonteiro C. Suppression of protein degradation in progressive cardiac hypertrophy of chronic aortic regurgitation. Circulation. 1993;87:1249–1257. doi: 10.1161/01.cir.87.4.1249. [DOI] [PubMed] [Google Scholar]
- 6.King RK, Magid NM, Opio G, Borer JS. Protein turnover in compensated chronic aortic regurgitation. Cardiology. 1997;88:518–525. doi: 10.1159/000177402. [DOI] [PubMed] [Google Scholar]
- 7.Parmacek MS, Magid NM, Lesch M, Decker RS, Samarel AM. Cardiac protein synthesis and degradation during thyroxine-induced left ventricular hypertrophy. Am J Physiol. 1986;251:C727–736. doi: 10.1152/ajpcell.1986.251.5.C727. [DOI] [PubMed] [Google Scholar]
- 8.Coleman PS, Parmacek MS, Lesch M, Samarel AM. Protein synthesis and degradation during regression of thyroxine-induced cardiac hypertrophy. J Mol Cell Cardiol. 1989;21:911–925. doi: 10.1016/0022-2828(89)90759-1. [DOI] [PubMed] [Google Scholar]
- 9.Drews O, Tsukamoto O, Liem D, Streicher J, Wang Y, Ping P. Differential regulation of proteasome function in isoproterenol-induced cardiac hypertrophy. Circ Res. 2010;107:1094–1101. doi: 10.1161/CIRCRESAHA.110.222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Razeghi P, Baskin KK, Sharma S, Young ME, Stepkowski S, Essop MF, Taegtmeyer H. Atrophy, hypertrophy, and hypoxemia induce transcriptional regulators of the ubiquitin proteasome system in the rat heart. Biochem Biophys Res Commun. 2006;342:361–364. doi: 10.1016/j.bbrc.2006.01.163. [DOI] [PubMed] [Google Scholar]
- 11.Willis MS, Rojas M, Li L, Selzman CH, Tang RH, Stansfield WE, Rodriguez JE, Glass DJ, Patterson C. Muscle ring finger 1 mediates cardiac atrophy in vivo. Am J Physiol Heart Circ Physiol. 2009;296:H997–H1006. doi: 10.1152/ajpheart.00660.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, Patterson C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hedhli N, Depre C. Proteasome inhibitors and cardiac cell growth. Cardiovasc Res. 2010;85:321–329. doi: 10.1093/cvr/cvp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang M, Li J, Huang W, Su H, Liang Q, Tian Z, Horak KM, Molkentin JD, Wang X. Proteasome functional insufficiency activates the calcineurin-NFAT pathway in cardiomyocytes and promotes maladaptive remodelling of stressed mouse hearts. Cardiovasc Res. 2010;88:424–433. doi: 10.1093/cvr/cvq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wildenthal K, Crie JS, Ord JM, Wakeland JR. The role of lysosomes and microtubules in cardiac protein degradation. Adv Myocardiol. 1985;5:137–144. doi: 10.1007/978-1-4757-1287-2_10. [DOI] [PubMed] [Google Scholar]
- 18.Wildenthal K, Wakeland JR, Ord JM, Stull JT. Interference with lysosomal proteolysis fails to reduce cardiac myosin degradation. Biochem Biophys Res Commun. 1980;96:793–798. doi: 10.1016/0006-291x(80)91424-2. [DOI] [PubMed] [Google Scholar]
- 19.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 20.Yitzhaki S, Huang C, Liu W, Lee Y, Gustafsson AB, Mentzer RM, Jr, Gottlieb RA. Autophagy is required for preconditioning by the adenosine A1 receptor-selective agonist CCPA. Basic Res Cardiol. 2009;104:157–167. doi: 10.1007/s00395-009-0006-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rothermel BA, Hill JA. Autophagy in load-induced heart disease. Circ Res. 2008;103:1363–1369. doi: 10.1161/CIRCRESAHA.108.186551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 24.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 25.Bukowska A, Lendeckel U, Bode-Boger SM, Goette A. Physiologic and Pathophysiologic Role of Calpain: Implications for the Occurrence of Atrial Fibrillation. Cardiovasc Ther. 2010 doi: 10.1111/j.1755-5922.2010.00245.x. [DOI] [PubMed] [Google Scholar]
- 26.Shiraha H, Glading A, Chou J, Jia Z, Wells A. Activation of m-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of m-calpain. Mol Cell Biol. 2002;22:2716–2727. doi: 10.1128/MCB.22.8.2716-2727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumamoto T, Kleese WC, Cong JY, Goll DE, Pierce PR, Allen RE. Localization of the Ca(2+)-dependent proteinases and their inhibitor in normal, fasted, and denervated rat skeletal muscle. Anat Rec. 1992;232:60–77. doi: 10.1002/ar.1092320108. [DOI] [PubMed] [Google Scholar]
- 28.Dayton WR, Schollmeyer JV. Immunocytochemical localization of a calcium-activated protease in skeletal muscle cells. Exp Cell Res. 1981;136:423–433. doi: 10.1016/0014-4827(81)90022-7. [DOI] [PubMed] [Google Scholar]
- 29.Azanza JL, Raymond J, Robin JM, Cottin P, Ducastaing A. Purification and some physico-chemical and enzymic properties of a calcium ion-activated neutral proteinase from rabbit skeletal muscle. Biochem J. 1979;183:339–347. doi: 10.1042/bj1830339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohrhauser DA, Underwood KR, Weaver AD. In vitro degradation of bovine myofibrils is caused by {micro}-calpain, not caspase-3. J Anim Sci. 2010 doi: 10.2527/jas.2010-3149. [DOI] [PubMed] [Google Scholar]
- 31.Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 32.Gao WD, Atar D, Liu Y, Perez NG, Murphy AM, Marban E. Role of troponin I proteolysis in the pathogenesis of stunned myocardium. Circ Res. 1997;80:393–399. [PubMed] [Google Scholar]
- 33.Sandmann S, Prenzel F, Shaw L, Schauer R, Unger T. Activity profile of calpains I and II in chronically infarcted rat myocardium--influence of the calpain inhibitor CAL 9961. Br J Pharmacol. 2002;135:1951–1958. doi: 10.1038/sj.bjp.0704661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, Robbins J, Lynch RA, Marreez Y, Dorn GW., 2nd Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res. 2007;100:1071–1078. doi: 10.1161/01.RES.0000261938.28365.11. [DOI] [PubMed] [Google Scholar]
- 35.Dargelos E, Poussard S, Brule C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90:359–368. doi: 10.1016/j.biochi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki K, Hata S, Kawabata Y, Sorimachi H. Structure, activation, and biology of calpain. Diabetes. 2004;53(Suppl 1):S12–18. doi: 10.2337/diabetes.53.2007.s12. [DOI] [PubMed] [Google Scholar]
- 37.Sato K, Kawashima S. Calpain function in the modulation of signal transduction molecules. Biol Chem. 2001;382:743–751. doi: 10.1515/BC.2001.090. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y, Wang KK. The calpain family and human disease. Trends Mol Med. 2001;7:355–362. doi: 10.1016/s1471-4914(01)02049-4. [DOI] [PubMed] [Google Scholar]
- 39.Carafoli E, Molinari M. Calpain: a protease in search of a function? Biochem Biophys Res Commun. 1998;247:193–203. doi: 10.1006/bbrc.1998.8378. [DOI] [PubMed] [Google Scholar]
- 40.Sorimachi H, Ishiura S, Suzuki K. Structure and physiological function of calpains. Biochem J. 1997;328(Pt 3):721–732. doi: 10.1042/bj3280721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh RB, Dandekar SP, Elimban V, Gupta SK, Dhalla NS. Role of proteases in the pathophysiology of cardiac disease. Mol Cell Biochem. 2004;263:241–256. doi: 10.1023/B:MCBI.0000041865.63445.40. [DOI] [PubMed] [Google Scholar]
- 42.Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D. Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion. J Mol Cell Cardiol. 2010;49:271–279. doi: 10.1016/j.yjmcc.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 43.Iwamoto H, Miura T, Okamura T, Shirakawa K, Iwatate M, Kawamura S, Tatsuno H, Ikeda Y, Matsuzaki M. Calpain inhibitor-1 reduces infarct size and DNA fragmentation of myocardium in ischemic/reperfused rat heart. J Cardiovasc Pharmacol. 1999;33:580–586. doi: 10.1097/00005344-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 44.Ke L, Qi XY, Dijkhuis AJ, Chartier D, Nattel S, Henning RH, Kampinga HH, Brundel BJ. Calpain mediates cardiac troponin degradation and contractile dysfunction in atrial fibrillation. J Mol Cell Cardiol. 2008;45:685–693. doi: 10.1016/j.yjmcc.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 45.McDonough JL, Arrell DK, Van Eyk JE. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ Res. 1999;84:9–20. doi: 10.1161/01.res.84.1.9. [DOI] [PubMed] [Google Scholar]
- 46.Van Eyk JE, Powers F, Law W, Larue C, Hodges RS, Solaro RJ. Breakdown and release of myofilament proteins during ischemia and ischemia/reperfusion in rat hearts: identification of degradation products and effects on the pCa-force relation. Circ Res. 1998;82:261–271. doi: 10.1161/01.res.82.2.261. [DOI] [PubMed] [Google Scholar]
- 47.Gao WD, Liu Y, Mellgren R, Marban E. Intrinsic myofilament alterations underlying the decreased contractility of stunned myocardium. A consequence of Ca2+-dependent proteolysis? Circ Res. 1996;78:455–465. doi: 10.1161/01.res.78.3.455. [DOI] [PubMed] [Google Scholar]
- 48.Saud ZA, Minobe E, Wang WY, Han DY, Horiuchi M, Hao LY, Kameyama M. Calpastatin binds to a calmodulin-binding site of cardiac Cav1.2 Ca2+ channels. Biochem Biophys Res Commun. 2007;364:372–377. doi: 10.1016/j.bbrc.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 49.Hao LY, Kameyama A, Kuroki S, Takano J, Takano E, Maki M, Kameyama M. Calpastatin domain L is involved in the regulation of L-type Ca2+ channels in guinea pig cardiac myocytes. Biochem Biophys Res Commun. 2000;279:756–761. doi: 10.1006/bbrc.2000.4040. [DOI] [PubMed] [Google Scholar]
- 50.Kameyama M, Kameyama A, Takano E, Maki M. Run-down of the cardiac L-type Ca2+ channel: partial restoration of channel activity in cell-free patches by calpastatin. Pflugers Arch. 1998;435:344–349. doi: 10.1007/s004240050521. [DOI] [PubMed] [Google Scholar]
- 51.Inserte J, Barrabes JA, Hernando V, Garcia-Dorado D. Orphan targets for reperfusion injury. Cardiovasc Res. 2009;83:169–178. doi: 10.1093/cvr/cvp109. [DOI] [PubMed] [Google Scholar]
- 52.Chen M, Won DJ, Krajewski S, Gottlieb RA. Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem. 2002;277:29181–29186. doi: 10.1074/jbc.M204951200. [DOI] [PubMed] [Google Scholar]
- 53.Yang D, Ma S, Tan Y, Li D, Tang B, Zhang X, Sun M, Yang Y. Increased expression of calpain and elevated activity of calcineurin in the myocardium of patients with congestive heart failure. Int J Mol Med. 2010;26:159–164. doi: 10.3892/ijmm_00000448. [DOI] [PubMed] [Google Scholar]
- 54.Razeghi P, Volpini KC, Wang ME, Youker KA, Stepkowski S, Taegtmeyer H. Mechanical unloading of the heart activates the calpain system. J Mol Cell Cardiol. 2007;42:449–452. doi: 10.1016/j.yjmcc.2006.08.114. [DOI] [PubMed] [Google Scholar]
- 55.Frey N, Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 56.Molkentin JD, Robbins J. With great power comes great responsibility: using mouse genetics to study cardiac hypertrophy and failure. J Mol Cell Cardiol. 2009;46:130–136. doi: 10.1016/j.yjmcc.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sugden PH, Fuller SJ, Weiss SC, Clerk A. Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br J Pharmacol. 2008;153(Suppl 1):S137–153. doi: 10.1038/sj.bjp.0707659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van der Heiden K, Cuhlmann S, Luong le A, Zakkar M, Evans PC. Role of nuclear factor kappaB in cardiovascular health and disease. Clin Sci (Lond) 2010;118:593–605. doi: 10.1042/CS20090557. [DOI] [PubMed] [Google Scholar]
- 59.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 60.Letavernier E, Perez J, Bellocq A, Mesnard L, de Castro Keller A, Haymann JP, Baud L. Targeting the calpain/calpastatin system as a new strategy to prevent cardiovascular remodeling in angiotensin II-induced hypertension. Circ Res. 2008;102:720–728. doi: 10.1161/CIRCRESAHA.107.160077. [DOI] [PubMed] [Google Scholar]
- 61.Shumway SD, Maki M, Miyamoto S. The PEST domain of IkappaBalpha is necessary and sufficient for in vitro degradation by mu-calpain. J Biol Chem. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- 62.Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, El-Jamali A, Dietz R, Scheidereit C, Bergmann MW. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. 2005;111:2319–2325. doi: 10.1161/01.CIR.0000164237.58200.5A. [DOI] [PubMed] [Google Scholar]
- 63.Rouet-Benzineb P, Gontero B, Dreyfus P, Lafuma C. Angiotensin II induces nuclear factor- kappa B activation in cultured neonatal rat cardiomyocytes through protein kinase C signaling pathway. J Mol Cell Cardiol. 2000;32:1767–1778. doi: 10.1006/jmcc.2000.1211. [DOI] [PubMed] [Google Scholar]
- 64.Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–7613. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burkard N, Becher J, Heindl C, Neyses L, Schuh K, Ritter O. Targeted proteolysis sustains calcineurin activation. Circulation. 2005;111:1045–1053. doi: 10.1161/01.CIR.0000156458.80515.F7. [DOI] [PubMed] [Google Scholar]
- 66.Heidrich FM, Zhang K, Estrada M, Huang Y, Giordano FJ, Ehrlich BE. Chromogranin B regulates calcium signaling, nuclear factor kappaB activity, and brain natriuretic peptide production in cardiomyocytes. Circ Res. 2008;102:1230–1238. doi: 10.1161/CIRCRESAHA.107.166033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heidrich FM, Ehrlich BE. Calcium, calpains, and cardiac hypertrophy: a new link. Circ Res. 2009;104:e19–20. doi: 10.1161/CIRCRESAHA.108.191072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cowan BR, Young AA. Left ventricular hypertrophy and renin-angiotensin system blockade. Curr Hypertens Rep. 2009;11:167–172. doi: 10.1007/s11906-009-0030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barry SP, Townsend PA. What causes a broken heart--molecular insights into heart failure. Int Rev Cell Mol Biol. 2010;284:113–179. doi: 10.1016/S1937-6448(10)84003-1. [DOI] [PubMed] [Google Scholar]
- 70.Bhuiyan MS, Shioda N, Fukunaga K. Chronic beta-AR activation-induced calpain activation and impaired eNOS-Akt signaling mediates cardiac injury in ovariectomized female rats. Expert Opin Ther Targets. 2009;13:275–286. doi: 10.1517/14728220902721312. [DOI] [PubMed] [Google Scholar]
- 71.Iizuka K, Kawaguchi H, Yasuda H. Calpain is activated by beta-adrenergic receptor stimulation under hypoxic myocardial cell injury. Jpn Circ J. 1991;55:1086–1093. doi: 10.1253/jcj.55.1086. [DOI] [PubMed] [Google Scholar]
- 72.Averna M, Stifanese R, De Tullio R, Passalacqua M, Salamino F, Pontremoli S, Melloni E. Functional role of HSP90 complexes with endothelial nitric-oxide synthase (eNOS) and calpain on nitric oxide generation in endothelial cells. J Biol Chem. 2008;283:29069–29076. doi: 10.1074/jbc.M803638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith IJ, Dodd SL. Calpain activation causes a proteasome-dependent increase in protein degradation and inhibits the Akt signalling pathway in rat diaphragm muscle. Exp Physiol. 2007;92:561–573. doi: 10.1113/expphysiol.2006.035790. [DOI] [PubMed] [Google Scholar]
- 74.Bellocq A, Doublier S, Suberville S, Perez J, Escoubet B, Fouqueray B, Puyol DR, Baud L. Somatostatin increases glucocorticoid binding and signaling in macrophages by blocking the calpain-specific cleavage of Hsp 90. J Biol Chem. 1999;274:36891–36896. doi: 10.1074/jbc.274.52.36891. [DOI] [PubMed] [Google Scholar]
- 75.Minami Y, Kimura Y, Kawasaki H, Suzuki K, Yahara I. The carboxy-terminal region of mammalian HSP90 is required for its dimerization and function in vivo. Mol Cell Biol. 1994;14:1459–1464. doi: 10.1128/mcb.14.2.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc Natl Acad Sci U S A. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 78.Johnston RK, Balasubramanian S, Kasiganesan H, Baicu CF, Zile MR, Kuppuswamy D. Beta3 integrin-mediated ubiquitination activates survival signaling during myocardial hypertrophy. FASEB J. 2009;23:2759–2771. doi: 10.1096/fj.08-127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Brancaccio M, Hirsch E, Notte A, Selvetella G, Lembo G, Tarone G. Integrin signalling: the tug-of-war in heart hypertrophy. Cardiovasc Res. 2006;70:422–433. doi: 10.1016/j.cardiores.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 80.Suryakumar G, Kasiganesan H, Balasubramanian S, Kuppuswamy D. Lack of beta3 integrin signaling contributes to calpain-mediated myocardial cell loss in pressure-overloaded myocardium. J Cardiovasc Pharmacol. 2010;55:567–573. doi: 10.1097/FJC.0b013e3181d9f5d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Y, Arnold JM, Pampillo M, Babwah AV, Peng T. Taurine prevents cardiomyocyte death by inhibiting NADPH oxidase-mediated calpain activation. Free Radic Biol Med. 2009;46:51–61. doi: 10.1016/j.freeradbiomed.2008.09.025. [DOI] [PubMed] [Google Scholar]
- 82.Franchini KG, Clemente CF, Marin TM. Focal adhesion kinase signaling in cardiac hypertrophy and failure. Braz J Med Biol Res. 2009;42:44–52. doi: 10.1590/s0100-879x2009000100008. [DOI] [PubMed] [Google Scholar]
- 83.Lal H, Verma SK, Foster DM, Golden HB, Reneau JC, Watson LE, Singh H, Dostal DE. Integrins and proximal signaling mechanisms in cardiovascular disease. Front Biosci. 2009;14:2307–2334. doi: 10.2741/3381. [DOI] [PubMed] [Google Scholar]
- 84.Lakshmikuttyamma A, Selvakumar P, Sharma AR, Anderson DH, Sharma RK. In vitro proteolytic degradation of bovine brain calcineurin by m-calpain. Neurochem Res. 2004;29:1913–1921. doi: 10.1023/b:nere.0000042218.27842.79. [DOI] [PubMed] [Google Scholar]
- 85.Yuan Y, Dopheide SM, Ivanidis C, Salem HH, Jackson SP. Calpain regulation of cytoskeletal signaling complexes in von Willebrand factor-stimulated platelets. Distinct roles for glycoprotein Ib-V-IX and glycoprotein IIb-IIIa (integrin alphaIIbbeta3) in von Willebrand factor-induced signal transduction. J Biol Chem. 1997;272:21847–21854. doi: 10.1074/jbc.272.35.21847. [DOI] [PubMed] [Google Scholar]
- 86.Cooray P, Yuan Y, Schoenwaelder SM, Mitchell CA, Salem HH, Jackson SP. Focal adhesion kinase (pp125FAK) cleavage and regulation by calpain. Biochem J. 1996;318(Pt 1):41–47. doi: 10.1042/bj3180041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sandmann S, Yu M, Unger T. Transcriptional and translational regulation of calpain in the rat heart after myocardial infarction--effects of AT(1) and AT(2) receptor antagonists and ACE inhibitor. Br J Pharmacol. 2001;132:767–777. doi: 10.1038/sj.bjp.0703860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sorimachi Y, Harada K, Saido TC, Ono T, Kawashima S, Yoshida K. Downregulation of calpastatin in rat heart after brief ischemia and reperfusion. J Biochem. 1997;122:743–748. doi: 10.1093/oxfordjournals.jbchem.a021818. [DOI] [PubMed] [Google Scholar]
- 89.Li X, Li Y, Shan L, Shen E, Chen R, Peng T. Over-expression of calpastatin inhibits calpain activation and attenuates myocardial dysfunction during endotoxaemia. Cardiovasc Res. 2009;83:72–79. doi: 10.1093/cvr/cvp100. [DOI] [PubMed] [Google Scholar]
- 90.Maekawa A, Lee JK, Nagaya T, Kamiya K, Yasui K, Horiba M, Miwa K, Uzzaman M, Maki M, Ueda Y, Kodama I. Overexpression of calpastatin by gene transfer prevents troponin I degradation and ameliorates contractile dysfunction in rat hearts subjected to ischemia/reperfusion. J Mol Cell Cardiol. 2003;35:1277–1284. doi: 10.1016/s0022-2828(03)00238-4. [DOI] [PubMed] [Google Scholar]
- 91.Mani SK, Shiraishi H, Balasubramanian S, Yamane K, Chellaiah M, Cooper G, Banik N, Zile MR, Kuppuswamy D. In vivo administration of calpeptin attenuates calpain activation and cardiomyocyte loss in pressure-overloaded feline myocardium. Am J Physiol Heart Circ Physiol. 2008;295:H314–326. doi: 10.1152/ajpheart.00085.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Greyson CR, Schwartz GG, Lu L, Ye S, Helmke S, Xu Y, Ahmad H. Calpain inhibition attenuates right ventricular contractile dysfunction after acute pressure overload. J Mol Cell Cardiol. 2008;44:59–68. doi: 10.1016/j.yjmcc.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arthur GD, Belcastro AN. A calcium stimulated cysteine protease involved in isoproterenol induced cardiac hypertrophy. Mol Cell Biochem. 1997;176:241–248. [PubMed] [Google Scholar]
- 94.Pietsch M, Chua KC, Abell AD. Calpains: attractive targets for the development of synthetic inhibitors. Curr Top Med Chem. 2010;10:270–293. doi: 10.2174/156802610790725489. [DOI] [PubMed] [Google Scholar]
- 95.Kemp CM, Sensky PL, Bardsley RG, Buttery PJ, Parr T. Tenderness--an enzymatic view. Meat Sci. 2010;84:248–256. doi: 10.1016/j.meatsci.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 96.Jiang ST. Contribution of muscle proteinases to meat tenderization. Proc Natl Sci Counc Repub China B. 1998;22:97–107. [PubMed] [Google Scholar]
- 97.Koohmaraie M. The role of Ca(2+)-dependent proteases (calpains) in post mortem proteolysis and meat tenderness. Biochimie. 1992;74:239–245. doi: 10.1016/0300-9084(92)90122-u. [DOI] [PubMed] [Google Scholar]
- 98.Villa PG, Henzel WJ, Sensenbrenner M, Henderson CE, Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J Cell Sci. 1998;111(Pt 6):713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- 99.Brown SB, Bailey K, Savill J. Actin is cleaved during constitutive apoptosis. Biochem J. 1997;323(Pt 1):233–237. doi: 10.1042/bj3230233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vaisid T, Kosower NS, Katzav A, Chapman J, Barnoy S. Calpastatin levels affect calpain activation and calpain proteolytic activity in APP transgenic mouse model of Alzheimer’s disease. Neurochem Int. 2007;51:391–397. doi: 10.1016/j.neuint.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 101.Siman R, Card JP, Davis LG. Proteolytic processing of beta-amyloid precursor by calpain I. J Neurosci. 1990;10:2400–2411. doi: 10.1523/JNEUROSCI.10-07-02400.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li QX, Evin G, Small DH, Multhaup G, Beyreuther K, Masters CL. Proteolytic processing of Alzheimer’s disease beta A4 amyloid precursor protein in human platelets. J Biol Chem. 1995;270:14140–14147. doi: 10.1074/jbc.270.23.14140. [DOI] [PubMed] [Google Scholar]
- 103.Nozaki K, Das A, Ray SK, Banik NL. Calpain inhibition attenuates intracellular changes in muscle cells in response to extracellular inflammatory stimulation. Exp Neurol. 2010;225:430–435. doi: 10.1016/j.expneurol.2010.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Thomas A, El Rouby S, Reed JC, Krajewski S, Silber R, Potmesil M, Newcomb EW. Drug-induced apoptosis in B-cell chronic lymphocytic leukemia: relationship between p53 gene mutation and bcl-2/bax proteins in drug resistance. Oncogene. 1996;12:1055–1062. [PubMed] [Google Scholar]
- 105.Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC, Newcomb EW. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 106.Chowdhury I, Tharakan B, Bhat GK. Caspases - an update. Comp Biochem Physiol B Biochem Mol Biol. 2008;151:10–27. doi: 10.1016/j.cbpb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 107.Wang KK, Roufogalis BD, Villalobo A. Further characterization of calpain-mediated proteolysis of the human erythrocyte plasma membrane Ca2+-ATPase. Arch Biochem Biophys. 1988;267:317–327. doi: 10.1016/0003-9861(88)90037-9. [DOI] [PubMed] [Google Scholar]
- 108.Salamino F, Sparatore B, Melloni E, Michetti M, Viotti PL, Pontremoli S, Carafoli E. The plasma membrane calcium pump is the preferred calpain substrate within the erythrocyte. Cell Calcium. 1994;15:28–35. doi: 10.1016/0143-4160(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 109.Molinari M, Anagli J, Carafoli E. PEST sequences do not influence substrate susceptibility to calpain proteolysis. J Biol Chem. 1995;270:2032–2035. doi: 10.1074/jbc.270.5.2032. [DOI] [PubMed] [Google Scholar]
- 110.McGinnis KM, Whitton MM, Gnegy ME, Wang KK. Calcium/calmodulin-dependent protein kinase IV is cleaved by caspase-3 and calpain in SH-SY5Y human neuroblastoma cells undergoing apoptosis. J Biol Chem. 1998;273:19993–20000. doi: 10.1074/jbc.273.32.19993. [DOI] [PubMed] [Google Scholar]
- 111.Jariel-Encontre I, Salvat C, Steff AM, Pariat M, Acquaviva C, Furstoss O, Piechaczyk M. Complex mechanisms for c-fos and c-jun degradation. Mol Biol Rep. 1997;24:51–56. doi: 10.1023/a:1006804723722. [DOI] [PubMed] [Google Scholar]
- 112.Hirai S, Kawasaki H, Yaniv M, Suzuki K. Degradation of transcription factors, c-Jun and c-Fos, by calpain. FEBS Lett. 1991;287:57–61. doi: 10.1016/0014-5793(91)80015-u. [DOI] [PubMed] [Google Scholar]
- 113.Carillo S, Pariat M, Steff AM, Roux P, Etienne-Julan M, Lorca T, Piechaczyk M. Differential sensitivity of FOS and JUN family members to calpains. Oncogene. 1994;9:1679–1689. [PubMed] [Google Scholar]
- 114.Murayama A, Fukai F, Murachi T. Action of calpain on the basic estrogen receptor molecule of porcine uterus. J Biochem. 1984;95:1697–1704. doi: 10.1093/oxfordjournals.jbchem.a134783. [DOI] [PubMed] [Google Scholar]
- 115.Shiba E, Kim S, Fujitani M, Kambayashi JI, Kawamura I, Tsujimoto S, Shimomura K, Tanji Y, Taguchi T, Kimoto Y, Izukura M, Takai SI. Possible involvement of calpain in the growth of estrogen receptor positive breast cancer cells. Anticancer Res. 1996;16:773–777. [PubMed] [Google Scholar]
- 116.Greenwood AF, Jope RS. Brain G-protein proteolysis by calpain: enhancement by lithium. Brain Res. 1994;636:320–326. doi: 10.1016/0006-8993(94)91031-6. [DOI] [PubMed] [Google Scholar]
- 117.Han Y, Weinman S, Boldogh I, Walker RK, Brasier AR. Tumor necrosis factor-alpha-inducible IkappaBalpha proteolysis mediated by cytosolic m-calpain. A mechanism parallel to the ubiquitin-proteasome pathway for nuclear factor-kappab activation. J Biol Chem. 1999;274:787–794. doi: 10.1074/jbc.274.2.787. [DOI] [PubMed] [Google Scholar]
- 118.Lin YC, Brown K, Siebenlist U. Activation of NF-kappa B requires proteolysis of the inhibitor I kappa B-alpha: signal-induced phosphorylation of I kappa B-alpha alone does not release active NF-kappa B. Proc Natl Acad Sci U S A. 1995;92:552–556. doi: 10.1073/pnas.92.2.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pfaff M, Du X, Ginsberg MH. Calpain cleavage of integrin beta cytoplasmic domains. FEBS Lett. 1999;460:17–22. doi: 10.1016/s0014-5793(99)01250-8. [DOI] [PubMed] [Google Scholar]
- 120.Meredith J, Jr, Mu Z, Saido T, Du X. Cleavage of the cytoplasmic domain of the integrin beta3 subunit during endothelial cell apoptosis. J Biol Chem. 1998;273:19525–19531. doi: 10.1074/jbc.273.31.19525. [DOI] [PubMed] [Google Scholar]
- 121.Xi X, Flevaris P, Stojanovic A, Chishti A, Phillips DR, Lam SC, Du X. Tyrosine phosphorylation of the integrin beta 3 subunit regulates beta 3 cleavage by calpain. J Biol Chem. 2006;281:29426–29430. doi: 10.1074/jbc.C600039200. [DOI] [PubMed] [Google Scholar]
- 122.Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci U S A. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.De Jongh KS, Colvin AA, Wang KK, Catterall WA. Differential proteolysis of the full-length form of the L-type calcium channel alpha 1 subunit by calpain. J Neurochem. 1994;63:1558–1564. doi: 10.1046/j.1471-4159.1994.63041558.x. [DOI] [PubMed] [Google Scholar]
- 124.Kubbutat MH, Vousden KH. Proteolytic cleavage of human p53 by calpain: a potential regulator of protein stability. Mol Cell Biol. 1997;17:460–468. doi: 10.1128/mcb.17.1.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pariat M, Carillo S, Molinari M, Salvat C, Debussche L, Bracco L, Milner J, Piechaczyk M. Proteolysis by calpains: a possible contribution to degradation of p53. Mol Cell Biol. 1997;17:2806–2815. doi: 10.1128/mcb.17.5.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Banno Y, Nakashima S, Hachiya T, Nozawa Y. Endogenous cleavage of phospholipase C-beta 3 by agonist-induced activation of calpain in human platelets. J Biol Chem. 1995;270:4318–4324. doi: 10.1074/jbc.270.9.4318. [DOI] [PubMed] [Google Scholar]
- 127.Park D, Jhon DY, Lee CW, Ryu SH, Rhee SG. Removal of the carboxyl-terminal region of phospholipase C-beta 1 by calpain abolishes activation by G alpha q. J Biol Chem. 1993;268:3710–3714. [PubMed] [Google Scholar]
- 128.Zimmerman UJ, Wang M, Nelson JB, Ekwunife FS, Liu L. Secretagogue-induced proteolysis of cAMP-dependent protein kinase in intact rat alveolar epithelial type II cells. Biochim Biophys Acta. 1996;1311:117–123. doi: 10.1016/0167-4889(95)00181-6. [DOI] [PubMed] [Google Scholar]
- 129.Kishimoto A, Mikawa K, Hashimoto K, Yasuda I, Tanaka S, Tominaga M, Kuroda T, Nishizuka Y. Limited proteolysis of protein kinase C subspecies by calcium-dependent neutral protease (calpain) J Biol Chem. 1989;264:4088–4092. [PubMed] [Google Scholar]
- 130.Dwyer LD, Miller AC, Parks AL, Jaken S, Malkinson AM. Calpain-induced downregulation of activated protein kinase C-alpha affects lung epithelial cell morphology. Am J Physiol. 1994;266:L569–576. doi: 10.1152/ajplung.1994.266.5.L569. [DOI] [PubMed] [Google Scholar]
- 131.Shea TB, Beermann ML, Griffin WR, Leli U. Degradation of protein kinase C alpha and its free catalytic subunit, protein kinase M, in intact human neuroblastoma cells and under cell-free conditions. Evidence that PKM is degraded by mM calpain-mediated proteolysis at a faster rate than PKC. FEBS Lett. 1994;350:223–229. doi: 10.1016/0014-5793(94)00769-1. [DOI] [PubMed] [Google Scholar]
- 132.Shevchenko S, Feng W, Varsanyi M, Shoshan-Barmatz V. Identification, characterization and partial purification of a thiol-protease which cleaves specifically the skeletal muscle ryanodine receptor/Ca2+ release channel. J Membr Biol. 1998;161:33–43. doi: 10.1007/s002329900312. [DOI] [PubMed] [Google Scholar]
- 133.Johnson GV. Tau phosphorylation and proteolysis: insights and perspectives. J Alzheimers Dis. 2006;9:243–250. doi: 10.3233/jad-2006-9s326. [DOI] [PubMed] [Google Scholar]
- 134.Xie HQ, Johnson GV. Calcineurin inhibition prevents calpain-mediated proteolysis of tau in differentiated PC12 cells. J Neurosci Res. 1998;53:153–164. doi: 10.1002/(SICI)1097-4547(19980715)53:2<153::AID-JNR4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 135.Takahashi M, Tanonaka K, Yoshida H, Koshimizu M, Daicho T, Oikawa R, Takeo S. Possible involvement of calpain activation in pathogenesis of chronic heart failure after acute myocardial infarction. J Cardiovasc Pharmacol. 2006;47:413–421. doi: 10.1097/01.fjc.0000210074.56614.3b. [DOI] [PubMed] [Google Scholar]